Introduction
Digitalis cardiac glycosides are well known drugs clinically used for treatment of congestive heart failure [
1]. Their action is mainly due to inhibition of Na
+,K
+-ATPase, an enzyme located in the cell membrane and promoting the outward transport of Na
+ and the inward transport of K
+ [
2]. Recently the existence of endogenous digitalis-like factors that may be responsible for essential hypertension [
3] has opened a new field in the study of compounds acting on the Na
+,K
+-ATPase. The most potent inhibitors of Na
+,K
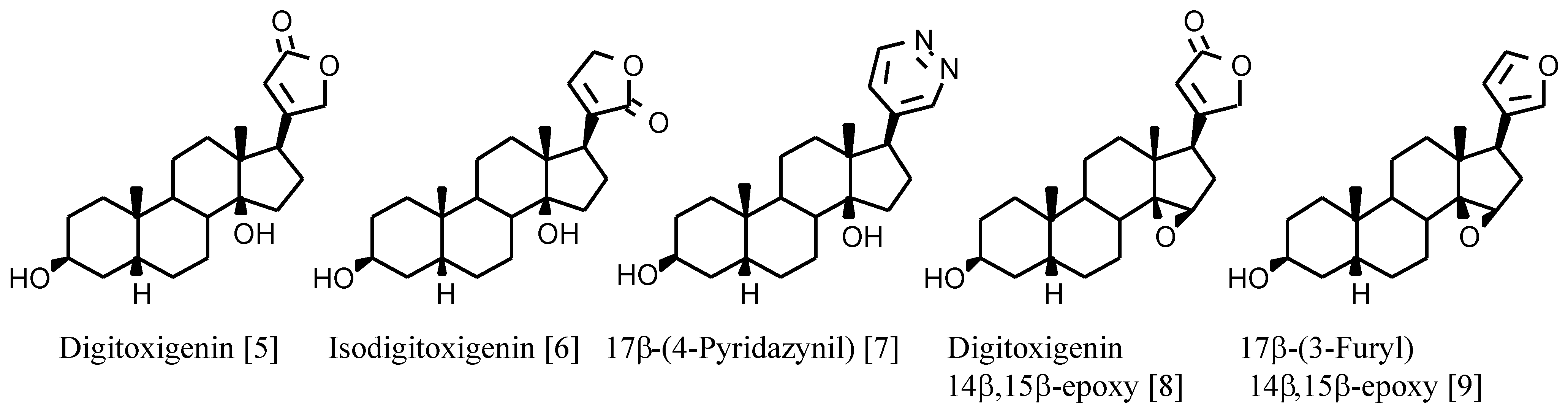
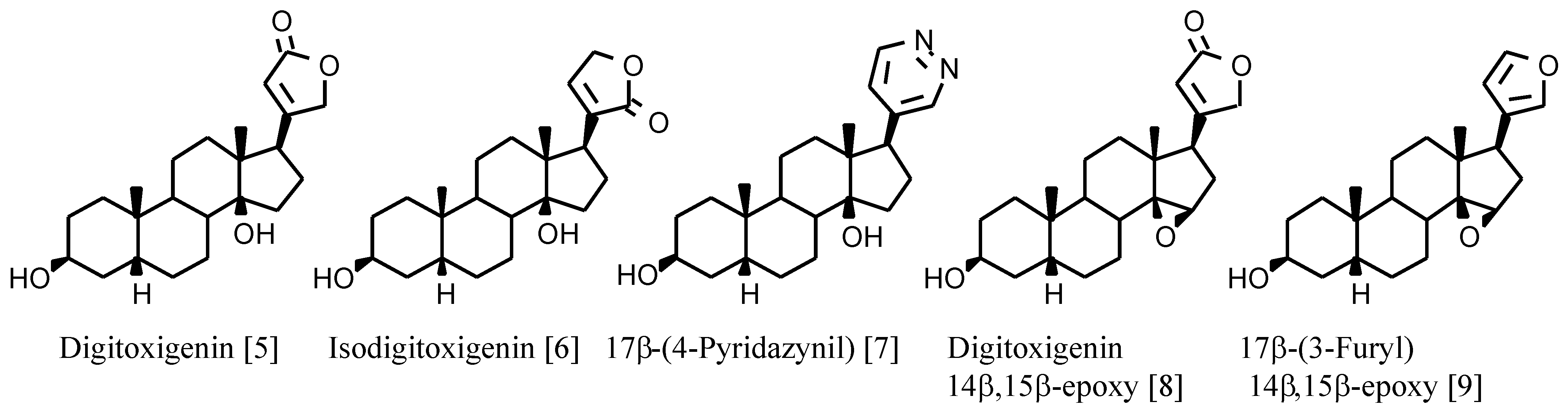
+-ATPase are cardenolides such as digitoxigenin (
Figure 1) with the following structural characteristics: 17β-unsaturated lactone, 3β- and 14β-hydroxy substituents and A/B and C/D
cis ring junctions. The 14β-hydroxy group is involved in hydrogen bonding with the receptor and plays an important role in binding digitalis compounds to Na
+,K
+-ATPase receptor; in fact compounds in which this group is absent show very low binding affinity or no affinity at all [
4]. However, the known derivatives with a 14β,15β-epoxy group (
Figure 1) show high binding affinities although not as high as the 14β-hydroxy analogues (
Table 1).
Herein, we report the synthesis and biological evaluation of novel 14β-methoxy derivatives of digitoxigenin and of other digitalis-like compounds. These compounds have a 14β-oxygen, which can be a hydrogen bonding acceptor, as is the case of 14β,15β-epoxide derivatives, but not a hydrogen bonding donor as is the case of 14β-hydroxy derivatives. Comparison of the binding values of these three classes of compounds could allow more insight into the requirements necessary for recognition by the receptor. Only a 3β-glucoside derivative of digitoxigenin-14β-methoxy has been described [
10], for which the inotropic activity was reported to be marginal, but no synthetic route was given.
Results and Discussion
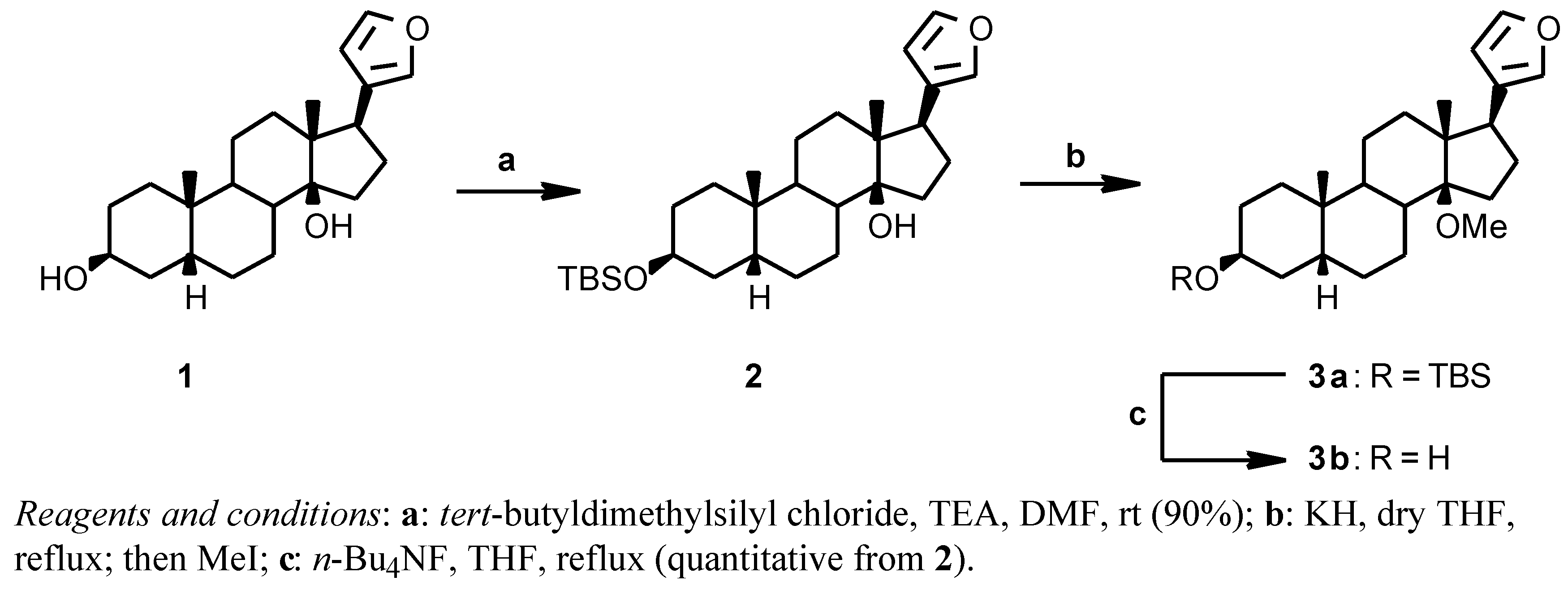
Attemps to introduce a methyl on the 14β-hydroxy group of digitoxigenin, with the secondary 3β-hydroxy protected, using diazomethane or dimethyl sulfate failed; diazomethane failed also when applied on the 17β-(3-furyl) analogue, while dimethyl sulfate gave a low yield. We then turned our attention to a Williamson reaction with MeI and, since the strongly basic reaction conditions proved incompatible with the presence of the α,β-unsaturated lactone of digitoxigenin, we tried the reaction on the 17 β-furyl derivative
2 (
Scheme 1). The known 17β-(3-furyl)-5β-androstane-3β,14β-diol
1 [
11] was reacted with
tert-butyldimethylsilyl chloride in DMF in the presence of triethylamine to give the protected derivative
2 (90%); this TBS derivative and KH were kept at reflux temperature for one hour in dry THF, the addition of MeI instantaneously gave the desired 14β-methoxy derivative
3a. The crude
3a was deprotected with
n-Bu
4NF in THF at reflux temperature to give
3b in quantitative yield from
2.
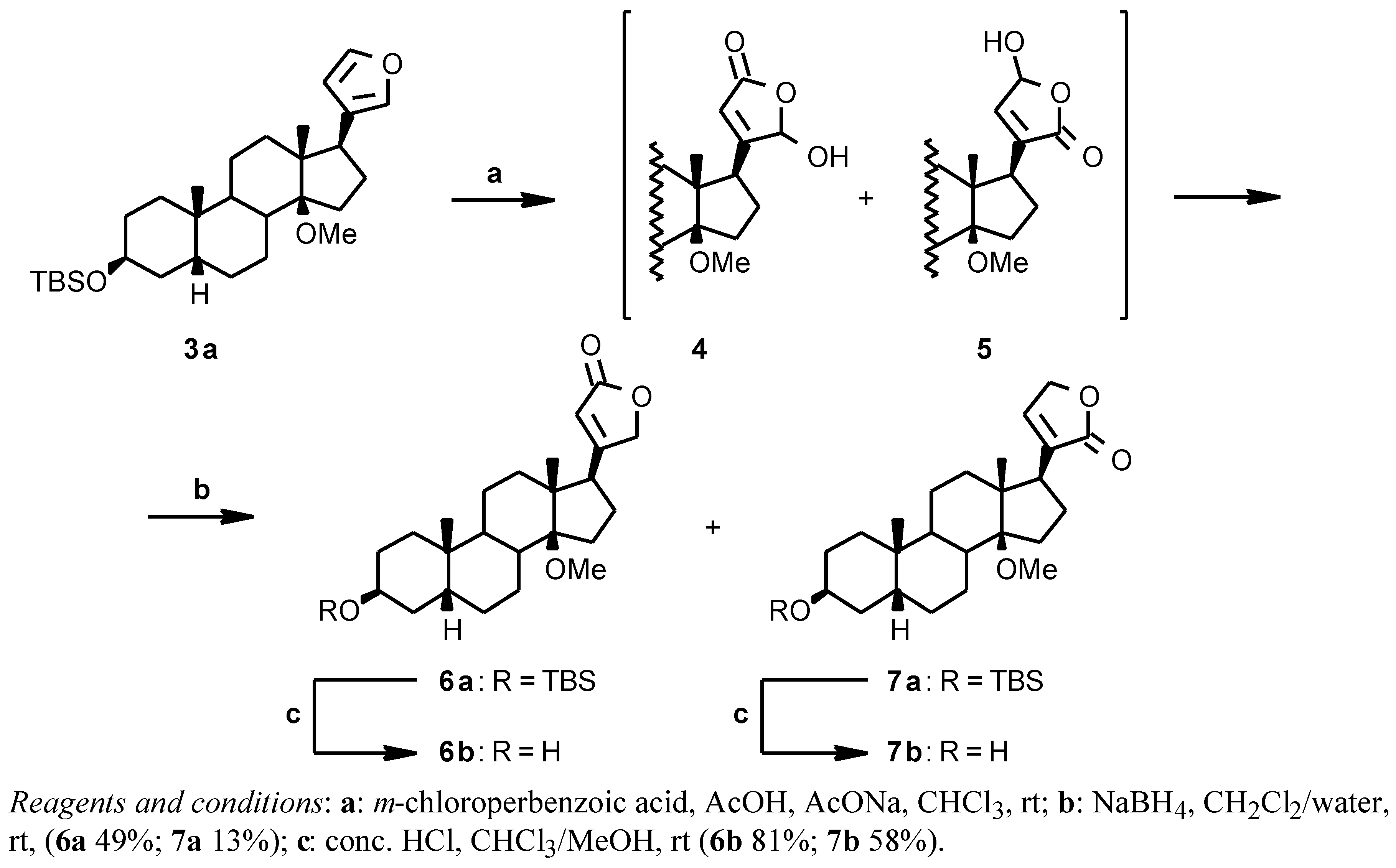
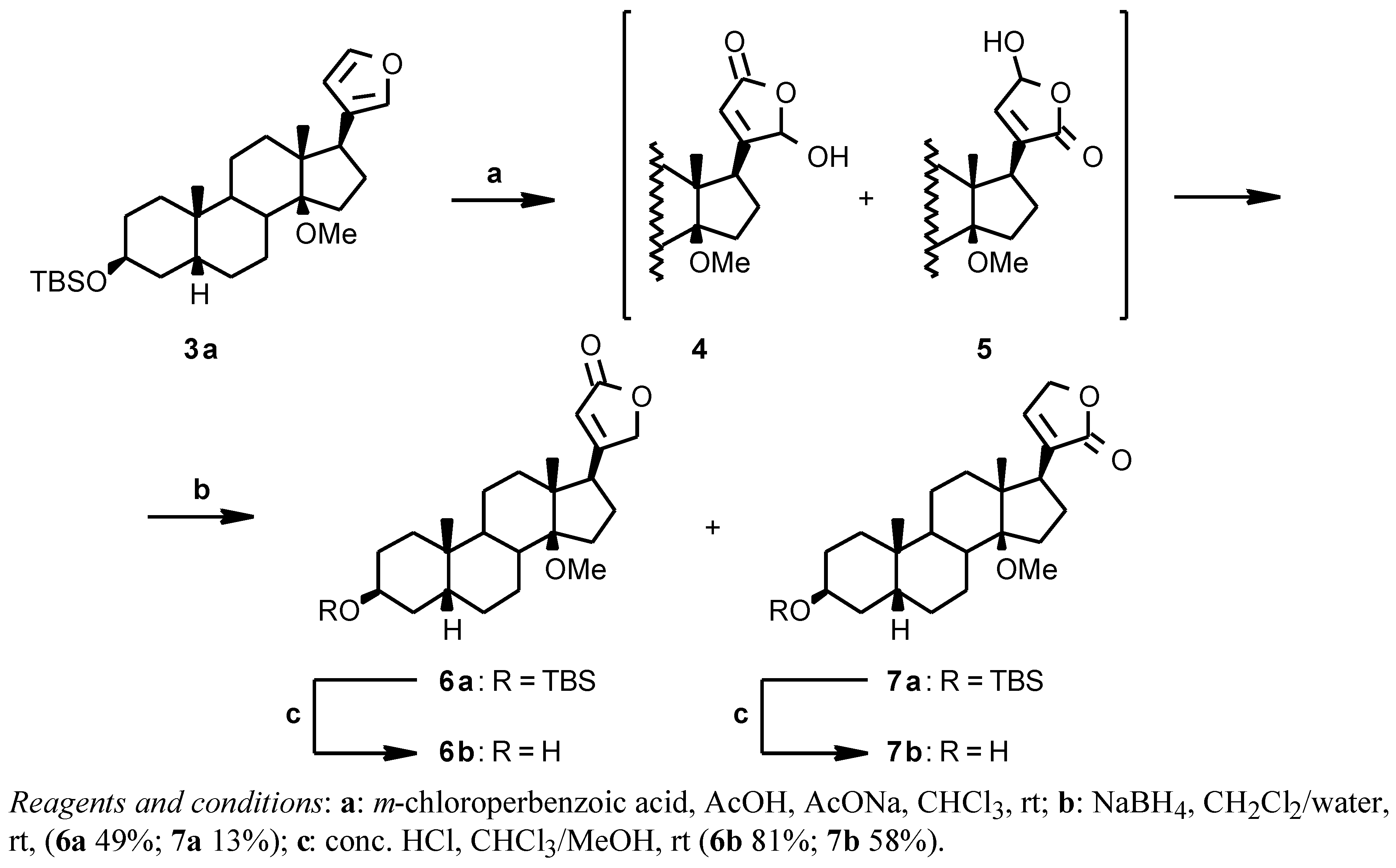
From the 14β-methoxy derivative
3a the 14β-methoxydigitoxigenin
6b could be obtained by the oxidative/reductive procedure [
6] shown in
Scheme 2. The crude
3a was reacted with
m-chloroperbenzoic acid in CHCl
3 in the presence of AcOH and AcONa; the crude hydroxy lactone intermediates
4 and
5 were reduced with NaBH
4 in CH
2Cl
2/water to give a mixture of the desired digitoxigenin derivative
6a and of the isomeric isodigitoxigenin derivative
7a in a 8:2 ratio. The two compounds were separated by flash chromatography to give
6a (49% from
2) and
7a (13% from
2) and then deprotected by acidic hydrolysis with HCl in a CHCl
3/MeOH mixture (
6b 81%;
7b 58%) [
12].
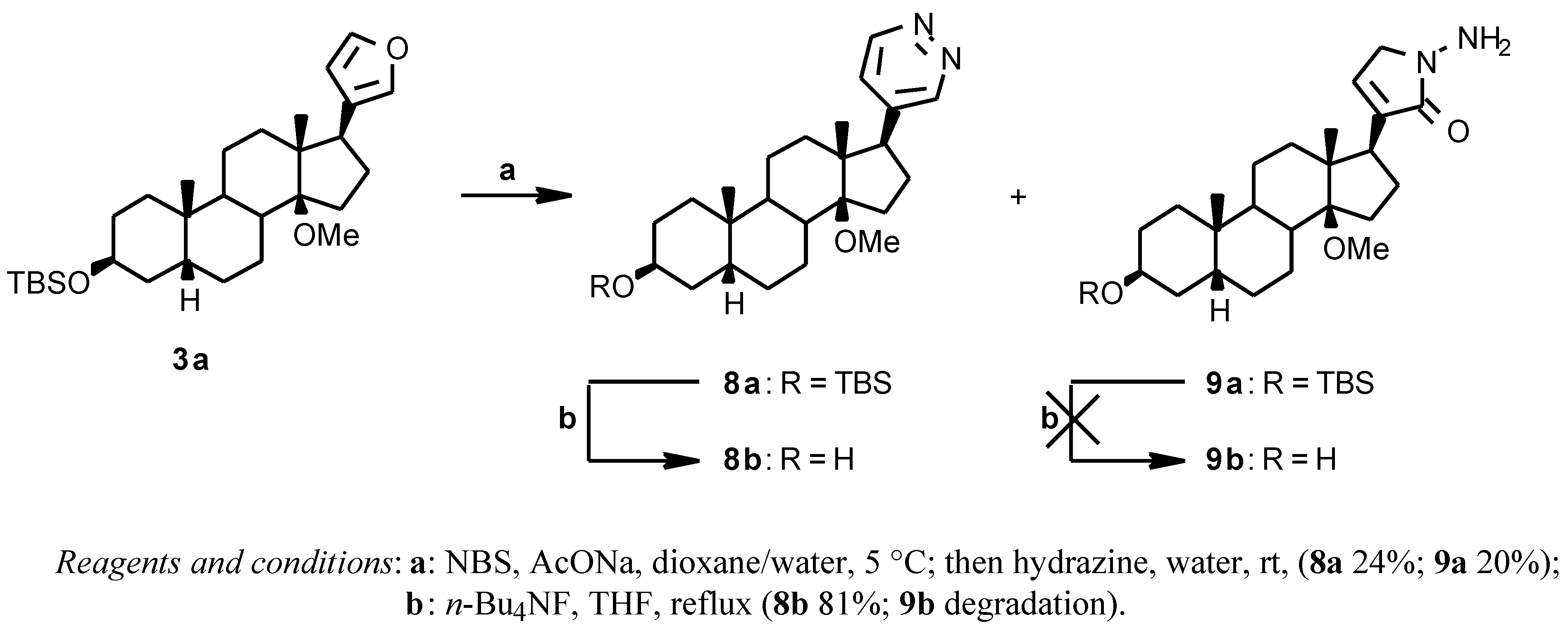
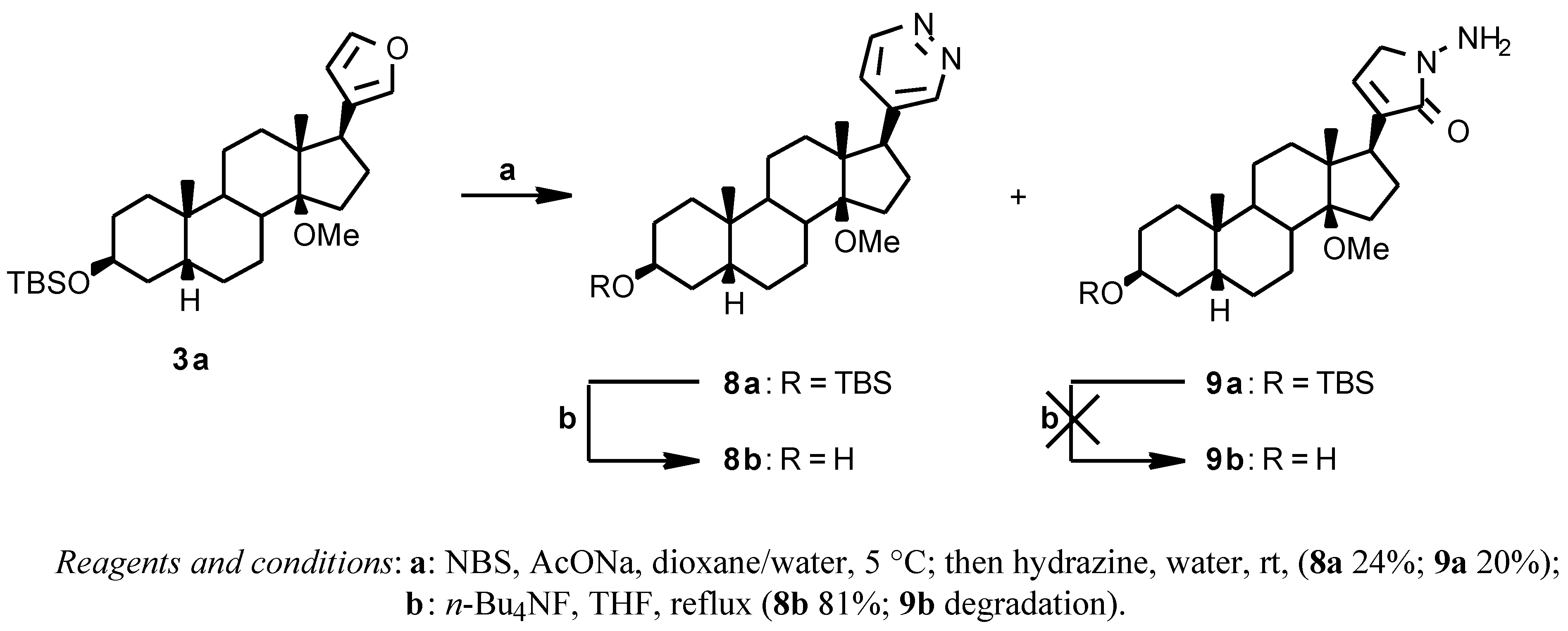
The 17 β-(4-pyridazinyl) derivative
8a (
Scheme 3) was prepared by reacting the crude 17β-(3-furyl) derivative
3a with NBS in dioxane/water in the presence of AcONa and then with hydrazine [
7] to give, after chromatographic purification, the desired
8a (24% from
2) and the N-amino lactam derivative
9a as a side product (20% from
2);
8a was deprotected with
n-Bu
4NF in THF at reflux temperature (81% yield), while
9a degraded to a complex mixture under the same conditions.
The synthesized compounds were evaluated, in comparison with 14β,15β-epoxy and/or 14β-hydroxy analogues, for displacement of the specific [
3H]-ouabain binding from the Na
+,K
+-ATPase receptor [
13a] isolated from dog kidney and purified according to Jørghensen [
13b]. The biological data are showed in
Table 1.
Experimental
General
Melting points were measured on a capillary melting point apparatus and are uncorrected. Elemental analyses were performed by Redox, Cologno Monzese, Italy. 1HNMR spectra were recorded on a Bruker AC-300 spectrometer at 300.13 MHz. Chemical shifts (δ) are given in ppm downfield from tetramethylsilane as internal standard. Chromatography was carried out on silica gel (Baker 7024-02). Solvents and reagents (Aldrich) were used as purchased.
3β-Hydroxy-14β-methoxy-17β-(3-furyl)-5β-androstane 3b
To a solution of 17β-(3-furyl)-5β-androstane-3β,14β-diol
1 [
11] (5.4 g, 15.08 mmol) in 47 mL of DMF and 18.6 mL of triethylamine,
tert-butyldimethylsilyl chloride (10.0 g, 66.34 mmol) were added at 0 °C and the mixture was allowed to warm to room temperature. After 4 hrs the mixture was poured into water and extracted with CH
2Cl
2. The organic layer was dried over Na
2SO
4 and evaporated to dryness under reduced pressure and the crude residue was filtered through a short pad of silica gel (cyclohexane) to give 3β-
tert-butyldimethylsilyloxy-17β-(3-furyl)-5β-androstan-14β-ol
2 (6.4 g, 90%) as an amorphous solid that was used for the next step without further purification.
A suspension of 2 (6.4 g, 13.56 mmol) and KH (2.7 g, 67.88 mmol) in 90 mL of dry THF was heated at 70 °C for 1 hr; then MeI (2.53 mL, 40.6 mmol) were added. After 30min the mixture was poured into water and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and evaporated to dryness under reduced pressure to give 3β-tert-butyldimethylsilyloxy-14β-methoxy-17β-(3-furyl)-5β-androstane 3a (6.59 g) as an amorphous solid; 1HNMR (300 MHz, CDCl3): 0.04 (6H, s, OSit-BuMe2), 0.78 (3H, s, CH3), 0.91 (9H, s, OSit-BuMe2), 0.98 (3H, s, CH3), 2.69 (1H, m, 17α-H), 3.39 (3H, s, OCH3), 4.07 (1H, brs, 3α-H), 6.39 (1H, brs, 22-H), 7.18 (1H, brs, 21-H), 7.32 (1H, brs, 23-H).
A solution of 3a (3.0 g, 6.35 mmol) in 57 mL of a 1.1 M solution of n-Bu4NF (63.5 mmol) in THF was heated at 70 °C under nitrogen for 1 hr and then poured into a saturated aq. solution of NaCl. The mixture was extracted with AcOEt and the organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and purified by flash-chromatography (n-hexane to n-hexane/AcOEt 80:20 v/v) to give 3b (2.3 g, quantitative from 2) as a white solid, m.p. 88-91°C (dec); 1H-NMR (300 MHz, CDCl3): 0.78 (3H, s, CH3), 1.01 (3H, s, CH3), 2.69 (1H, m, 17α-H), 3.39 (3H, s, OCH3), 4.14 (1H, brs, 3α-H), 6.39 (1H, brs, 22-H), 7.18 (1H, brs, 21-H), 7.32 (1H, brs, 23-H). Analysis calculated for C24H36O3: C, 77.38; H, 9.74. Found: C, 77.38; H, 9.76.
3β-Hydroxy-14β-methoxy-5β-card-20(22)-enolide 6b and 3β-hydroxy-14β-methoxy-5β-24-norchol-20(22)-en-21,23-lactone 7b
To a solution of 3β-tert-butyldimethylsilyloxy-14β-methoxy-17β-(3-furyl)-5β-androstane 3a (6.5 g, 13.4 mmol; prepared as described above) in 320 mL of CHCl3, AcONa (2.8 g, 33.9 mmol), AcOH (1.48 mL, 33.9 mmol) and m-chloroperbenzoic acid (75%, 6.85 g, 29.8 mmol) were added. The reaction mixture was stirred at room temperature for 1.5 hrs, then diluted with 500 mL of CHCl3, washed with a 5% aq. solution of Na2SO3 and with a 5% aq. solution of NaHCO3; the organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and the residue used for the next reduction step.
The residue obtained above was dissolved in 1.6 L of CH2Cl2 and 320 mL of water and to the well stirred biphasic mixture NaBH4 (6.0 g, 158.72 mmol) was added in two portions, the second after 4 hrs. After another 18 hrs the reaction mixture was added to a 5% aq. solution of citric acid (1 L) and the organic layer extracted, separated, washed with a 5% aq. solution of NaHCO3 and a saturated aq. solution of NaCl. The organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and purified by flash-chromatography (CH2Cl2) to give the protected derivatives 6a (3.3 g, 49% from 2) and 7a (0.88 g, 13% from 2).
The silyloxy derivative 6a (2.4 g, 4.78 mmol) was dissolved in 100 mL of CHCl3/MeOH 1:1, few drops of conc. HCl were added and the reaction mixture was stirred at room temperature for 24 h. The acidic solution was neutralized with solid NaHCO3 and evaporated at reduced pressure; the residue was extracted with CH2Cl2 and water; the organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and crystallized from Et2O to give 6b (1.5 g, 81%) as a white solid: m.p.: 179-182 °C; 1H-NMR (300 MHz, CDCl3): 0.91 (3H, s, CH3), 0.99 (3H, s, CH3), 2.71 (1H, m, 17α-H), 3.34 (3H, s, OCH3), 4.12 (1H, brs, 3α-H), 4.82 (2H, m, 21-H), 5.83 (1H, brs, 22-H). Analysis calculated for C24H36O4: C, 74.19; H, 9.34. Found: C, 73.85; H, 9.36.
The silyloxy derivative 7a (0.54 g, 1.07 mmol) were dissolved in 25 mL of CHCl3/MeOH 1:1, few drops of conc. HCl were added and the reaction mixture was stirred at room temperature for 24 h. The acidic solution was neutralized with solid NaHCO3 and evaporated at reduced pressure; the residue was extracted with CH2Cl2 and water; the organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and purified by flashchromatography (CH2Cl2/AcOEt 90:10 v/v) to give 7b (0.24 g, 58%) as a white solid: m.p.: 163-166 °C; 1H-NMR (300 MHz, CDCl3): 0.90 (3H, s, CH3), 0.99 (3H, s, CH3), 2.77 (1H, m, 17α-H), 3.32 (3H, s, OCH3), 4.14 (1H, brs, 3α-H), 4.82 (2H, m, 23-H), 7.24 (1H, brs, 22-H). Analysis calculated for C24H36O4: C, 74.19; H, 9.34. Found: C, 74.25; H, 9.33.
3β-Hydroxy-14β-methoxy-17β-(4-pyridazinyl)-5β-androstane 8b
To a solution of 3β-tert-butyldimethylsilyloxy-14β-methoxy-17β-(3-furyl)-5β-androstane 3a (1.0 g, 2.0 mmol; prepared as described above) and AcONa (0.36 g, 2.2 mmol) in 70 mL of a dioxane/water 10:1 (v/v) mixture, NBS (0.39 g, 2.2 mmol) in 7 mL of a dioxane/water 9:1 v/v mixture were slowly added at 5 °C. After 0.5 hrs a solution of hydrazine hydrate (4 mL, 82.4 mmol) in 4 mL of water was slowly dropped while maintaining the temperature at 5 °C. The resulting mixture was kept at room temperature for 36 hrs, and then was poured into a saturated aq. solution of NaCl and extracted with Et2O. The organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and purified by flashchromatography (AcOEt/ cyclohexane 95:5 v/v, then AcOEt 100) to give 8a (0.24 g, 24% from 2) and 9a (0.21 g, 20% from 2).
9a: 1H-NMR (300 MHz, CDCl3): 0.04 (6H, s, OSit-BuMe2), 0.87 (3H, s, CH3), 0.91 (9H, s, OSit-BuMe2), 0.96 (3H, s, CH3), 2.81 (1H, m, 17α-H), 3.32 (3H, s, OCH3), 3.9-4.15 (5H, m, NH2, 3α-H and 23-H), 6.72 (1H, brs, 22-H).
A solution of 8a (0.24 g, 0.48 mmol) in 5 mL of a 1.1 M solution of n-Bu4NF (5.5 mmol) in THF was heated at 70 °C under nitrogen for 1 hr and then poured into a saturated aq. solution of NaCl and extracted with EtOAc. The organic layer was dried over Na2SO4, evaporated to dryness under reduced pressure and purified by flashchromatography (AcOEt) to give 8b (0.16 g, 81%) as a white foam. An analytical sample was obtained as the hzdrogen fumarate, a white solid, m.p. 157-159 °C (dec); 1H-NMR (300 MHz, DMSO-d6): 0.53 (3H, s, CH3), 0.91 (3H, s, CH3), 2.71 (1H, m, 17α-H), 3.28 (3H, s, OCH3), 3.89 (1H, brs, 3α-H), 6.62 (1H, s, fumarate), 7.49 (1H, m, 21-H), 9.03 (2H, m, 22-H and 23-H). Analysis calculated for C24H36N2O2 · 0.5 C4H4O4: C, 70.55; H, 8.65 N 6.33. Found: C, 70.28; H, 8.60; N 6.28.
When 9a was reacted with n-Bu4NF in the same conditions described above, a complex mixture of unidentified products was obtained.


{kind=link}
{kind=link}
{kind=link}
{kind=link}