Synthesis of N-7-Substituted Purines from Imidazole Precursors

Institute of Organic Chemistry, Polish Academy of Sciences, ul. Kasprzaka 44/52, PL-01-224 Warszawa, Poland
Molecules 1999, 4(10), 287-309; https://doi.org/10.3390/41000287
Submission received: 23 February 1999
/
Accepted: 4 June 1999
/
Published: 25 September 1999
Abstract
:Synthesis of purines and purine mono-1-oxides from the respective 5-substituted 4-nitroimidazole derivatives is described. The commercially available 4-nitroimidazole, in a few simple steps (including Vicarious Nucleophilic Substitution of Hydrogen), was transformed into: 4-aminoimidazoyl-5-carboximes, N-[1-(4-amino-1H-imidazol-5-yl)methylidene]-N-(aryl)amine derivatives or into 4-amino-5-aminomethyl-imidazole. These intermediates were cyclized to the final products.

1. Introduction
In the past years we have reported new and efficient approaches to fused pyrimidines [1,2,3], using aromatic nitrocompounds which were commercially or very readily available as starting materials. In these publications, the synthesis of many fused pyrimidines based on carbocyclic (benzene, naphthalene) and heterocyclic rings (quinoline, thiophene, imidazole) was described. Among them, occasionally a few examples of purine synthesis were also reported. In this methodology, the appropriate nitroaromatic compounds were transformed into the desired ortho-substituted derivatives, and the crucial step of these syntheses was realized by Vicarious Nucleophilic Substitution of Hydrogen (VNS) [4]. Hence, the use of VNS reaction has been found to be an excellent tool for introduction of the dihalomethyl group [5] and isocyanomethyl group [3,6] in the ortho position of aromatic or heteroaromatic nitrocompounds, which thus giving an opportunity for further transformations leading to cyclizable intermediates.
Here, we would like to present the application of these approaches to pyrimidines for synthesis of purines. As outlined on the general scheme in the Abstract, the starting material in this synthesis is the commercially available 4-nitroimidazole, in which the presence of the nitro group allows introduction of the desired substituents Y at the carbon atom bearing a hydrogen in position 5-.
Synthesis of purines from imidazole precursors has been described in the literature [7,8]. Among these works, the latest reviews [7b,7c] should be mentioned, as they cover most of the synthetic aspects of purines with the use of this method. However, thus far, a serious limitation for these syntheses was the rather difficult access to suitable key-intermediates or to their precursors. Now, based on the VNS methodology, the new approach to purines presented here seems to be easy and efficient. We tried to fill the gap concerning the preparation of intermediates, and to illustrate this development by several new syntheses of purines.
2. Results and Discussion
2.1. Synthesis of Purines via Oximes (3)
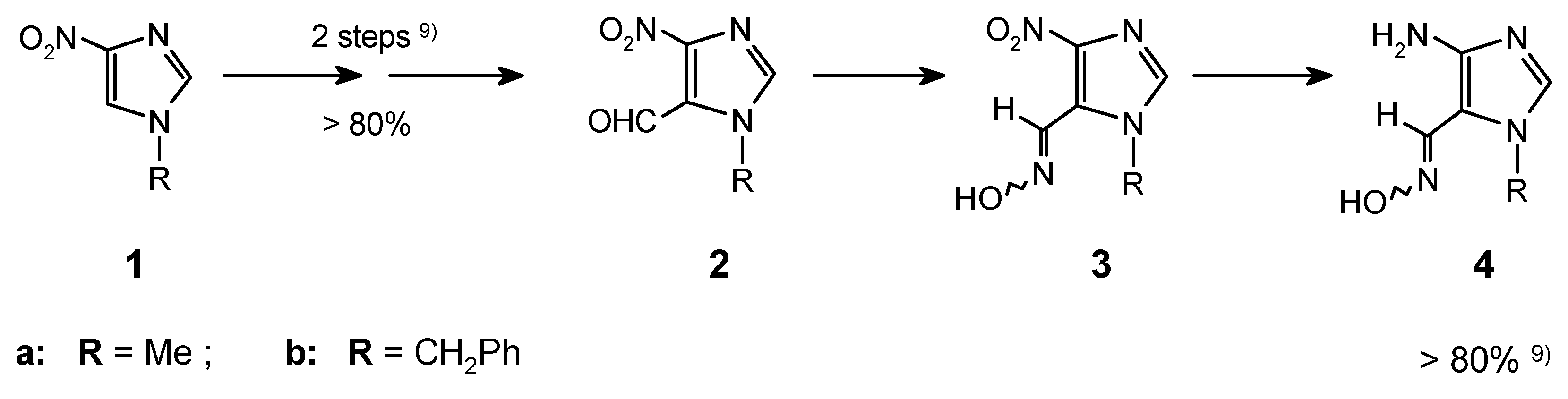
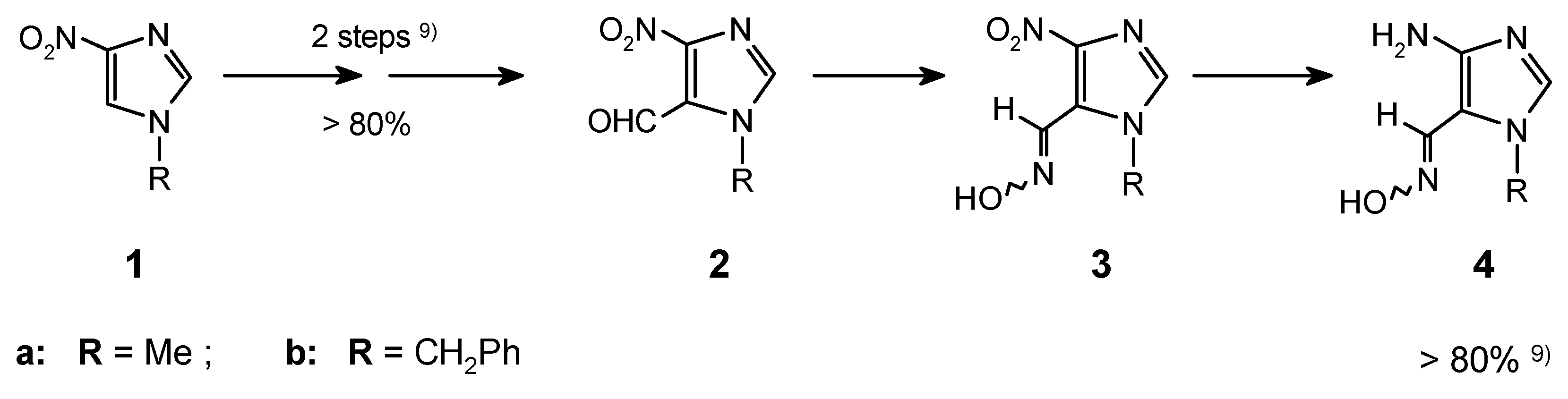
7-Substituted purines can be efficiently obtained from the 4-aminoimidazole-5-carbaldehyde oximes (4). Appropriate oximes were easily prepared from 4-nitroimidazole via the VNS in the reactions of 1a,b with chloroform followed by subsequent standard transformations [1,9] (Scheme 1).
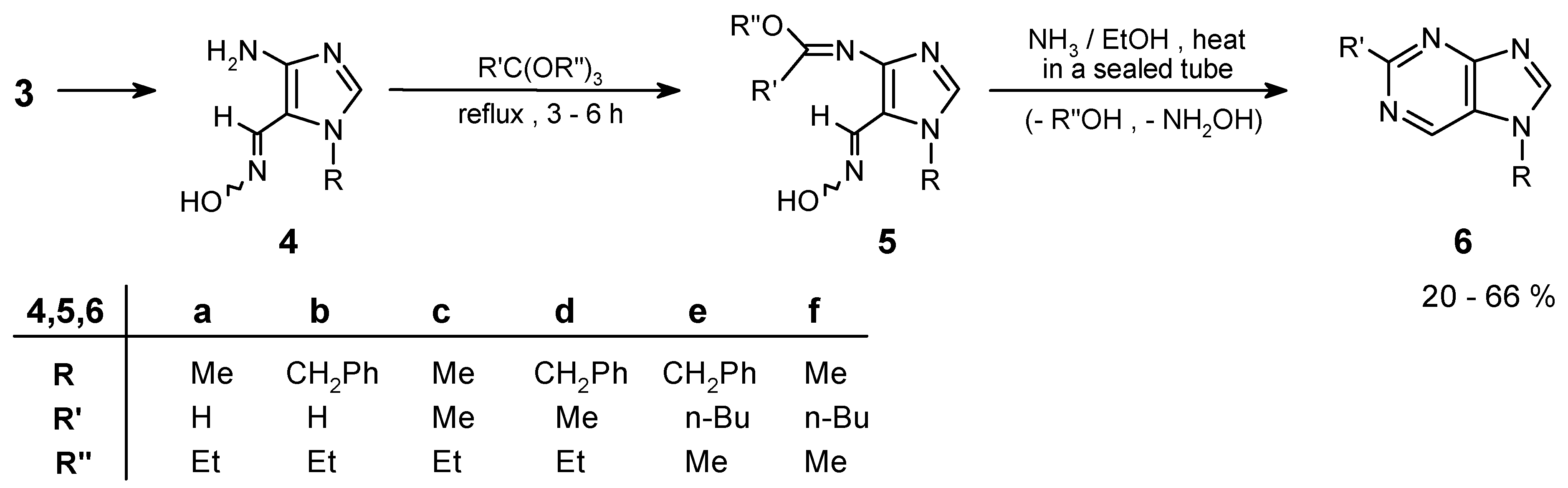
Because of their instability, the aminooximes 4 should be directly used in the next steps after preparation from the corresponding 4-nitroimidazole-5-carboximes (3a,b). Condensation of the amino group in oximes 4a,b with orthoesters resulted in formation of iminoethers 5. These imidates underwent the previously unknown cyclocondensation of the ortho-neighbouring iminoether and oxime groups with ammonia (carried out in a sealed tube), and led to the purine ring systems 6 in good overall yield. This approach (Scheme 2) was exemplified by preparation of a few purines and it was described in the previous communication [1]. Full data for the all synthesized compounds is included in this experimental.
Preparation of purines of this type is rather a difficult task. In the earlier work [7,8], in the synthesis from imidazole derivatives, substituted purines (6-amino-, hydroxy-, etc.) or dihydro-compounds were usually obtained. The yields for the last step of these syntheses varied from 40 to 70%. On the other hand, compounds 6a and 6b, thus far described in the literature [23], were obtained via catalytic dehalogenation of already prepared purine derivatives (i.e. chloropurines) in yields of 60-80% (given for the last step as well).
In the method presented here, synthesis of purines which do not contain any substituent in position 6- is possible directly from imidazole precursors, in overall yields (calculated for the last four steps) 20-66%; depending on R in position 2-.
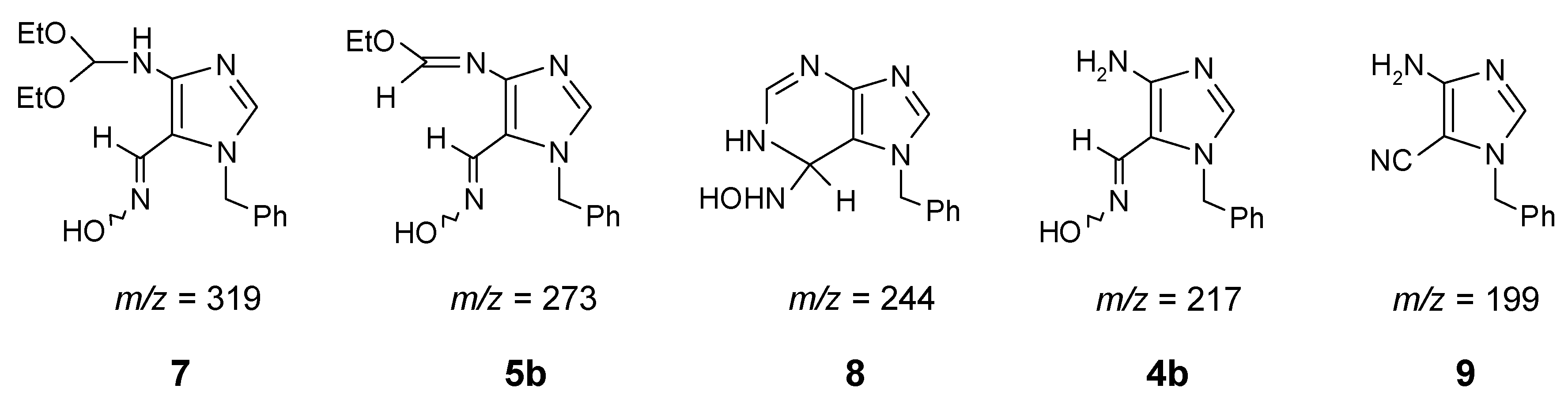
In order to study this reaction further, the crude mixture was examined by Liquid Secondary Ions Mass Spectrometry [LSIMS (+)] and 5 other molecular ions were identified. They may originate from the following intermediates and the side-products: 7 [m/z = 319 (M+H, relative intensity - 10%)], the starting imidate 5b [m/z = 273 (M+H, 3%)], intermediate 8 [m/z = 244 (M+H, 12%)], 4b [m/z = 217 (M+H, 8%); hydrolysis of imidate moiety to amino group], and 9 [m/z = 199 (M+H, 9%); hydrolysis of imidate and dehydration of oxime group to nitrile]. We were not able to isolate these products in a pure form, but some of them (4b, 5b, and 9) were additionally identified by comparison with authentic samples [1,9], using TLC method.
2.2. Purine mono-N-Oxides
2.2.1. Synthesis of Purine mono-N-Oxides via Oximes (3)
Aromatic or heteroaromatic ortho-aminocarboximes are very valuable intermediates for preparation of fused pyrimidine mono-N-oxides by the cyclization reaction with the use of orthoesters [10]. This is a method for the selective introduction of the N->O function into the desired position in heteroaromatic compounds having more than one nitrogen atom. However, only a few examples of application of this method (or its modifications) have been described in the literature [11,12,13], probably because of the limited availability of the proper ortho-aminocarboximes. Now, easy access to these intermediates by the VNS methodology allows us to develop this very useful cyclization, as it was demonstrated for synthesis of many heterocyclic systems [2].
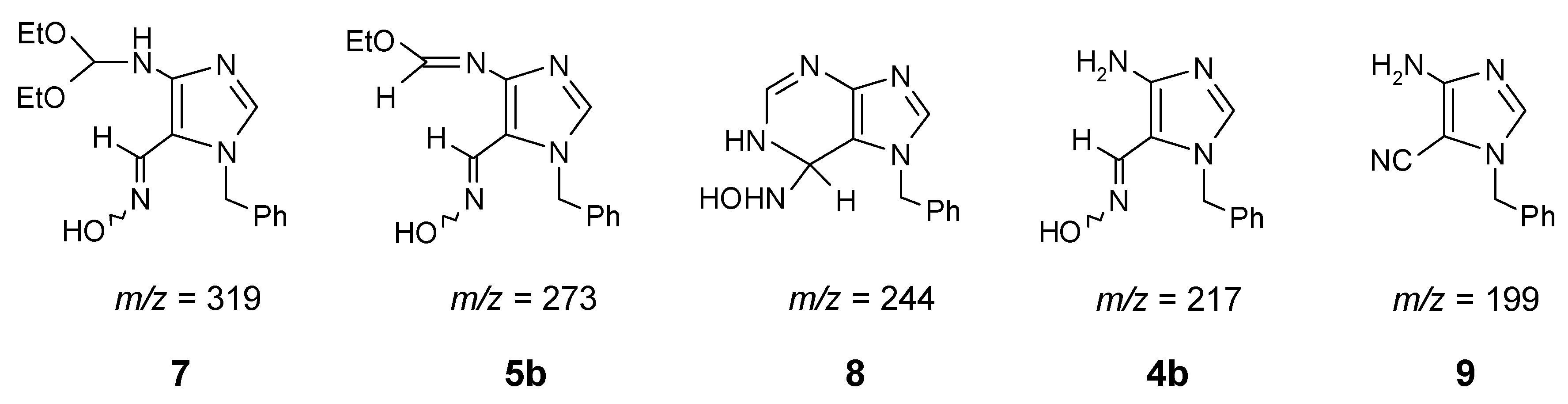
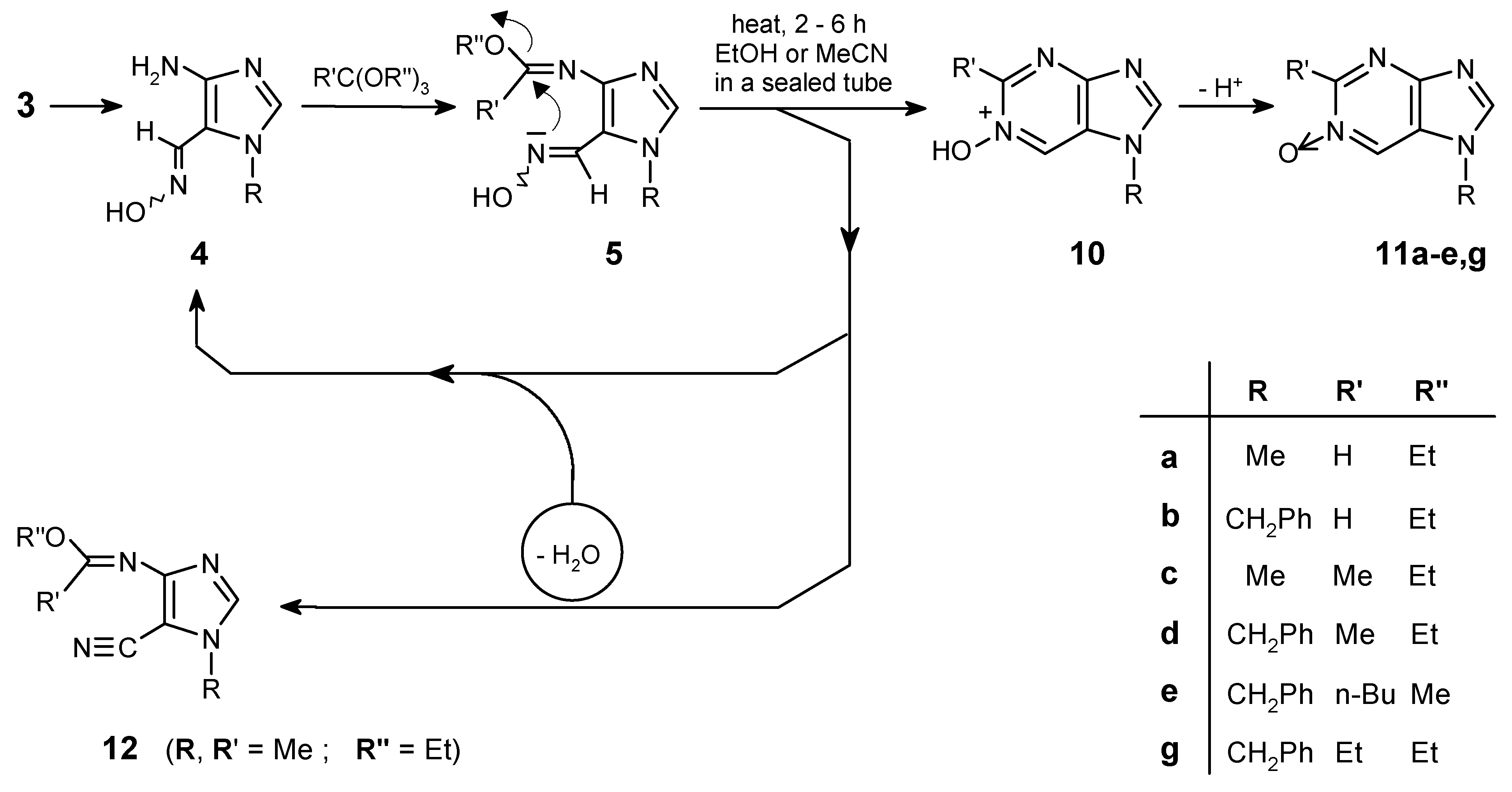
Aminooximes such as 4 are proper key-intermediates for synthesis of [7H]-purine mono-N-oxides. These substrates can easily cyclize to purine 1-oxides, preparation of which would be a difficult task and practically impossible to realize by direct N-oxidation of the corresponding purine.
Condensation of 4 with various orthoesters resulted in formation of ortho oxime-imidates 5. Then nitrogen attack of the oxime moiety on the imidate electrophilic ”C=N” double bond, followed by the loss of a proton, leads selectively to the desired purine mono-N-oxides. However, for purine derivatives this cyclization was very erratic and the yields were changeable, because of a few competitive reactions observed, i.e.: back-hydrolysis of imidate 5 to the starting amine 4, and the formation of nitrile 12. It can be explained by the fact that ethanol (solvent for this reaction) always contains traces of water - hence the hydrolysis to starting amine 4 was possible. Moreover, this imidate (5) even when heated in dry ethanol can be hydrolyzed by alcohol moiety. When a dry acetonitrile has been used instead of ethanol, the decomposition was also observed, because the water which is necessary for this hydrolysis, at higher temperature can be produced in the competitive dehydration of oxime 5 to nitrile 12. So, for each case of the purine N-oxide preparation, optimization of the conditions was necessary. All these possible transformations are outlined in Scheme 3 and the optimized conditions were listed in Table 1.
In addition, the method presented here allows not only for selective introduction of the N->O function into the 1- position of the target N-oxides, but it can also be applied for functionalization at C-2 (introduction of alkyl substituents) by using diverse orthoesters. For some reactions the attempts to convert 5 into 11 failed. For example, the optimization in the case of orthopropionate triethyl ester, while reacting with 4b, was unsuccessful. Finally in ethanol, at temperature 200°C small amounts (<5%) of the proper N-oxide 11g were detected by 1H NMR.
2.2.2. 13C NMR Spectra of Purine mono-N-Oxides (11)
The structures of N-oxides 11a-e,g were confirmed by 1H NMR and 13C NMR spectra. According to our knowledge 13C NMR spectra have not so far been published for any of this type of purine mono-N-oxides and this is the first attempt of the assignment of chemical shifts to carbon atoms of the structure. First, C-8 signals were easily identified due to the almost identical chemical shifts in all compounds. Also the assignment of C-2.1, C-2.2, C-2.3, C-2.4 and C-7.1 was a trivial problem. However, it was difficult to assign correctly the remaining values on the basis of 13C decoupled spectra because of a considerable upfield shifts of C-6 (in compounds 11b,d,e) and the closeness of these signals to aromatic peaks of benzyl group. Also a pair of very weak signals of quaternary carbons C-4 and C-5, despite significant difference in their chemical shifts (over 25 ppm), was difficult to assign correctly (in the case of 11a,c some even were not detected). In addition, a theoretical calculations of the chemical shifts for all of the investigated purine N-oxides using the ACD/Labs program gave very poor agreement between the calculated data and those determined experimentally.
Therefore, for the selected compound 11d two-dimensional HETCOR spectrum (GHMBC technique) was recorded. The proton chemical shifts of H-6 and H-8 were differentiated easily as the signal of H-8 always appears at higher field [7]. However, the direct proof came from its strong correlation with already identified C-8 signal. Therefore, it was assigned definitively: δ(H-8) = 8.22 ppm and δ(H-6) = 8.43 ppm. We found that the proton signal at δ = 8.43 ppm (H-6) corresponds to carbon atom δ = 129.6 ppm (C-6!) and its strong correlation with carbon atom ca δ = 151 ppm was also observed. In this spectral technique it is typical to find correlation via three-bond distance, while the two-bond correlation with carbon atom at δ = 123.4 ppm is very weak. Such a situation usually occurs due to a smaller two-bond coupling constant as compared to 3JH-C. On the other hand, proton H-8 (δ = 8.22 ppm) also shows the correlation with the carbon atom at δ = 151.3 ppm and with another one at δ = 123.4 ppm (characteristic correlations via three bonds). Hence, it was assigned definitively: δ(C-4) = 151.3 ppm and δ(C-5) = 123.4 ppm. The remaining correlations: H-6/C-2, CH3/C-2, CH3/C-5 (5 bonds, effective correlation), CH3/C-6 (4 bonds, weak), CH2/C-5 (3 bonds, effective), CH2/C-8 (3 bonds, effective) confirmed the correct assignment in purine N-oxide skeleton of 11d.
In the same manner the chemical shifts in aromatic ring of benzyl group (C-7.2, C-7.3, C-7.4, C-7.5) were assigned, where the major problem was to differentiate the strongest peaks originating from carbon atoms C-7.3 (C-7.3’) and C-7.4 (C-7.4’). In this case, the correlations between CH2 group and the respective aromatic carbon atoms were the most diagnostic. Therefore, on the basis of this N-oxide GHMBC spectrum we assigned the values for the remaining structures in this series of compounds. All the 13C NMR parameters are listed in Table 2.
Analysis of the spectra, reported in this paper, revealed a high regularity in the chemical shifts at C-4, C-5, C-6, C-8 of the corresponding purine bicyclic system of all investigated structures. While, at C-2 small signal deviations in C-2 substituted products (11c,d,e) occurred. However, a considerably upfield shift was observed in the C-2 substituted products when compared to with the corresponding unsubstituted compounds at C-2 (ca 10 ppm for 11d,e as compared to 11b and ca 2 ppm for 11c as compared to 11a).
In these spectra a considerable influence of the substituent at N-7 on the chemical shifts of C-6 was also observed. Replacement of methyl group at this position by a benzyl group (compounds 11b,d,e) is reflected in the upfield shift (ca 10 ppm) of the corresponding δ(C-6) values as compared to C-6 in N-methyl derivatives 11a and 11c. It can be an effect of the steric bulk of the CH2Ph group or of a local aromatic ring current effect of the phenyl group.
2.3. Synthesis of Purines via Schiff bases (13)
In the previous case (Chapter 2.1) the efficient synthesis of 7-substituted purines starting from 4-nitroimidazole-5-carboxime derivatives was presented. In that method along with the proper products, even if obtained with good yields, the formation of small amounts of the respective N-oxides was occasionally observed. It was due to a condensation (without ammonia) between the ortho-situated iminoether and the oxime groups. This type of cyclization was utilized independently in the synthesis of N-oxides and it was discussed in Chapter 2.2.1 (Scheme 3).
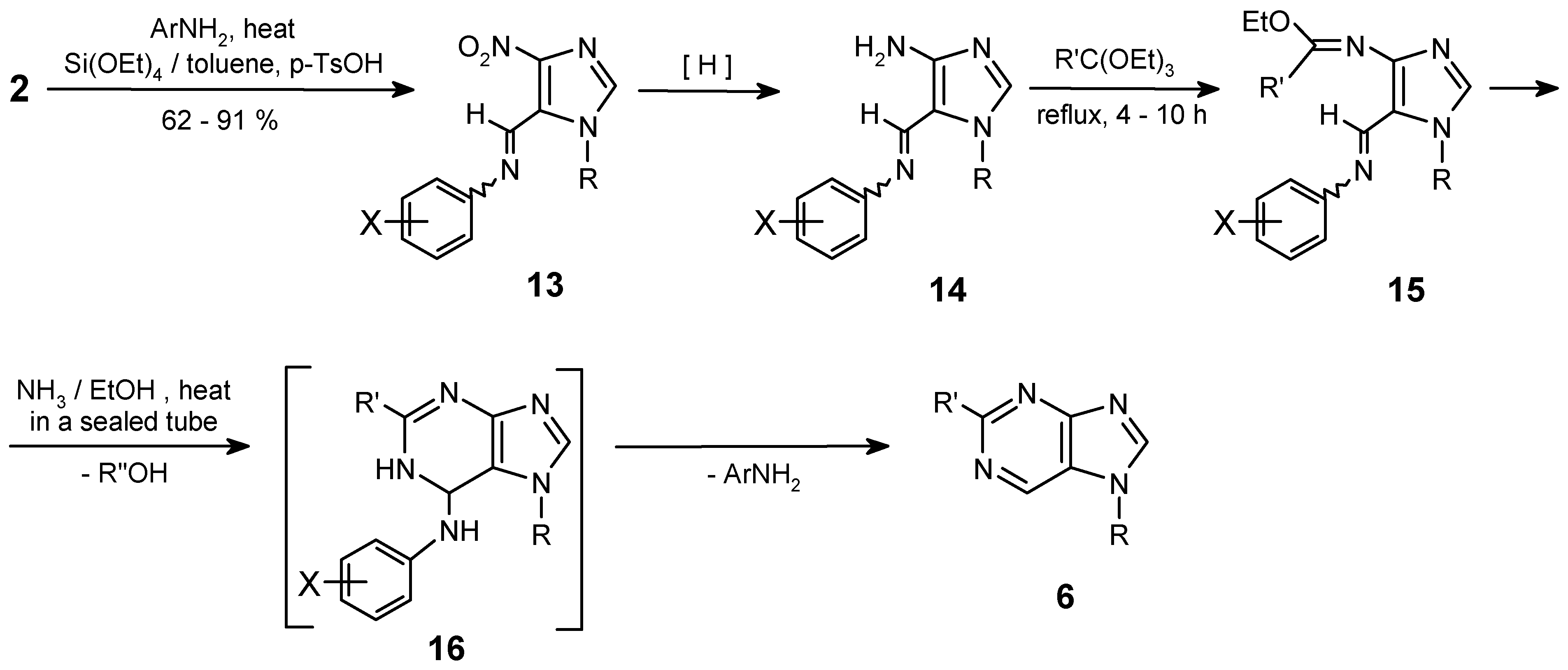
Herein, an alternative approach to the purine skeleton is reported, because the aldehydes 2 can be also easily transformed into Schiff bases of N-protected (R = CH2Ph, CH3) 4-nitroimidazolecarb-aldehydes (13) and they were used (instead of oximes) in the condensation with ammonia. So N-oxide formation is avoided with this method.
Selective reduction of the nitro group in imines 13 on 10% Pd/C in ethanol (25-40 psi, ca 4 h) followed by condensation with orthoesters results in the formation of imidates (15). Their heating in a sealed tube with an excess of ammonia in ethanol (80-140°C, 3-6 h) gives purines 6. Perhaps the first step of this cyclocondensation is the addition of the ammonia moiety to the carbon atom on the >C=N double bond of the Schiff base, and then replacement of the OEt group by the amino species by addition-elimination.
If this postulated reaction sequence operates as drawn on Scheme 4, then the last step of this transformation must be the elimination of aryloamino- moiety from the dihydro-intermediate 16. This could indeed be the case, as a small amounts of 16 (e.g. 16b) were identified in the reaction mixture along with the main products 6. It is worth to mention that the ArNH- moiety, in basic conditions, is rather a poor leaving group. Hence, one can expect difficulties on the elimination step leading to the corresponding purine ring.
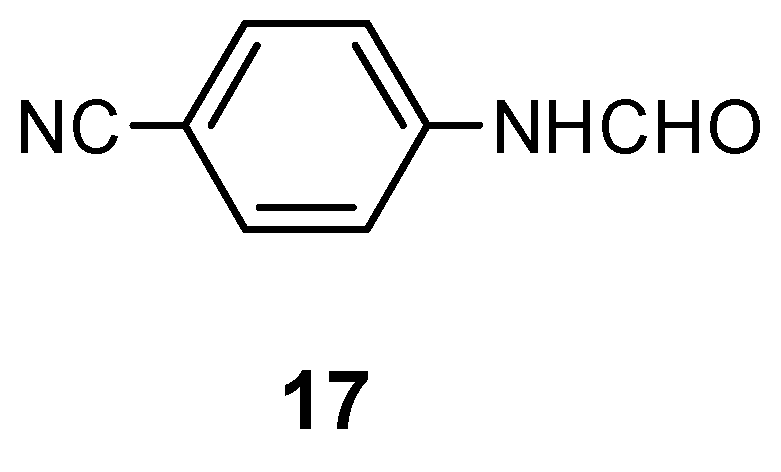
The reaction of the Schiff base prepared from the 1-methyl-4-nitro-1H-imidazole-5-carbaldehyde and para-toluidine (13a; X = para-Me) afforded the purine 6a with moderate total yield (20%). One can expect, that electron-withdrawing groups in the aryl substituents should make this elimination easier. Looking for a good arylamine to prepare the desired Schiff base, a para-cyanoaniline (X = para-CN) was selected to check this postulated substituent effect and to improve the yields of purines. Initially, it was found that the CN group was resistant to reduction in these catalytic conditions. However, in preliminary reactions when the methyl substituent in para-position (Hammett constant: σpara(CH3) = -0.17 [14]) was replaced by the strong electron-withdrawing CN group (σpara(CN) = 0.66 [14c]), the yield of purines 6a or 6b decreased dramatically (< 5%), while in the post-reaction mixture, the starting para-cyanoaniline and its N-formylated derivative 17 were found (40-45%).
Similar results were obtained, or no product was detected for para-chloro-substituent (this was examined on other model compounds [15]), for a 2,4,6-tribromo-derivative (13e) and for the bulky trityl moiety (13f). It was found that these strong electron-withdrawing groups increase a reactivity of the imine >C=N double bond; hence, its hydrogenation on the reduction step (13 -> 14) probably occurred, decreasing the overall yield. Additionally, the degradation of this labile >C=N double bond during the cyclocondensation (15 -> 6) is also possible. This problem was investigated carefully on model compounds (benzene and naphthalene derivatives) and the results will be published soon [15].
On the other hand, the best results were obtained in the case of the moderate electron-withdrawing group X = para-Br, σ = 0.23 [14] (imine 13h, 36%). Optimization of this reaction amounted the yield of 6b up to 47% and the use of para-bromoarylimine 13g gave satisfactorily results as well. However, the introduction of alkyl substituents into position 2- (entries i,j; Table 3), probably due to an additional steric effects, gave poor yields. One can expect that this is a limitation for the presented synthesis.
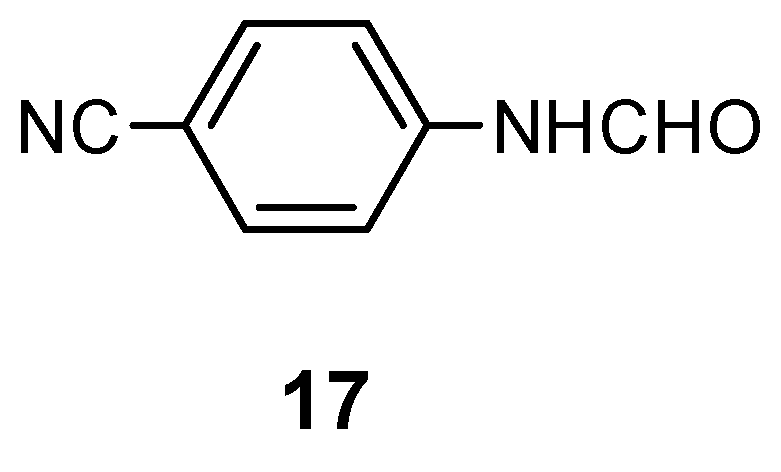
2.4. Synthesis of Purines via Isonitriles
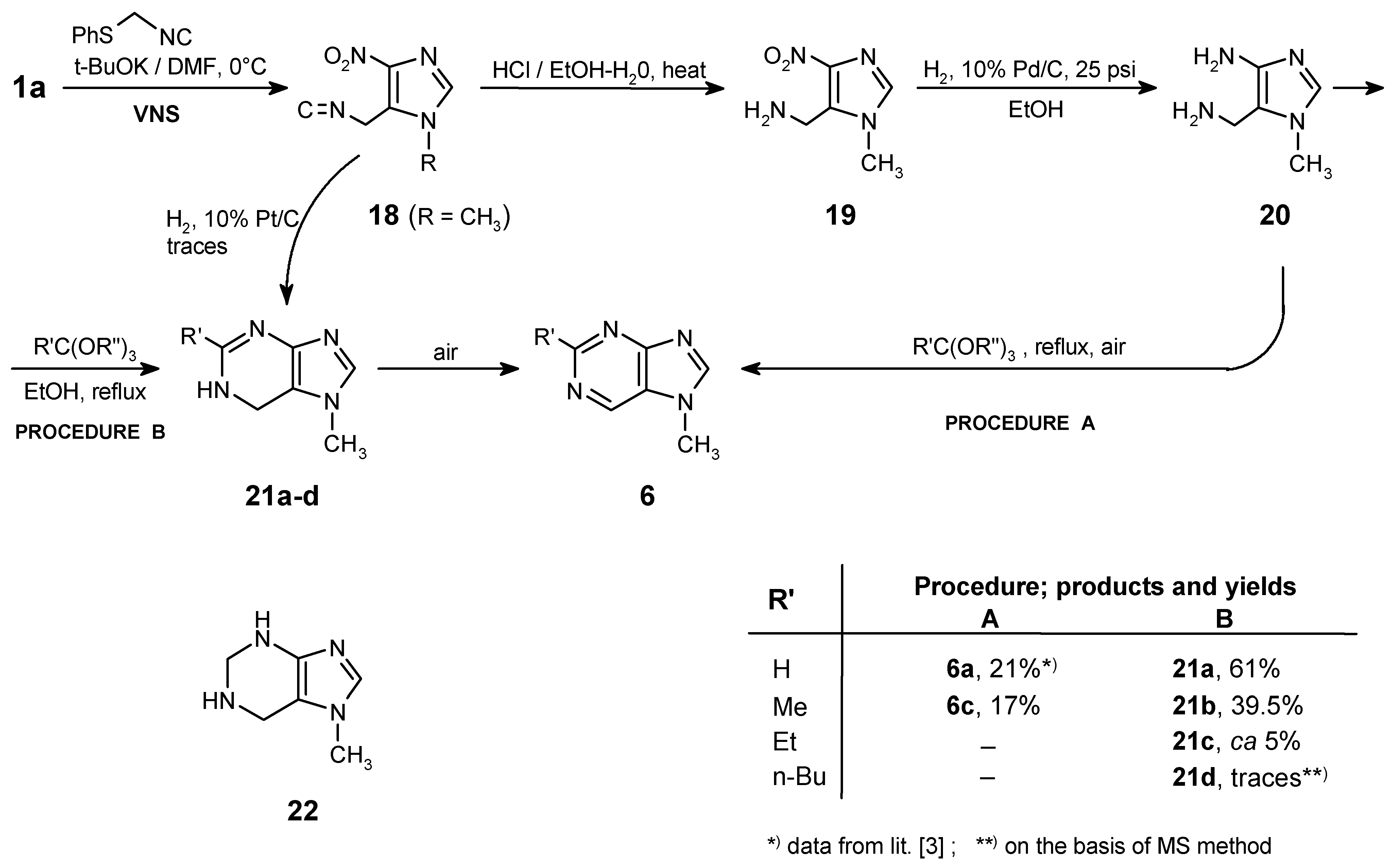
We have described lately [6] a general method for the synthesis of ortho-(isocyanomethyl)nitro-aromatic or nitroheteroaromatic compounds. As well, a practical application of this method for the synthesis of fused pyrimidines has been published already [3]. In this chapter an approach to purines via the 4-nitro-5-isocyanomethylimidazole derivative (18) is presented. The above isonitrile (18) was efficiently obtained from the 1-methyl-4-nitro-1H-imidazole 1a by the Vicarious Nucleophilic Substitution of Hydrogen (VNS) in 86% yield.
The attempts to cyclize isonitrile 18 to purine 21a (R’ = H) in various catalytic conditions was unsuccessful. It could be due to inhibition of the catalyst by the isocyano moiety [16]. However, using platinum as catalyst (10% Pt/C) traces of the desired purine were detected.
The exhaustive hydrolysis of 18 (concentrated HCl, EtOH-H2O, reflux, 3 h) afforded the hydro-chloride of 19, which after recrystallization from methanol is a very valuable and stable intermediate for the next steps of this synthesis. Its catalytic reduction (10% Pd/C, 25 psi, 4 h, in EtOH) and treatment of the crude diamine formed (20) with appropriate orthoesters (reflux, 6 h) gave purines 6a,c in ca 20% overall yields (path A). The last step, an aromatization of the dihydro-derivative 21 occurred spontaneously while exposed to air. However, this may be a more complicated red-ox process, because in the post-reaction mixture traces of a compound with molecular mass equal 138 was detected by MS (m/z = 138, 100%). This could be the tetrahydro-compound 22, but the attempts to isolate this product failed, therefore we do not have any additional spectroscopic evidence for this structure.
To improve the yields of the corresponding purines the condensation with orthoesters was carried out in boiling ethanol to assure lower temperature (path B). At this temperature the reaction was stopped at the stage of the dihydro-compounds 21a,b giving also considerably higher overall yields (61% and 39.5% respectively). However, the condensation of 20 with orthoesters of carboxylic acids bearing a longer carbon chain (orthopropionate, orthovalerate) or an aromatic substituent (orthobenzoate) failed or only traces of the expected products were detected. In these cases the condensation is probably a slower process and the degradation of the relatively unstable amino-derivative occurred [17].
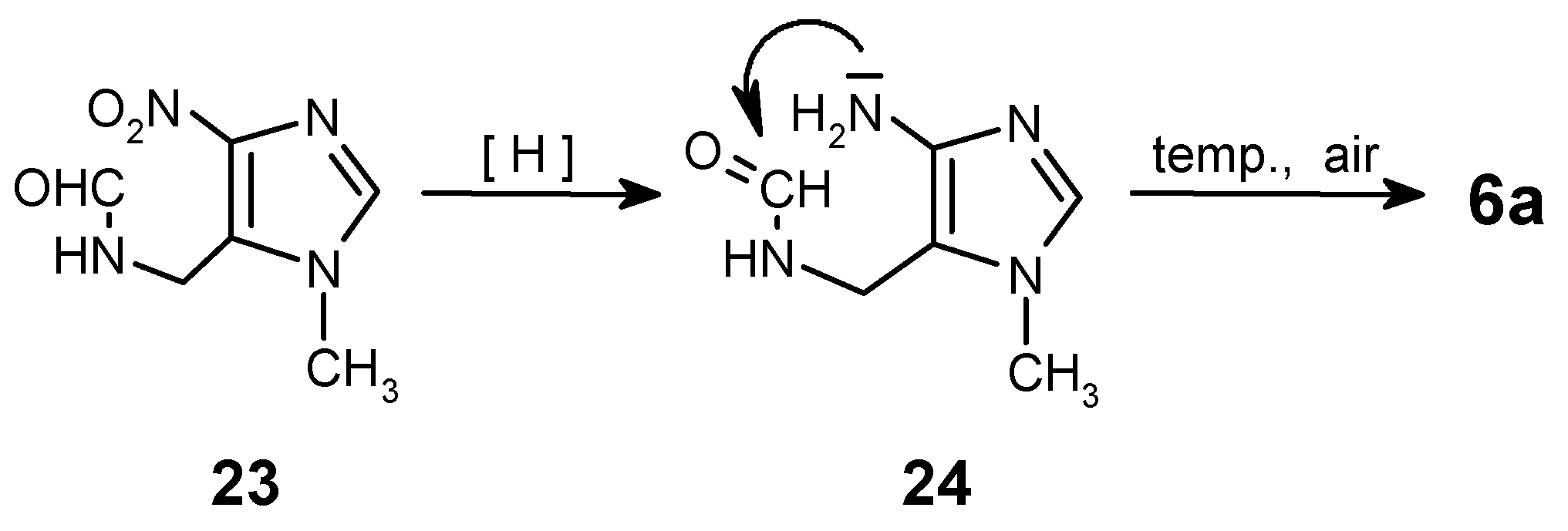
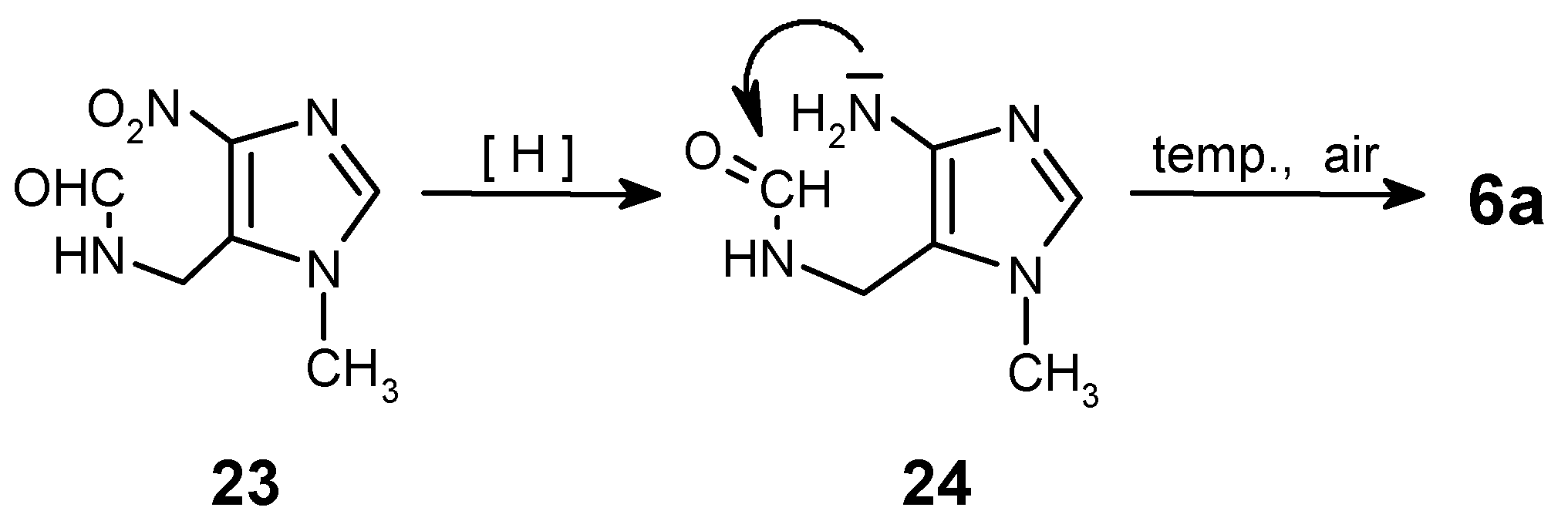
The hydrolysis of the isocyano group must be carried-out exhaustively, because after incomplete hydrolysis of isonitrile 18, followed by catalytic reduction and cyclocondensation of the crude mixture with triethyl orthoacetate, two products 6c and 6a (!) can be obtained unexpectedly. It was a case in a one of our preliminary experiments. The explanation of this fact is given on Scheme 6. Partial hydrolysis of N=C group gave the formamido-derivative 23, which after reduction of the nitro group followed by cyclization and spontaneous oxidation [reflux in CH3C(OC2H5)3] leads to purine 6a.
To assure full conversion to amine 19, the hydrolysis had to be carried out during 3 h in boiling ethanol. Otherwise, formation of small amounts of compound 23 was always observed!
3. Conclusions
New and efficient approaches to fused pyrimidines, previously described in several papers from our laboratory [1,2,3], can also be applied for the synthesis of purines. The scope and limitations of the above methods for this synthesis is presented. With these methods, the preparation of purines and their N-oxides which do not contain any substituent in position 2- and/or 6- is possible directly from imidazole precursors. We used the new reaction (Vicarious Nucleophilic Substitution of Hydrogen) and the new substituent pattern to achieve this goal. Until now, this was rather a difficult task, because in earlier work [7,8] substituted purines (6-amino-, hydroxy, etc.) or dihydro-compounds were obtained usually in the synthesis from imidazole derivatives.
Some of the approaches presented herein, i.e. synthesis via cyclocondensation of iminoether and oxime groups with ammonia, or selective preparation of purine mono-N-oxides, are of substantial value. Taking into account commercially available starting material (4-nitroimidazole) these approaches probably can receive more attention.
By chance we also determined 13C NMR spectra for a series of newly prepared purine N-oxides.
Experimental
NMR spectra were recorded with a Varian GEMINI-200 spectrometer operating at 200 MHz for 1H and 50 MHz for 13C. Coupling constants J are expressed in hertz [Hz]. Two-dimensional HETCOR GHMBC spectra were recorded with a Varian INOVA-500 spectrometer (500 MHz / 125 MHz). The assignments of the chemical shifts accompanied by asterisks*) may be exchanged. 13C NMR spectra were assigned on the basis of literature data and theoretical calculations using the ACD/Labs program. 13C NMR of purine N-oxides 11a-e are discussed in Chapter 2.2.2. IR spectra were measured with a Perkin Elmer 1600 FTIR spectrometer and mass spectra with an AMD 604 (AMD Intectra GmbH, Germany) spectrometer (electron impact and LSIMS methods); m/z values are given as a % of relative intensity. Melting points are uncorrected. TLC analysis was performed on aluminum foil plates precoated with silica gel 60F 254 Merck. Silica gel 200-300 mesh (Merck AG) was used for column chromatography.
N-Benzyl- and N-methyl-4-nitroimidazoles (1a,b) were prepared from commercial 4-nitroimidazole (Fluka AG) by alkylation with CH3I and PhCH2Br in t-BuOK/DMSO system ([1H]-products were formed selectively and the yields were higher as compared to those described earlier in the literature [18,19]; see procedures below). The starting Schiff bases 13a-h were prepared from imidazolecarbaldehydes according to typical procedures [20, 21], optimized in our laboratory. para-Cyanoaniline was obtained by catalytic reduction of para-cyanonitrobenzene (10% Pd/C, 40 psi, 2 h, in EtOH); quantitatively, mp 90-91°C (EtOH), lit. mp 86°C [22]. Other aniline derivatives used are commercially available.
Alkylation of 4-nitro-1H-imidazole. General Procedure
To a stirred solution of 4-nitroimidazole (5.66 g, 0.050 mol) and t-BuOK (5.70 g, 0.051 mol) in DMSO (100 mL) an alkyl halide (CH3I - 28.40 g, 0.200 mol; PhCH2Br - 17.10 g, 0.100 mol) was added dropwise at room temperature. The reaction was continued for 6-10 h, then poured into water with ice (200 mL) and extracted with CHCl3 (3 x 50 mL). The combined organic layers were washed with water (150 mL), dried over MgSO4, concentrated and purified by chromatography to give the desired products (1a, 1b); eluent: from n-hexane/CHCl3 - 1:1 to CHCl3.
1-Methyl-4-nitro-1H-imidazole (1a)
Yield 92%, mp 141-142°C (EtOH), lit.[18] mp 134-135°C. - 1H NMR (acetone-d6): δ = 8.11 (d, J = 1.3 Hz, 1 H, H-5), 7.64 (d, J = 1.3 Hz, 1 H, H-2), 3.89 (s, 3 H, CH3). - IR (KBr): v = 3141, 1556, 1496, 1433, 1381, 1253 cm-1. - MS, m/z (% rel. int.): 127 (M+ ·, 100), 111 (5), 97 (3), 81 (6), 69 (9), 66 (6), 54 (7), 42 (60).
1-Benzyl-4-nitro-1H-imidazole (1b)
Yield 91%, mp 84-85°C (EtOH), lit.[19] mp 78-79°C. - 1H NMR (CDCl3): δ = 7.72 (s, 1 H, H-5), 7.49 (s, 1 H, H-2), 7.48-7.37 & 7.29-7.18 (2 x m, 5 H, H-Ph), 5.17 (s, 2 H, CH2). - 13C NMR (CDCl3): δ = 167.2 (C-4), 136.0, 133.7, 129.4, 129.2, 127.8, 119.3, 52.1 (CH2). - MS, m/z (% rel. int.): 203 (M+ ·, 22), 105 (4), 91 (100), 65 (12), 39 (3).
Preparation of Purines 6a-f. General Procedure
The 4-nitro-5-imidazole carbaldehyde oxime derivative (3a, 3b; 0.5 mmol) [9] was dissolved in ethanol (5 mL) and it was hydrogenated in Parr Apparatus using a catalytic amount of 10% Pd/C (ca 20 mg) at 40 psi during 4 h. The catalyst was filtered off, washed with alcohol and the filtrate after evaporation to dryness gave crude aminooximes 4a, 4b quantitatively.
The above aminooximes were suspended in the appropriate orthoester (HC(OEt)3, MeC(OEt)3, n-BuC(OMe)3, 5 mL) and the reaction mixture was heated in reflux until completion (1.5-4 h; except for 5e: 6 h). The excess of the orthoester was removed under reduced pressure and the residue (crude imidates 5a-f) was dissolved in ethanol saturated with ammonia (4 mL). The reaction mixture was heated in a sealed tube at 120-130°C for 2-3 h (except for 6e: 200°C, 6 h). After cooling and evaporation of the solvent the purines were isolated by column chromatography (eluent: from CHCl3 to CHCl3/MeOH - 20:1; except for 6a: from CHCl3 to CHCl3/MeOH - 8:1).
Examples of 1H NMR data for crude imidates 5b-d are given below. Another imidate (5g) is described in the next paragraph.
N-[1-Benzyl-5-(hydroxyiminomethyl)-1H-imidazol-4-yl]-formimidic acid ethyl ester (5b)
Crude (E/Z isomers). - 1H NMR (CDCl3, main isomer): δ = 8.49 (s, 1H, (EtO)CH=N), 8.32 (s, 1 H, CH-oxime), 7.37 (s, 1 H, H-2), 7.35-7.12 (m, 5 H, H-Ph), 5.44 (s, 2 H, CH2Ph), 4.30 (q, J = 7.2 Hz, 2 H, CH2CH3), 1.35 (t, J = 7.2 Hz, 3 H, CH2CH3), OH - undetected.
N-[5-(Hydroxyiminomethyl)-1-methyl-1H-imidazol-4-yl]-acetimidic acid ethyl ester (5c)
Crude, ca 80% purity (E/Z isomers). - 1H NMR (CDCl3, main isomer): δ = 8.10 (s, 1H, CH-oxime), 7.29 (s, 1 H, H-2), 4.23 (q, J = 7.1 Hz, 2 H, CH2CH3), 3.78 (s, 3 H, NCH3), 2.10 (s, 3 H, CH3), 1.31 (t, J = 7.1 Hz, 3 H, CH2CH3), OH - undetected. - IR (CHCl3): v = 1664 (C=N imidate), 1621 cm-1 (C=N oxime). - LSIMS (+), m/z (% rel. int.): 211 (M+H, 52).
N-[1-Benzyl-5-(hydroxyiminomethyl)-1H-imidazol-4-yl]-acetimidic acid ethyl ester (5d)
Crude (E/Z isomers). - 1H NMR (CDCl3, main isomer): δ = 8.15 (s, 1H, CH-oxime), 7.42-7.10 (m, 6 H, H-Ph & H-2), 5.46 (s, 2 H, CH2Ph), 4.23 (q, J = 7.1 Hz, 2 H, CH2CH3), 2.15 (s, 3 H, CH3), 1.31 (t, J = 7.1 Hz, 3 H, CH2CH3), OH - undetected.
7-Methyl-7H-purine (6a)
Yield 60%, mp 176-178°C (CHCl3), lit. [23a] mp 183-184°C. - 1H NMR (CDCl3): δ = 9.17 (s, 1 H, H-2), 8.98 (s, 1 H, H-6), 8.20 (s, 1 H, H-8), 4.00 (s, 3 H, NCH3). - MS, m/z (% rel. int.): 134 (M+ ·, 100), 107 (21), 81 (16), 80 (16), 57 (25), 56 (23), 42 (67); HR-MS calcd. for C6H8N4 - 134.0593, found - 134.0593.
7-Benzyl-7H-purine (6b)
Yield 66%, data for this compound - see lit. [1].
2,7-Dimethyl-7H-purine (6c)
Yield 49%, mp 85°C (CHCl3/MeOH). - 1H NMR (CDCl3): δ = 8.85 (s, 1 H, H-6), 8.13 (s, 1 H, H-8), 3.95 (s, 3 H, NCH3), 2.84 (s, 3 H, CH3). - 13C NMR (CDCl3): δ = 162.8 (C-2), 157.3 (C-4), 148.3 (C-8), 139.4 (C-6), 125.1 (C-5), 32.0 (NCH3), 25.9 (CH3). - MS, m/z (% rel. int.): 148 (M+ ·, 100), 147 (23), 134 (8), 121 (11), 120 (20), 106 (10), 94 (13), 80 (12), 67 (9), 53 (17), 52 (17), 42 (35); HR-MS calcd. for C7H8N4 - 148.0749, found - 148.0744. - LSIMS (+), m/z (% rel. int.): 149 (M+H, 100).
7-Benzyl-2-methyl-7H-purine (6d)
Yield 20%, mp 146-149°C (CHCl3/ether). - 1H NMR (CDCl3): δ = 8.62 (s, 1 H, H-6), 8.23 (s, 1 H, H-8), 7.42-7.18 (m, 5 H, H-Ph), 5.41 (s, 2 H, CH2), 2.83 (s, 3 H, CH3). - 13C NMR (CDCl3): δ = 162.7 (C-2), 161.6 (C-4), 147.8 (C-8), 140.1 (C-6), 133.7, 129.3 (2xC), 128.9, 127.4 (2xC) [C-Ph], 123.0 (C-5), 50.2 (CH2), 25.9 (CH3). - MS, m/z (% rel. int.): 224 (M+ ·, 36), 223 (17), 91 (100), 65 (19); HR-MS calcd. for C13H12N4 - 224.1062, found - 224.1062.
7-Benzyl-2-butyl-7H-purine (6e)
Yield 31%, mp 103-106°C (CHCl3/MeOH). - 1H NMR (CDCl3): δ = 8.67 (s, 1 H, H-6), 8.25 (s, 1 H, H-8), 7.43-7.32 & 7.30-7.15 (2 x m, 5 H, H-Ph), 5.41 (s, 2 H, CH2Ph), 3.08 (t, J = 7.7 Hz, 2 H, CH2C3H7), 1.94-1.30 (m, 4 H, 2 x CH2), 0.93 (t, J = 7.2 Hz, 3 H, CH3). - 13C NMR (CDCl3): δ = 166.2 (C-2), 147.9 (C-8), 139.9 (C-6), 133.7, 129.4 (2xC), 129.1, 127.5 (2xC) [C-Ph], 123.2 (C-5), 50.3 (CH2Ph), 39.1, 31.3, 22.6, 14.0 [C4H9], C-4 - undetected. - MS, m/z (% rel. int.): 266 (M+ ·, 7), 251 (3), 237 (8), 224 (38), 91 (100), 65 (12); HR-MS calcd. for C16H18N4 - 266.1531, found - 266.1539. - LSIMS (+), m/z (% rel. int.): 267 (M+H, 59), 91 (100). - Elemental analysis for C16H18N4 (266.35): calcd. C 72.15, H 6.81, N 21.04; found C 71.61, H 6.42, N 21.16.
2-Butyl-7-methyl-7H-purine (6f)
Yield 35%, semi-crystalline. - 1H NMR (acetone-d6): δ = 9.01 (s, 1 H, H-6), 8.37 (s, 1 H, H-8), 4.04 (s, 3 H, NCH3), 2.97 (t, J = 7.0 Hz, 2 H, CH2C3H7), 1.92-1.20 (m, 4 H, 2 x CH2), 0.87 (t, J = 7.2 Hz, 3 H, CH3). - 13C NMR (CDCl3): δ = 166.2 (C-2), 161.3 (C-4), 148.3 (C-8), 139.4 (C-6), 123.8 (C-5), 39.2, 32.0, 31.4, 22.6, 14.0 [C4H9 & NCH3]. - MS, m/z (% rel. int.): 190 (M+ ·, 6), 175 (12), 162 (7), 161 (33), 149 (10), 148 (100), 147 (8), 42 (9); HR-MS calcd. for C10H14N4 - 190.1218, found - 190.1219.
Preparation of Purine mono-N-Oxides (11a-e, g). General Procedure
The intermediates 5a-e,g were prepared according to the procedure used previously for preparation of purines 6a-f.
The appropriate imidate 5a-e,g was dissolved in a dry ethanol or dry acetonitrile (4 mL for ca 50 mg) and the reaction mixture was heated in a sealed tube for 2-4 h at 100-200°C (see Table 1). After cooling and evaporation of the solvent the residue was purified by chromatography to give the desired purine N-oxides (eluent: from CHCl3 to CHCl3/MeOH - 20:1; except for 11a: from CHCl3 to CHCl3/MeOH - 5:1). Yields are given in Table 1, 13C NMR spectra - in Table 2.
As the transformation of 5g to N-oxide 11g proved somewhat troublesome, an analytical sample of 5g was purified by column chromatography for characterization (the yield was not determined).
N-[1-Benzyl-5-(hydroxyiminomethyl)-1H-imidazol-4-yl]-propionimidic acid ethyl ester (5g)
Thick oil. - 1H NMR (CDCl3): δ = 8.06 (s, 1 H, CH-oxime), 7.37-7.09 (m, 6 H, H-Ph & H-2), 5.40 (s, 2 H, CH2Ph), 4.25 (q, J = 7.1 Hz) & 4.24 (q, J = 7.1 Hz) [2 H, OCH2CH3, E/Z isomers], 2.52 (q, J = 7.6 Hz) & 2.51 (q, J = 7.6 Hz) [2 H, CH2CH3, E/Z isomers], 1.33 (t, J = 7.1 Hz) & 1.32 (t, J = 7.1 Hz) [3 H, OCH2CH3, E/Z isomers], 1.134 (t, J = 7.6 Hz) & 1.127 (t, J = 7.6 Hz) [3 H, CH2CH3, E/Z isomers], OH - undetected. - MS, m/z (% rel. int.): 300 (M+ ·, 31), 284 (6), 283 (27), 272 (2), 271 (2), 255 (9), 239 (7), 238 (9), 237 (12), 216 (7), 199 (9), 198 (6), 197 (5), 135 (6), 121 (8), 106 (5), 92 (10), 91 (100), 65 (9), 57 (10); HR-MS calcd. for C16H20N4O2 - 300.1586, found - 300.1586.
7-Methyl-7H-purine 1-oxide (11a)
Semi-crystalline. - 1H NMR (CDCl3/DMSO-d6 - 5:1): δ = 8.96 (s, 2 H, H-2 & H-6), 8.32 (s, 1 H, H-8), 3.93 (s, 3 H, NCH3). - MS, m/z (% rel. int.): 150 (M+ ·, <1), 135 (9), 134 (100), 107 (10), 106 (5), 80 (4), 79 (3), 66 (2), 53 (2), 52 (2), 42 (10); HR-MS calcd. for C6H6N4O - 150.0542, found - 150.0537.
7-Benzyl-7H-purine 1-oxide (11b)
Mp 223-224°C (subl.). - 1H NMR (CDCl3): δ = 8.91 (d, J = 1.8 Hz, 1 H, H-2), 8.43 (d, J = 1.8 Hz, 1 H, H-6), 8.28 (s, 1 H, H-8), 7.45-7.20 (m, 5 H, H-Ph), 5.34 (s, 2 H, CH2). - MS, m/z (% rel. int.): 226 (M+ ·, 12), 210 (38), 209 (13), 91 (100), 65 (12); HR-MS calcd. for C12H10N4O - 226.0855, found - 226.0854. - Elemental analysis for C12H10N4O x 1/2H2O (235.24): calcd. C 61.27, H 4.71, N 23.82; found C 62.34, H 4.69, N 22.41.
2,7-Dimethyl-7H-purine 1-oxide (11c)
Mp 190-192°(CHCl3/MeOH). - 1H NMR (CDCl3): δ = 8.85 (s, 1 H, H-6), 8.12 (s, 1 H, H-8), 3.95 (s, 3 H, NCH3), 2.85 (s, 3 H, CH3). - MS, m/z (% rel. int.): 164 (M+ ·, 2), 163 (5), 162 (4), 148 (100), 147 (23), 134 (35), 121 (10), 120 (16), 107 (9), 106 (10), 94 (8), 80 (10), 67 (4), 53 (8), 52 (7), 42 (24); HR-MS calcd. for C7H8N4O - 164.0698, found - 164.0696.
7-Benzyl-2-methyl-7H-purine 1-oxide (11d)
Data - see lit. [2].
7-Benzyl-2-butyl-7H-purine 1-oxide (11e)
Mp 225-226°C (CHCl3 ). - 1H NMR (CDCl3): δ = 8.47 (s, 1 H, H-6), 8.22 (s, 1 H, H-8), 7.45-7.15 (m, 5 H, H-Ph), 5.31 (s, 2 H, CH2Ph), 3.15 (t, J = 7.7 Hz, 2 H, CH2C3H7), 1.96-1.78 & 1.55-1.33 (2 x m, 4 H, 2 x CH2), 0.96 (t, J = 7.2 Hz, 3 H, CH3). - MS, m/z (% rel. int.): 282 (M+ ·, 2), 266 (12), 265 (42), 224 (19), 198 (6), 91 (100), 65 (8); HR-MS calcd. for C16H18N4O - 282.1481, found - 282.1479. - LSIMS (+), m/z (% rel. int.): 283 (M+H, 100), 91 (91).
7-Benzyl-2-ethyl-7H-purine 1-oxide (11g)
Semi-crystalline. - 1H NMR (CDCl3): δ = 8.65 (s, 1 H, H-6), 8.24 (s, 1 H, H-8), 7.50-7.18 (m, 5 H, H-Ph), 5.41 (s, 2 H, CH2Ph), 3.09 (q, J = 7.6 Hz, 2 H, CH2CH3), 1.40 (t, J = 7.6 Hz, 3 H, CH2CH3). - MS, m/z (% rel. int.): 254 (M+ ·, 6), 253 (5), 252 (4), 238 (22), 237 (14), 210 (5), 147 (9), 134 (10), 106 (15), 91 (100), 77 (14), 65 (17), 44 (9), 39 (8); HR-MS calcd. for C14H14N4O - 254.1168, found - 254.1165.
N-(5-Cyano-1-methyl-1H-imidazol-4-yl)-acetimidic acid ethyl ester (12)
This was obtained as a by-product in the reaction leading to purine N-oxide 11c (yield less than 10%); semi-crystalline, unstable. - 1H NMR (CDCl3): δ = 7.36 (s, 1 H, H-2), 4.28 (q, J = 7.1 Hz, 2 H, CH2), 3.73 (s, 3 H, NCH3), 2.09 (s, 3 H, CH3), 1.33 (t, J = 7.1 Hz, 3 H, CH2CH3). - IR (CHCl3): v = 2219 (CN), 1662 cm-1 (C=N). - MS, m/z (% rel. int.): 192 (M+ ·, 39), 177 (3), 164 (11), 151 (11), 150 (24), 149 (51), 148 (15), 147 (19), 135 (14), 122 (100), 107 (18), 95 (6), 67 (14), 43 (63), 42 (18); HR-MS calcd. for C9H12N4O - 192.1011, found - 192.1026. - LSIMS (+), m/z (% rel. int.): 193 (M+H, 100).
Transformations to Schiff bases
Procedure A. - To a solution of aldehyde (2a, 2b correspondingly; 0.50 mmol) and aniline derivative (0.70 mmol) in toluene (5 mL) a catalytic amount of p-TsOH acid (5 mg, 0.03 mmol) was added and the mixture was heated to reflux for 2.5-3 h. After cooling and evaporation to dryness, crude products were purified by chromatography (CHCl3 as eluent) to give a mixture of E/Z isomers (products 13a, 13b and 13h were obtained).
Procedure B. - Imidazole aldehyde (2a, 2b correspondingly; 1.0 mmol), the proper aniline derivative (1.20 mmol), a catalytic amount of p-TsOH acid (5 mg, 0.03 mmol) and Si(OEt)4 (7.0 g) were dissolved in toluene (30 mL) and the reaction mixture was heated to reflux for 3-20 h until the aldehyde disappeared (TLC monitoring). After cooling, the solvent and most of the Si(OEt)4 was evaporated. The residue was recrystallized from ethanol or purified by chromatography (eluent CHCl3 or CHCl3/MeOH - 20:1) to give pure Schiff bases (mixture of E/Z isomers). Products 13c-g were obtained.
Data for products 13a and 13b were described earlier [9].
4-{[(E/Z)-1-(1-Methyl-4-nitro-1H-imidazol-5-yl)methylidene]amino}benzonitrile (13c)
Solid, yield 66%. - 1H NMR (CDCl3, main isomer in E/Z mixture): δ = 9.19 (s, 1 H, CH=N), 7.74 (apparent d, J = 8.6 Hz, 2 H, H-Ar), 7.56 (s, 1 H, H-2), 7.31 (apparent d, J = 8.6 Hz, 2 H, H-Ar), 4.15 (s, 3 H, CH3). - MS, m/z (% rel. int.): 255 (M+ ·, 53), 239 (15), 238 (100), 225 (27), 210 (73), 208 (62), 207 (38), 196 (10), 184 (15), 183 (10), 182 (12), 181 (14), 180 (12), 168 (15), 167 (40), 155 (13), 141 (13), 138 (33), 129 (18), 128 (10), 114 (22), 108 (29), 102 (45), 84 (21), 81 (13), 67 (23), 42 (73). - Elemental analysis for C12H9N5O2 (255.24): calcd. C 56.47, H 3.55, N 27.44; found C 56.38, H 3.42, N 27.35.
4-{[(E/Z)-1-(1-Benzyl-4-nitro-1H-imidazol-5-yl)methylidene]amino}benzonitrile (13d)
Solid, yield 62%. - 1H NMR (CDCl3 main isomer in E/Z mixture): δ = 9.12 (s, 1 H, CH=N), 7.70 (apparent d, J = 8.3 Hz, 2 H, H-ArCN), 7.61 (s, 1 H, H-2), 7.42-7.35 (m, 2 H, H-Ar), 7.23-7.13 (m, 5 H, H-Ar), 5.79 (s, 2 H, CH2). - MS, m/z (% rel. int.): 331 (M+ ·, 20), 314 (13), 284 (27), 226 (14), 225 (100), 180 (17), 129 (9), 91 (72), 65 (10). - Elemental analysis for C18H13N5O2 x 1/2H2O (340.34): calcd. C 63.52, H 4.15, N 20.58; found C 63.51, H 4.03, N 19.98.
N-[(E/Z)-1-(1-Benzyl-4-nitro-1H-imidazol-5-yl)methylidene]-N-(2,4,6-tribromophenyl)-amine (13e)
Yield 67%, one isomer only, mp 173-174°C (CHCl3). - 1H NMR (CDCl3): δ = 9.04 (s, 1 H, CH=N), 7.73 (s, 2 H, H-Ar(Br)3), 7.57 (s, 1 H, H-2), 7.43-7.18 (m, 5 H, H-Ph), 5.86 (s, 2 H, CH2). - MS, m/z (% rel. int.): 546 (4), 544 (12), 542 (12), and 540 (4) [isotopic M+ ·], 527 (6), 525 (6), 497 (8), 496 (5), 495 (8), 465 (6), 463 (11), 461 (6), 433 (7), 418 (8), 416 (7), 359 (48), 357 (100), 355 (50), 91 (80), 65 (10). - Elemental analysis for C17H11N4Br3O2 (543.01): calcd. C 37.60, H 2.04, N 10.32; found C 37.69, H 2.18, N 10.34.
N-[(E/Z)-1-(1-Benzyl-4-nitro-1H-imidazol-5-yl)methylidene]-N-tritylamine (13f)
Yield 71%, one isomer only, mp 167-168°C (EtOH). - 1H NMR (CDCl3): δ = 8.59 (s, 1 H, CH=N), 7.52 (s, 1 H, H-2), 7.33-7.24 & 7.07-6.94 (2 x m, 20 H, H-Ph), 5.85 (s, 2 H, CH2). - MS, m/z (% rel. int.): 472 (M+ ·, 0.1), 244 (20), 243 (100), 228 (6), 165 (28), 91 (5). - Elemental anal. for C30H24N4O2 (472.55): calcd. C 76.25, H 5.12, N 11.86; found C 76.07, H 5.11, N 11.75.
N-(4-Bromophenyl)-N-[(E/Z)-1-(1-methyl-4-nitro-1H-imidazol-5-yl)methylidene]-amine (13g)
Solid, yield 90%. - 1H NMR (CDCl3, main isomer in E/Z mixture, >95%): δ = 9.20 (s, 1 H, CH=N), 7.56 (apparent d, J = 7.5 Hz, 2 H, H-Ar), 7.51 (s, 1 H, H-2), 7.16 (apparent d, J = 7.5 Hz, 2 H, H-Ar), 4.14 (s, 3 H, CH3). - MS, m/z (% rel. int.): 310 (99) & 308 (100) [isotopic M+ ·], 293 (74), 291 (74), 265 (18), 264 (10), 263 (54), 262 (20), 261 (38), 260 (14), 239 (12), 237 (18), 236 (13), 235 (9), 234 (8), 229 (27), 222 (42), 220 (40), 212 (31), 199 (26), 198 (27), 184 (67), 182 (16), 169 (10), 157 (27), 155 (28), 138 (11), 108 (13), 84 (15), 81 (10), 76 (16), 75 (14), 67 (18), 42 (34); HR-MS calcd. for C11H9N4BrO2 - 307.9909, found - 307.9908. - Elemental analysis for C11H9N4BrO2 (309.12): calcd. C 42.74, H 2.93; found C 43.14, H 3.15.
N-[(E/Z)-1-(1-Benzyl-4-nitro-1H-imidazol-5-yl)methylidene]-N-(4-bromophenyl)-amine (13h)
Solid, yield 91%. - 1H NMR (CDCl3, main isomer in E/Z mixture): δ = 9.15 (s, 1 H, CH=N), 7.60-7.00 (m, 10 H, H-Ar & H-2), 5.81 (s, 2 H, CH2). - MS, m/z (% rel. int.): 386 (12) & 384 (12) [isotopic M+ ·], 369 (11), 367 (11), 339 (16), 338 (12), 337 (16), 336 (9), 280 (63), 278 (65), 262 (9), 260 (11), 235 (9), 233 (9), 199 (12), 183 (7), 172 (5), 171 (5), 157 (9), 155 (9), 129 (9), 91 (100), 65 (15); HR-MS calcd. for C17H13N4BrO2 - 384.0222, found - 384.0219. - LSIMS (+), m/z (% rel. int.): 387 (47) & 385 (49) [isotopic M+H], 91 (100). - Elemental analysis for C17H13N4BrO2 (385.22): calcd. C 53.01, H 3.40, N 14.54; found C 52.40, H 3.38, N 14.18.
Catalytic Hydrogenation of Schiff Bases 13a-h
The nitrocompound was dissolved in ethanol (5-10 mL for 100 mg) and it was hydrogenated in Parr apparatus using 10-30% of catalyst (10% Pd/C): 13a, 13b, 13g, 13h - 40 psi, 4 h; 13c, 13d - 25 psi, 4 h; 13e - 15 psi, 1 h; 13f - 25 psi, 5 h. The reactions were monitored on TLC (CHCl3/MeOH - 20:1). After the reduction, the catalyst was filtered off, washed with methanol, and the solvent was evaporated to give the crude products 14a-h.
Compounds 14a, 14b were described in the previous paper [9]. For crude compound 14d MS spectrum was recorded and for compound 14g - 1H NMR and MS spectra are given.
4-{[(E/Z)-1-(4-Amino-1-benzyl-1H-imidazol-5-yl)methylidene]amino}benzonitrile (14d)
Crude, main product in the mixture (ca 90% purity). - MS, m/z (% rel. int.): 301 (M+ ·, 4), 210 (1), 192 (3), 118 (100), 91 (49), 64 (16); HR-MS calcd. for C18H15N5 - 301.1327, found - 301.1331.
N-[(E/Z)-1-(4-Amino-1-methyl-1H-imidazol-5-yl)methylidene]-N-(4-bromophenyl)-amine (14g)
Crude, ca 80% purity. - 1H NMR (DMSO-d6): δ = 8.98 (s, 1 H, CH=N), 8.06 (s, 1 H, H-2), 7.70-7.20 (m, 4 H, H-Ar), 3.70 (s, 3 H, CH3). - MS, m/z (% rel. int.): 280 (5) & 278 (5) [isotopic M+ ·]. - LSIMS (+), m/z (% rel. int.): 281 (91) & 279 (100) [isotopic M+H].
Transformation of Imines 14a-h to Purines
The above imines 14a-h (ca 100 mg) were suspended in the appropriate orthoester (HC(OEt)3, MeC(OEt)3, 5 mL) and the reaction mixture was heated to reflux until the reaction was complete (with HC(OEt)3: 14a, 14b, 14h - 4 h; 14c, 14e, 14f - 10 h; 14d, 14g - 6 h; with MeC(OEt)3: 14g - 4 h, 14h - 6 h).
The excess of the orthoester was removed under reduced pressure and the residue (crude imidates 15a-j) was dissolved in ethanol saturated with ammonia (5 mL). The reaction mixture was heated in a sealed tube to give the desired purines (conditions for highest yields: 15a, 15b - 130°C, 4 h; 15c - 80°C, 10 h; 15d - 80°C, 4 h; 15e, 15f - 110°C, 6 h; 15g, 15i, 15j - 130°C, 3 h; 15h -140°C, 4 h. The products were isolated as it was described in synthesis via oximes. Yields - see Table 3.
N-(7-Benzyl-6,7-dihydro-1H-purin-6-yl)-N-(4-methylphenyl)-amine (16b)
The formation of this compound was observed occasionally (from 13b) with the yields less than 5-10%; attempts to isolate and purify this intermediate gave the sample of ca 80% purity, still contaminated with other by-products. - 1H NMR (acetone-d6, diagnostic signals): δ = 8.30 (s, broad, 1 H, NH), 8.28 (s, 1 H, H-2*)), 8.10 (s, 1 H, H-8*)), 7.52 & 7.12 (AA’XX’, H-Tol), 7.40-7.18 (m, H-Ph & CH*)), 5.68 (s, 2 H, CH2), 2.41 (s, 3 H, CH3). - MS, m/z (% rel. int.): 317 (M+ ·, 10), 316 (44), 315 (24), 225 (4), 91 (100), 65 (26), 59 (24), 57 (27), 55 (26), 43 (51).
N-(4-Cyanophenyl)-formamide (17)
Mp 186-190°C (CHCl3), lit. [24] mp 189-190°C. - 1H NMR (CDCl3): δ = 8.45 (s, 1 H, CHO), 7.80- 7.60 (AA’XX’, 4 H, H-Ar), NH - undetected. - MS, m/z (% rel. int.): 147 (M+1, 7), 146 (M+ ·, 63), 119 (12), 118 (100), 117 (7), 91 (51), 90 (13), 64 (12), 63 (9).
5-Isocyanomethyl-1-methyl-4-nitro-1H-imidazole (18)
To a stirred solution of t-BuOK (616 mg, 5.50 mmol) in anhydrous DMF (4 mL), cooled to 0°C, a solution of 1-methyl-4-nitro-1H-imidazole 1a (254 mg, 2.00 mmol) and phenylthiomethylisocyanide (333 mg, 2.23 mmol) in DMF (3 mL) was added dropwise via syringe - keeping the temperature at ca 0-3°C. After additional 0.5 h of stirring at this temperature the mixture was poured into crushed dry ice (ca 5 g) followed by slow addition of ethyl acetate (40 mL). The mixture was washed with cold brine (3 x 15 mL) and the organic layer was dried with anhydrous Na2SO4. After evaporation of the solvent the product was isolated by column chromatography using at the beginning CCl4, then CHCl3 as eluent. Yield 285 mg (86%); mp 68-70°C (CHCl3). - 1H NMR (CDCl3): δ = 7.50 (s, 1 H, H-2), 5.19 (s, 2 H, CH2), 3.87 (s, 3 H, NCH3). - MS, m/z (% rel. int.): 166 (M+ ·, 37), 140 (5), 134 (5), 120 (10), 119 (32), 109 (12), 95 (19), 94 (30), 93 (27), 83 (16), 81 (30), 68 (16), 67 (40), 66 (15), 55 (14), 54 (26), 53 (12), 52 (15), 42 (100); HR-MS calcd. for C6H6N4O2 - 166.0491, found - 166.0490.
Hydrolysis of Isocyanide 18
The isocyanide 18 (1.0 mmol) was dissolved in aqueous 80% ethanol (5 mL) and a four drops of concd. HCl were added. The mixture was refluxed for 3 h, evaporated to dryness and the crude product (as hydrochloride, quantitative yield) was recrystallized from methanol.
(1-Methyl-4-nitro-1H-imidazol-5-yl)methylamine (19)
As hydrochloride, mp 230-245°C (MeOH, decomp.). - 1H NMR (DMSO-d6): δ = 8.54 (s, broad, 3 H, NH3 +), 7.92 (s, 1 H, H-2), 4.34 (s, 2 H, CH2), 3.85 (s, 3 H, NCH3). - MS, m/z (% rel. int.): 157 (MH+, 4), 156 (1), 155 (3), 153 (3), 139 (49), 138 (58), 121 (31), 120 (17), 111 (8), 110 (12), 109 (37), 108 (79), 94 (28), 93 (15), 83 (29), 82 (28), 81 (23), 69 (14), 68 (19), 67 (24), 58 (12), 56 (12), 55 (17), 54 (24), 42 (100), 38 (30) & 36 (94) [isotopic HCl+ ·]; HR-MS calcd. for C5H7N4O (M-OH) - 139.0620, found - 139.0621. - LSIMS (+), m/z (% rel. int.): 157 (MH+, 100).
N-[(1-Methyl-4-nitro-1H-imidazol-5-yl)methyl]-formamide (23)
This side-product (small amounts, less than 5%; usually obtained due to a partial hydrolysis of isocyano group in 18) was isolated by column chromatography for characterization (eluent: CHCl3/MeOH - 20:1); ca 90% purity. - 1H NMR (CDCl3): δ = 8.18 (s, 1 H, CHO), 7.34 (s, 1 H, H-2), 6.78 (s, broad, 1 H, NH), 4.71 (d, J = 6.6 Hz, 2 H, CH2), 3.92 (s, 3 H, NCH3). - MS, m/z (% rel. int.): 184 (M+ ·, 5), 167 (6), 138 (100), 121 (21), 120 (65), 111 (51), 110 (16), 109 (33), 108 (53), 94 (17), 93 (17), 83 (23), 82 (22), 81 (16), 67 (17), 54 (12), 42 (57); HR-MS calcd. for C6H8N4O3 - 184.0596, found - 184.0571. - LSIMS (+), m/z (% rel. int.): 185 (M+H, 100).
(4-Amino-1-methyl-1H-imidazol-5-yl)methylamine (20)
This was obtained from hydrochloride of 19 by catalytic hydrogenation in Parr Apparatus according to known procedure (10% Pd/C, 25 psi, 4 h, in EtOH) [2,3,9]. Hydrochloride of 19 was neutralized by alcoholic solution of ammonia before hydrogenation. After the reduction the crude residue was used directly for the next steps.
For crude intermediate 20 (as hydrochloride) MS spectrum was recorded.
- MS, m/z (% rel. int.): 127 (MH+, 15), 110 (12), 109 (13), 108 (18), 94 (7), 81 (9), 68 (13), 58 (16), 42 (56), 38 (37) & 36 (100) [isotopic HCl+ ·].
Cyclocondensation of Diamine 20 with Orthoesters
Procedure A: The crude 5-aminomethyl-1-methyl-1H-imidazole-4-yl amine (20; ca 0.3 mmol) was suspended in the appropriate orthoester (HC(OEt)3, MeC(OEt)3, 3 mL) and the reaction mixture was heated to reflux for 6 h. Under these conditions the intermediates 21a, 21b underwent spontaneous oxidation to 6a, 6c. The excess of the orthoester was removed under reduced pressure and the products (6a, 6c) were isolated by column chromatography (eluent: from CHCl3 to CHCl3/MeOH - 20:1).
Procedure B: The crude diamine (20; ca 0.3 mmol) and the appropriate orthoester (HC(OEt)3, MeC(OEt)3, EtC(OEt)3, n-BuC(OMe)3; 2 mL) were dissolved in ethanol (10 mL) and the mixture was heated to reflux for 6 h. The ethanol and orthoester were evaporated and the products (21a-d) were isolated by preparative TLC (eluent: CHCl3/MeOH - 10:1). The yields are given in Table on Scheme 5.
7-Methyl-6,7-dihydro-1H-purine (21a)
Unstable, decomposed slowly yielding 6a. - 1H NMR (CDCl3/DMSO-d6 - 2:1): δ = 8.08 (s, 1 H, H-2), 7.54 (s, 1 H, H-8), 4.77 (s, 2 H, CH2), 3.72 (s, 3 H, NCH3), NH - undetected. - MS, m/z (% rel. int.): 136 (M+ ·, 5), 135 (6), 134 (46), 122 (14), 107 (14), 69 (24), 44 (84), 42 (100); HR-MS calcd. for C6H8N4 - 136.0749, found - 136.0742. - LSIMS (+), m/z (% rel. int.): 137 (M+H, 26).
2,7-Dimethyl-6,7-dihydro-1H-purine (21b)
Unstable, decomposed slowly yielding 6c. - 1H NMR (CDCl3): δ = 8.13 (s, 1 H, H-8), 5.33 (s, broad, 1 H, NH), 4.00 (s, 2 H, CH2), 3.96 (s, 3 H, NCH3), 2.87 (s, 3 H, CH3). - MS, m/z (% rel. int.): 150 (M+ ·, 3), 149 (24), 148 (100), 147 (24), 134 (59), 120 (16), 107 (13), 106 (11), 80 (11), 59 (21), 57 (25), 42 (33); HR-MS calcd. for C7H10N4 - 150.0905, found - 150.0954; calcd. for C7H9N4 (M-H) - 149.0827, found -149.0841.
2-Ethyl-7-methyl-6,7-dihydro-1H-purine (21c)
This was identified by MS analysis only, unstable. - MS, m/z (% rel. int.): 164 (M+ ·, 5), 163 (14), 162 (78), 161 (100), 134 (48), 110 (43), 107 (17), 80 (14), 56 (28), 42 (59); HR-MS calcd. for C8H11N4 (M-H) - 163.0984, found - 163.0984.
2-Butyl-7-methyl-6,7-dihydro-1H-purine (21d)
This was not isolated; the structure was proposed on the basis of MS (the molecular ion was detected while the post-reaction mixture was analyzed); m/z (% rel. int.) = 192 (M+ ·, 9), 191 (M-H, 38).
Acknowledgements
This work was supported (in part) by the State Committee for Scientific Research, Grant 2 P303 087 07.
References and Notes
- Ostrowski, S. Synlett 1995, 253–254.
- Ostrowski, S. Heterocycles 1996, 43, 389–396.
- Ostrowski, S. J. Chem. Res. (S), 1998, 14-15; (M), 0180-0187.
- Makosza, M. Synthesis 1991, 103–111. Makosza, M.; Wojciechowski, K. Liebigs Ann. Chem. 1997, 1805–1816.
- Makosza, M.; Owczarczyk, Z. J. Org. Chem. 1989, 54, 5094–5100.
- Makosza, M.; Ostrowski, S.; Kinowski, A.J. Synthesis 1993, 1215–1217.
- Brown, D.J. The Chemistry of Heterocyclic Compounds; (Weissberger, A. & Taylor, E.C. ed.); vol. 24/2, Brown, D.J. Wiley-Interscience: New York - London - Sydney - Toronto, 1971. [Google Scholar] Lister, J.H. ibid., The Purines; Supplement 1, New York - Chichester - Brisbane - Toronto - Singapore, 1996; pp. 61–82. [Google Scholar] Lythgoe, D.J.; Ramsden, Ch.A. Adv. Heterocycl. Chem. 1994, 61, 1–58.
- Sarasin, J.; Wegmann, E. Helv. Chim. Acta 1924, 7, 713–719. Cook, A.H.; Smith, E. J. Chem. Soc. 1949, 2329-2333, 3001-3007. Shaw, G.; Butler, D.N. J. Chem. Soc. 1959, 4040–4045. Naylor, R.N.; Shaw, G.; Wilson, D.V.; Butler, D.N. J. Chem. Soc. 1961, 4845–4850. Kadir, K.; Shaw, G.; Wright, D. J. Chem. Soc., Perkin Trans. 1 1980, 2728–2731. Andersen, K.E.; Pedersen, E.B. Liebigs Ann. Chem. 1985, 921–928. Birkett, P.R.; King, H.; Chapleo, Ch.B.; Ewing, D.F.; Mackenzie, G. Tetrahedron 1993, 49, 11029–11036.
- Ostrowski, S. Pol. J. Chem. 1994, 68, 2237–2247.
- Katritzky, A.R.; Lagowski, J.M. Chemistry of the Heterocyclic N-oxides; Blomquist, A.T., Ed.; Academic Press: London - New York, 1971; pp. 19–141. [Google Scholar]
- Adachi, K. Yakugaku Zasshi 1957, 77, 507-510, 514-516, Chem. Abstr. 1957, 51, 14744i, 14745i. Sternbach, L.H.; Kaiser, S.; Reeder, E. J. Am. Chem. Soc. 1960, 82, 475–480. Sternbach, L.H.; Reeder, E.; Keller, O.; Matlesics, W. J. Org. Chem. 1961, 26, 4488–4497.
- Schöpf, C.; Hartmann, A.; Koch, K. Chem. Ber. 1936, 69, 2766–2769. Hansen, S.B.; Petrow, V. J. Chem. Soc. 1953, 350–351. Braithwaite, R.S.W.; Holt, P.F.; Hughes, A.N. J. Chem. Soc. 1958, 4073–4077.
- Taylor, E.C.; Cheng, C.C.; Vogl, O. J. Org. Chem. 1959, 24, 2019–2021. Cresswell, R.M.; Brown, G.-B. J. Org. Chem. 1963, 28, 2560–2563.
- Hammet, L.P. J. Am. Chem. Soc. 1937, 59, 96–103. Shorter, J. Chem. in Britain 1969, 5, 269–274. Brown, H.C.; Okamoto, Y. J. Am. Chem. Soc. 1958, 80, 4979–4987.
- Ostrowski, S.; Wolniewicz, A.M. Proceedings of “5th Polish Symposium on Organic Chemistry”, Konstancin-Jeziorna, Poland, 1998; p. P-63, to be published soon.
- Malatesta, L. Progress in Inorganic Chemistry; Cotton, F.A., Ed.; vol. 1, Interscience: New York, 1959; pp. 283–379. [Google Scholar] Cotton, F.A.; Zingales, F. J. Am. Chem. Soc. 1961, 83, 351–355.
- A possible explanation of this instability is that 4-aminoimidazole derivatives can exist as the imino tautomeric form and are sufficiently reactive which may lead to ring-opening products: Al-Shaar, A.H.M.; Gilmour, D.W.; Lythgoe, D.J.; McClenaghan, I.; Ramsden, Ch.A. J. Chem. Soc., Perkin Trans. 1 1992, 2779–2788. Al-Shaar, A.H.M.; Chambers, R.K.; Gilmour, D.W.; Lythgoe, D.J.; McClenaghan, I.; Ramsden, Ch.A. ibid. 1992, 2789–2811. Bodor, N.; Dewar, M.J.S.; Harget, A.J. J. Am. Chem. Soc. 1970, 92, 2929–2936.
- Bergman, J.; Sand, P. Tetrahedron Lett. 1984, 25, 1957–1960.
- Iradyan, M.A.; Iradyan, N.S.; Avetyan, Sh.A. Arm. Khim. Zh. 1978, 31, 435–440, Chem. Abstr. 1978, 89, 197409g.
- Layer, R.W. Chem. Rev. 1963, 63, 489–510.
- Love, B.E.; Ren, J. J. Org. Chem. 1993, 58, 5556–5557.
- Sensi, P.; Gallo, G.G. Gazz. Chim. Ital. 1955, 85, 235–244.
- Bendich, A.; Russell, P.J., Jr.; Fox, J.J. J. Am. Chem. Soc. 1954, 76, 6073–6077. Montgomery, J.A.; Temple, C., Jr. J. Am. Chem. Soc. 1961, 83, 630–635.
- Huot, J.-Y.; Serve, D.; Desjardins, S.; Lessard, J. Can. J. Chem. 1988, 66, 35–44.
- Samples Availability: Available from the authors.
Scheme 1.
Scheme 2.
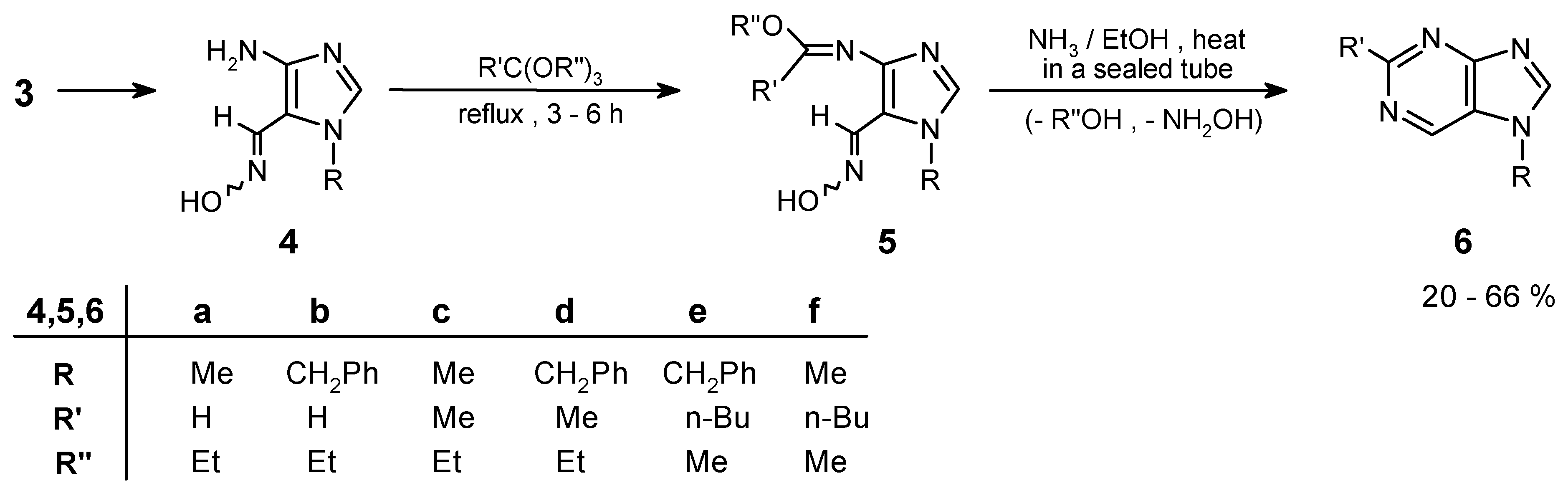
Figure 1.
Scheme 3.
Figure 2.
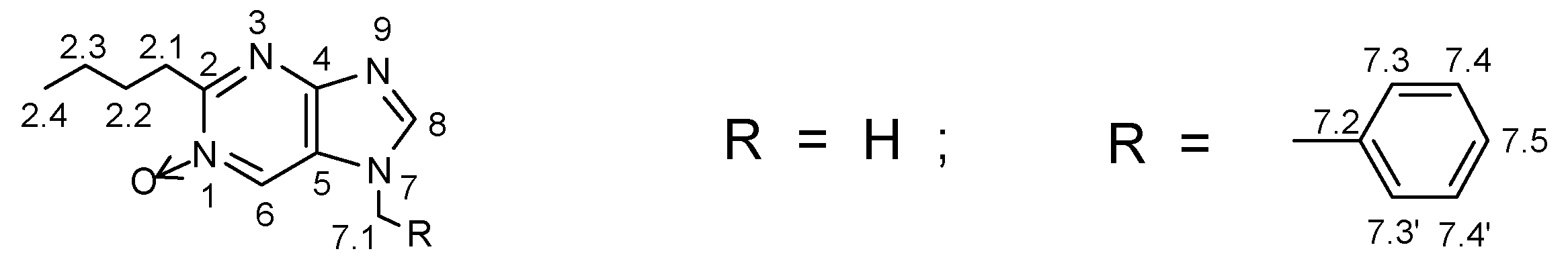
Scheme 4.
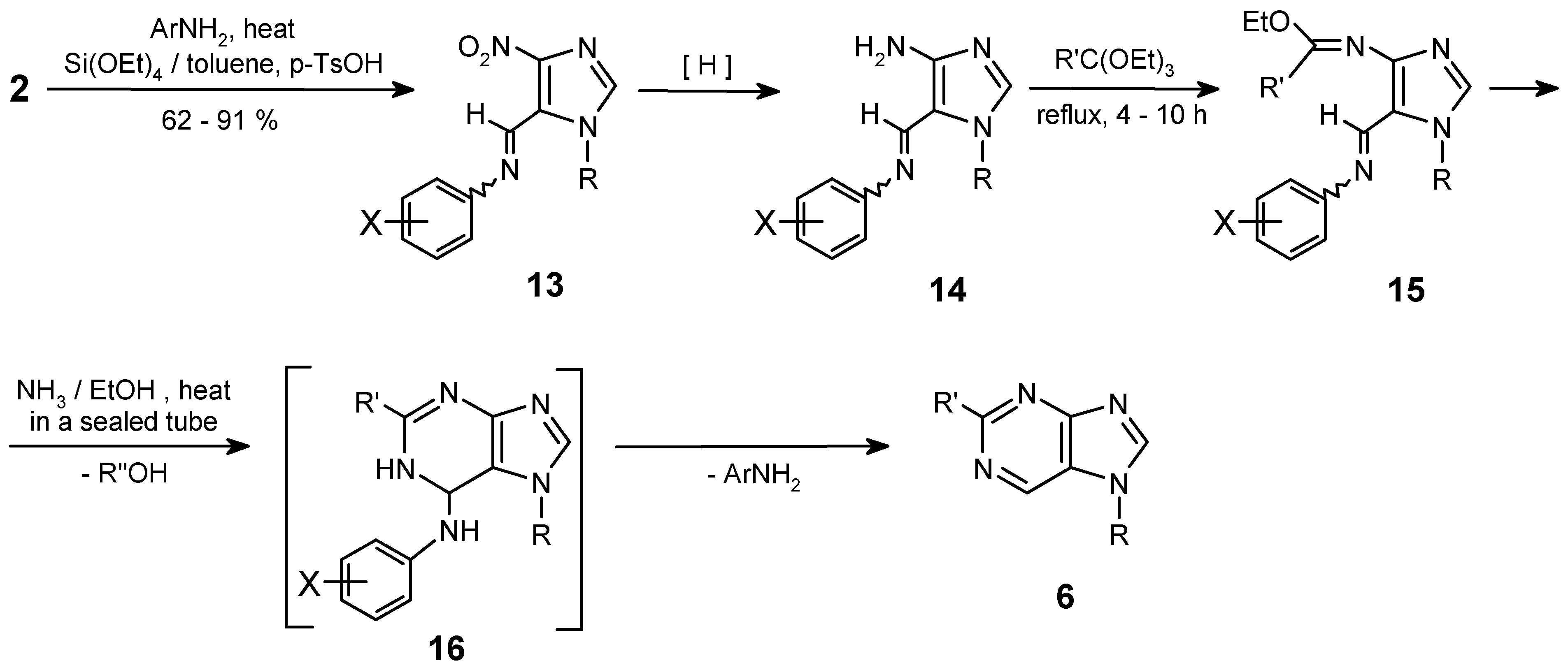
Figure 3.
Scheme 5.
Scheme 6.

{kind=link}
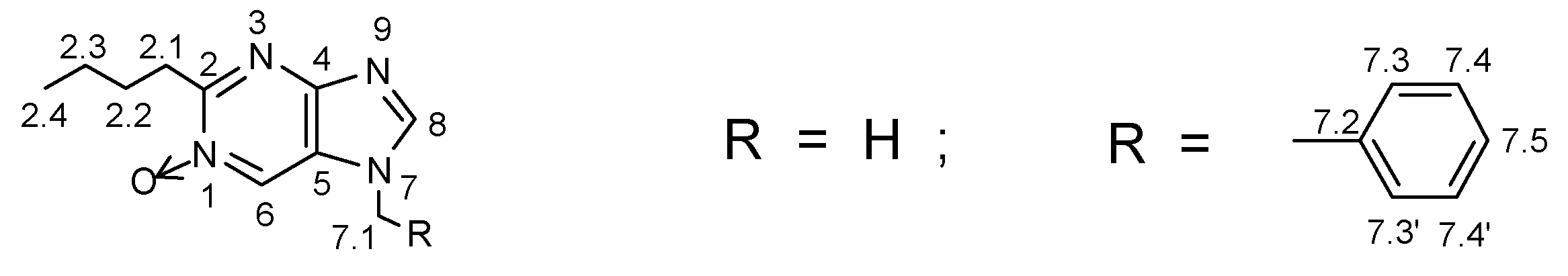{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Conditions | Yield [%] |
|---|---|---|
| 11a | 140°C, acetonitrile, 4 h | 52 |
| 11b | 140°C, acetonitrile, 4 h | 58 |
| 11c | 200°C, acetonitrile, 3 h | 13 |
| 11d | 100°C, ethanol, 3 h | 83*) |
| 11e | 160°C, ethanol, 2 h | 73 |
| 11g | 200°C, ethanol, 3 h | < 5 |
*) data from lit. [2].
| Compound & No | C-2 | C-4 | C-5 | C-6 | C-8 | C-2.1 | C-2.2 | C-2.3 | C-2.4 | C-7.1 | C-7.2 | C-7.3 (C-7.3’) | C-7.4 (C-7.4’) | C-7.5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 11a 11a | 151.8 | **) | 125.0 | 139.1 | 148.3 | _ | _ | _ | _ | 31.1 | _ | _ | _ | - |
 11c 11c | 153.4 | **) | **) | 139.5 | 148.4 | 26.0 | _ | _ | _ | 32.0 | _ | _ | _ | _ |
 11b 11b | 146.0 | 151.9 | 124.3 | 129.8 | 148.9 | _ | _ | _ | _ | 50.7 | 132.6 | 127.6 | 129.7 | 129.6 |
 11d 11d | 156.1 | 151.3 | 123.4 | 129.6 | 148.3 | 20.1 | _ | _ | _ | 50.5 | 132.9 | 127.5 | 129.5 | 129.3 |
 11e 11e | 159.0 | 151.4 | 123.0 | 129.8 | 148.3 | 31.9 | 27.5 | 22.4 | 13.9 | 50.5 | 133.0 | 127.5 | 129.6 | 129.4 |
*) compound 11a in CDCl3/DMSO-d6 (5:1); **) undetected.
| 13-16 | X | R | R’ | Product, No &Yields*) |
|---|---|---|---|---|
| a | 4-Me | Me | H | 6a, 20% |
| b | 4-Me | CH2Ph | H | 6b, 28% |
| c | 4-CN | Me | H | 6a, < 5% |
| d | 4-CN | CH2Ph | H | 6b, < 5% |
| e | 2,4,6--tribromo | CH2Ph | H | 6b, traces |
| f | Ar = CPh3 | CH2Ph | H | 6b, < 5% |
| g | 4-Br | Me | H | 6a, 36% |
| h | 4-Br | CH2Ph | H | 6b, 36 (47**)) |
| i | 4-Br | Me | Me | 6c, 13% |
| j | 4-Br | CH2Ph | Me | 6d, <5% |
*) yields calcd. for the 13 (4 steps); **) optimized yield.
© 1999 by the authors. Reproduction of this article, by any means, is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Ostrowski, S. Synthesis of N-7-Substituted Purines from Imidazole Precursors. Molecules 1999, 4, 287-309. https://doi.org/10.3390/41000287
AMA Style
Ostrowski S. Synthesis of N-7-Substituted Purines from Imidazole Precursors. Molecules. 1999; 4(10):287-309. https://doi.org/10.3390/41000287
Chicago/Turabian StyleOstrowski, Stanislaw. 1999. "Synthesis of N-7-Substituted Purines from Imidazole Precursors" Molecules 4, no. 10: 287-309. https://doi.org/10.3390/41000287