Photooxidation of n-Hexanal in Air

Abstract
:Introduction
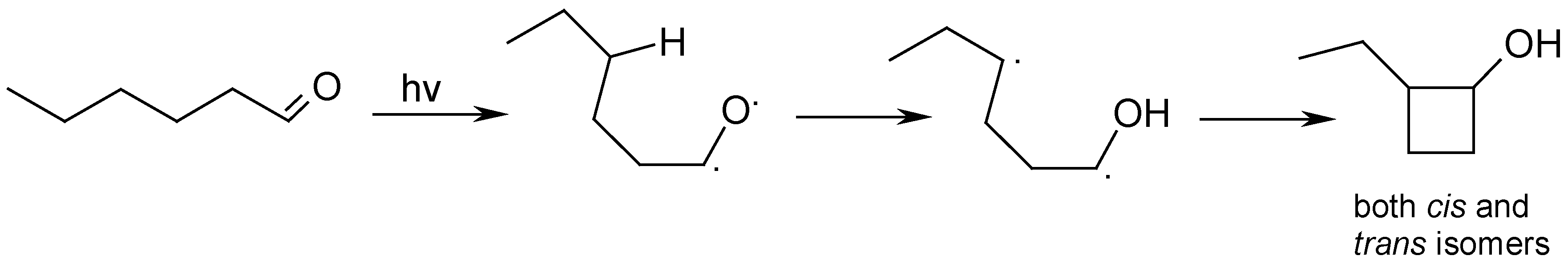
Results and Discussion
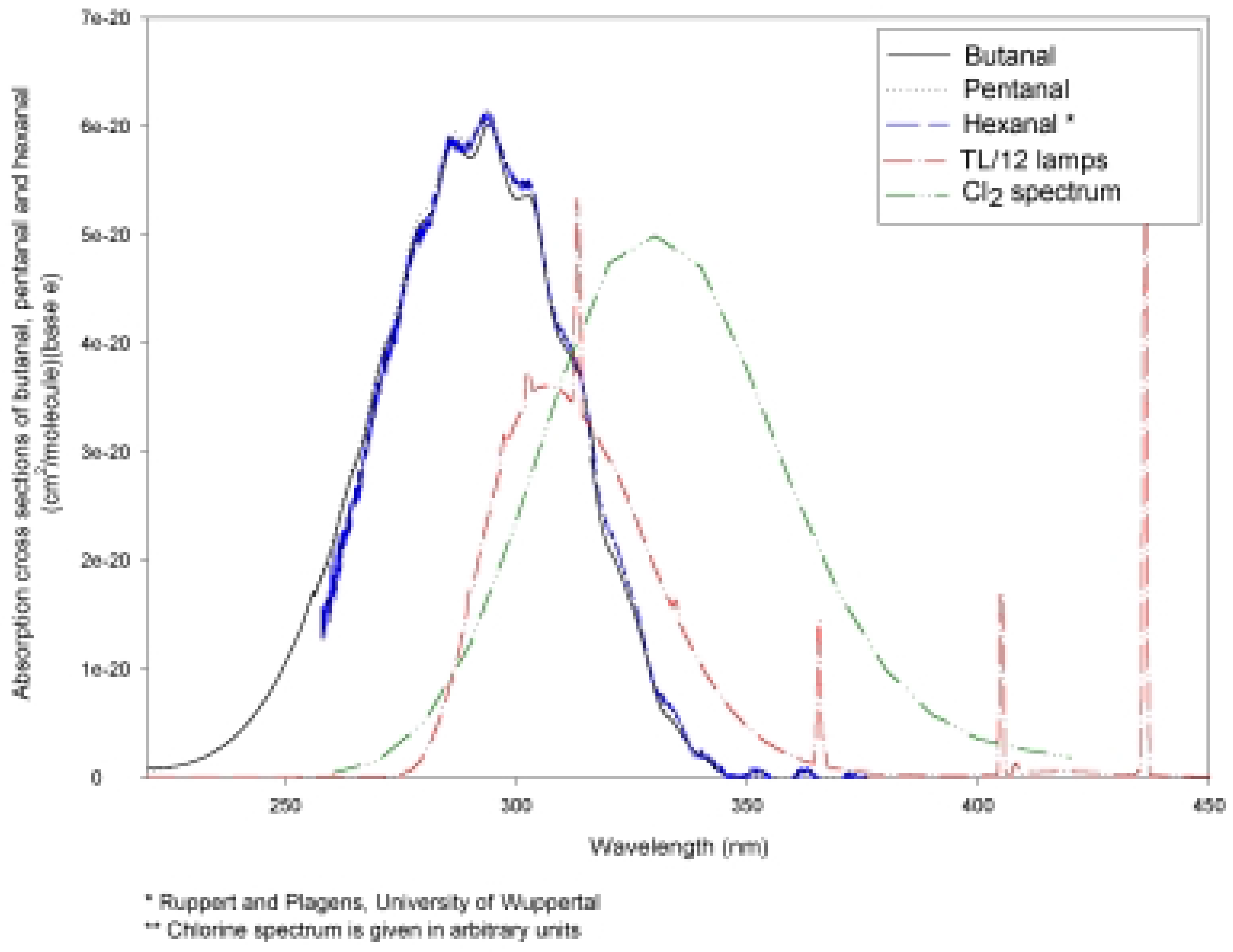
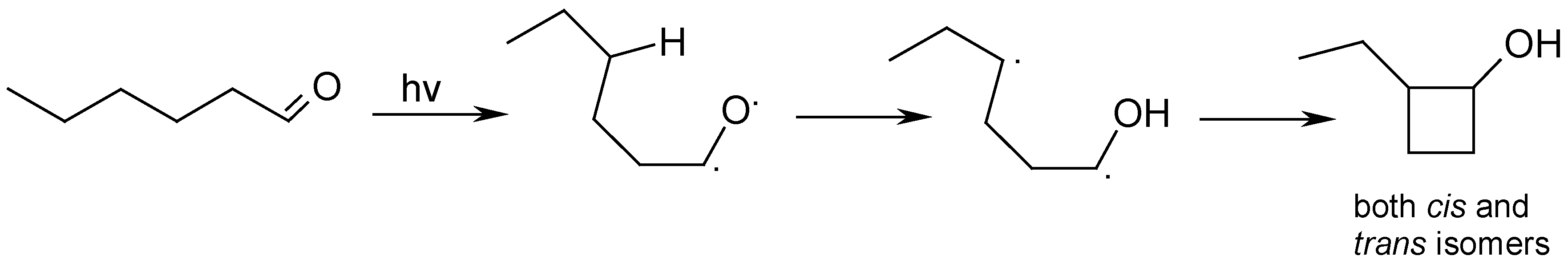
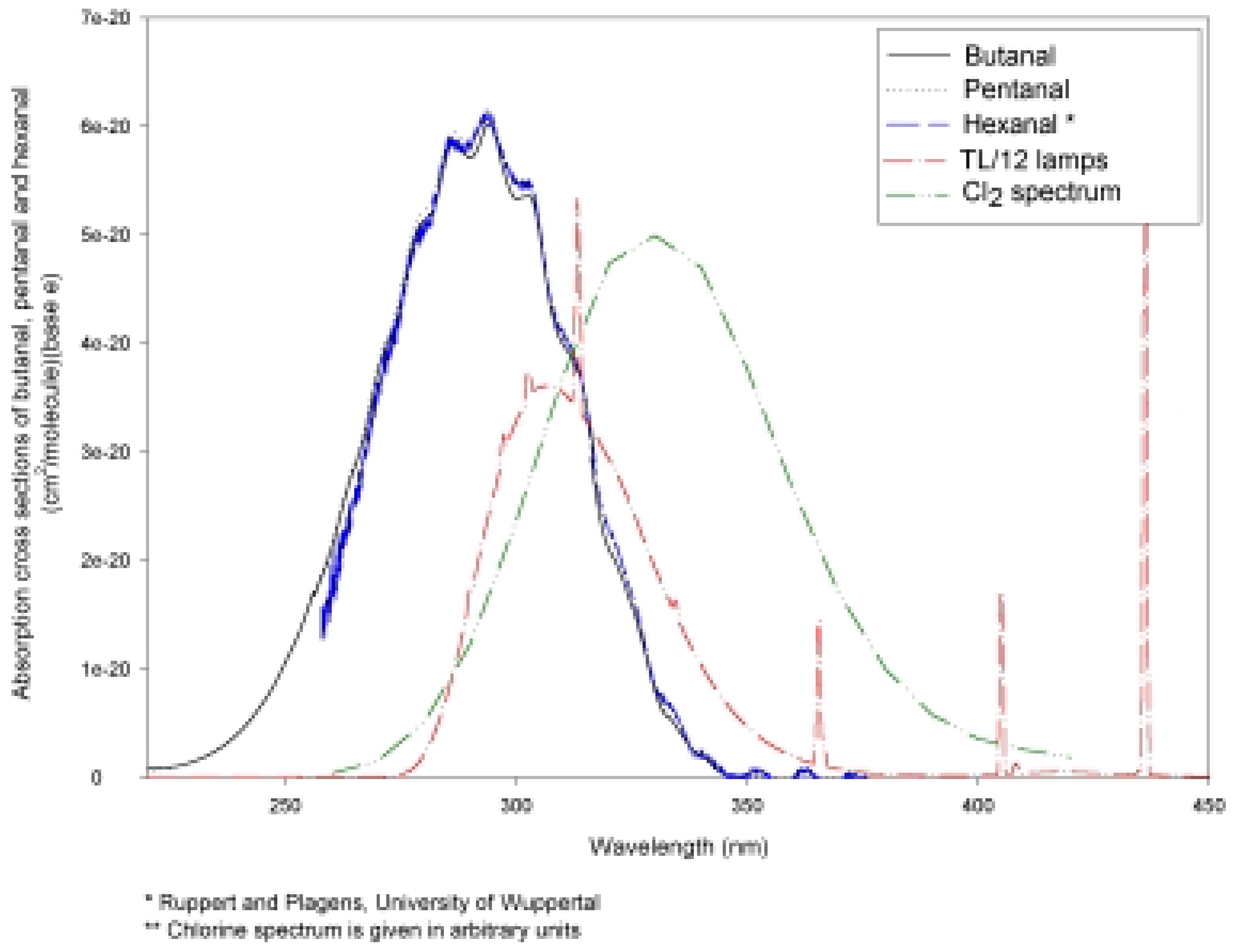
Photolysis of n-Hexanal
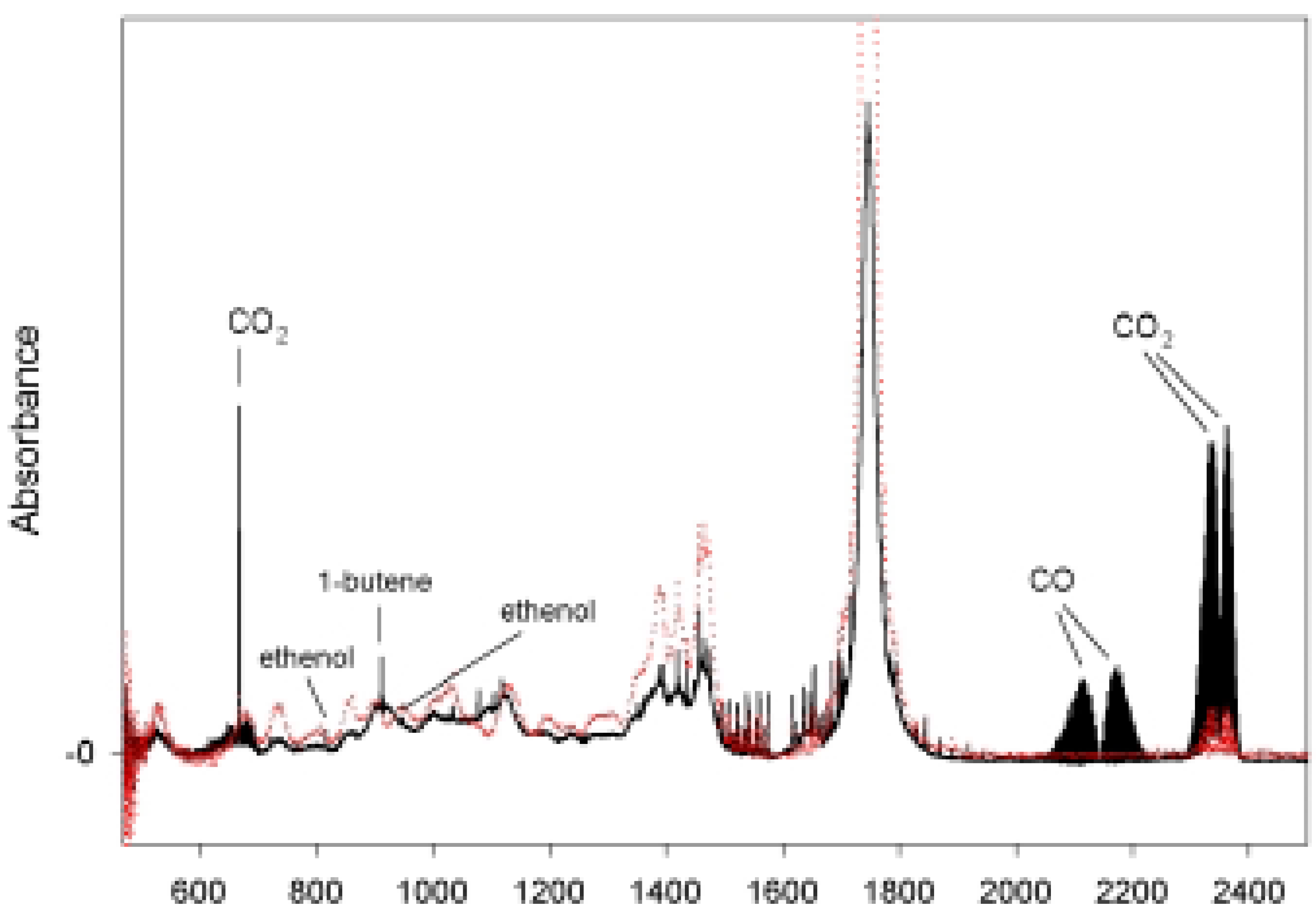
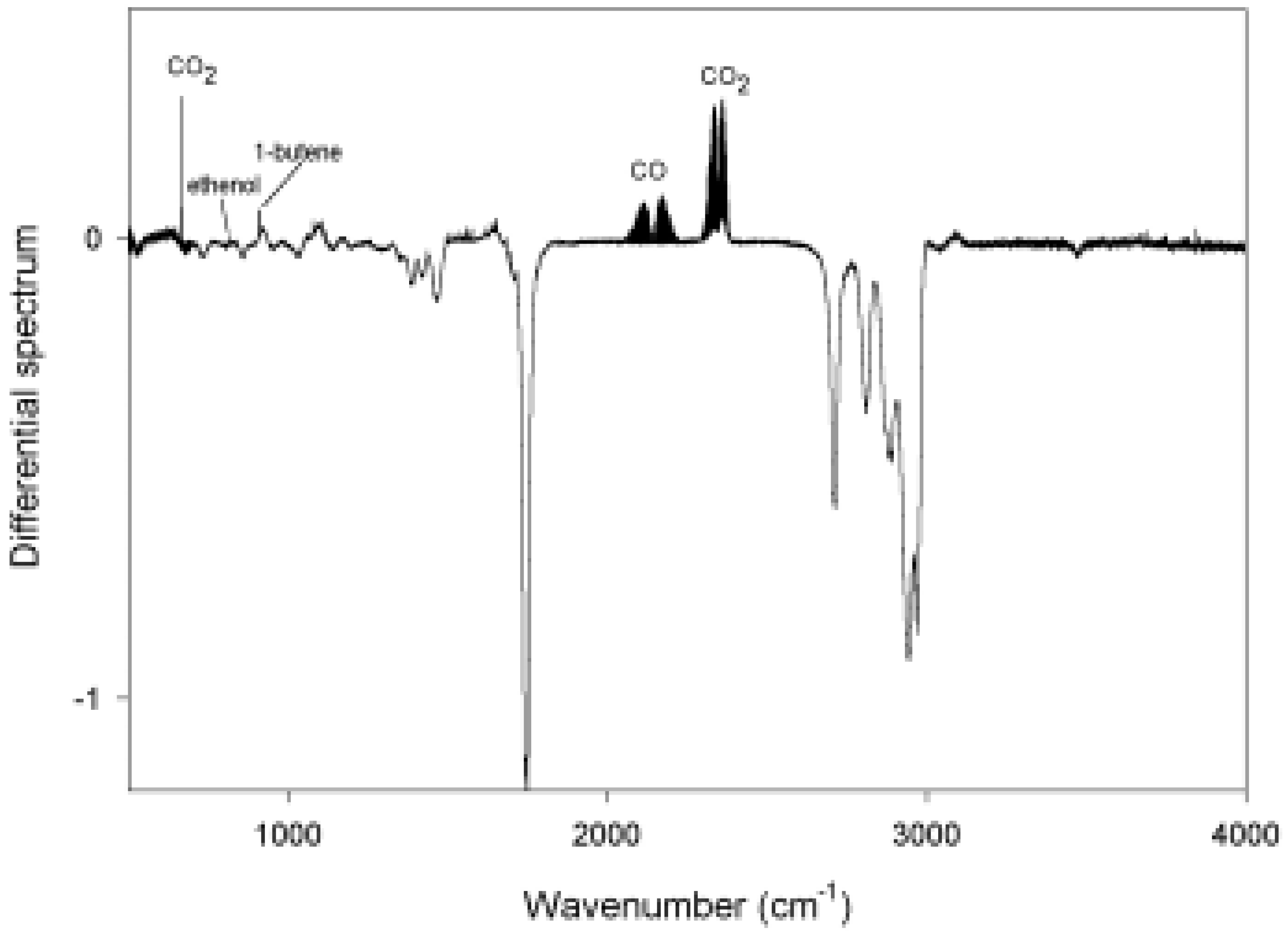
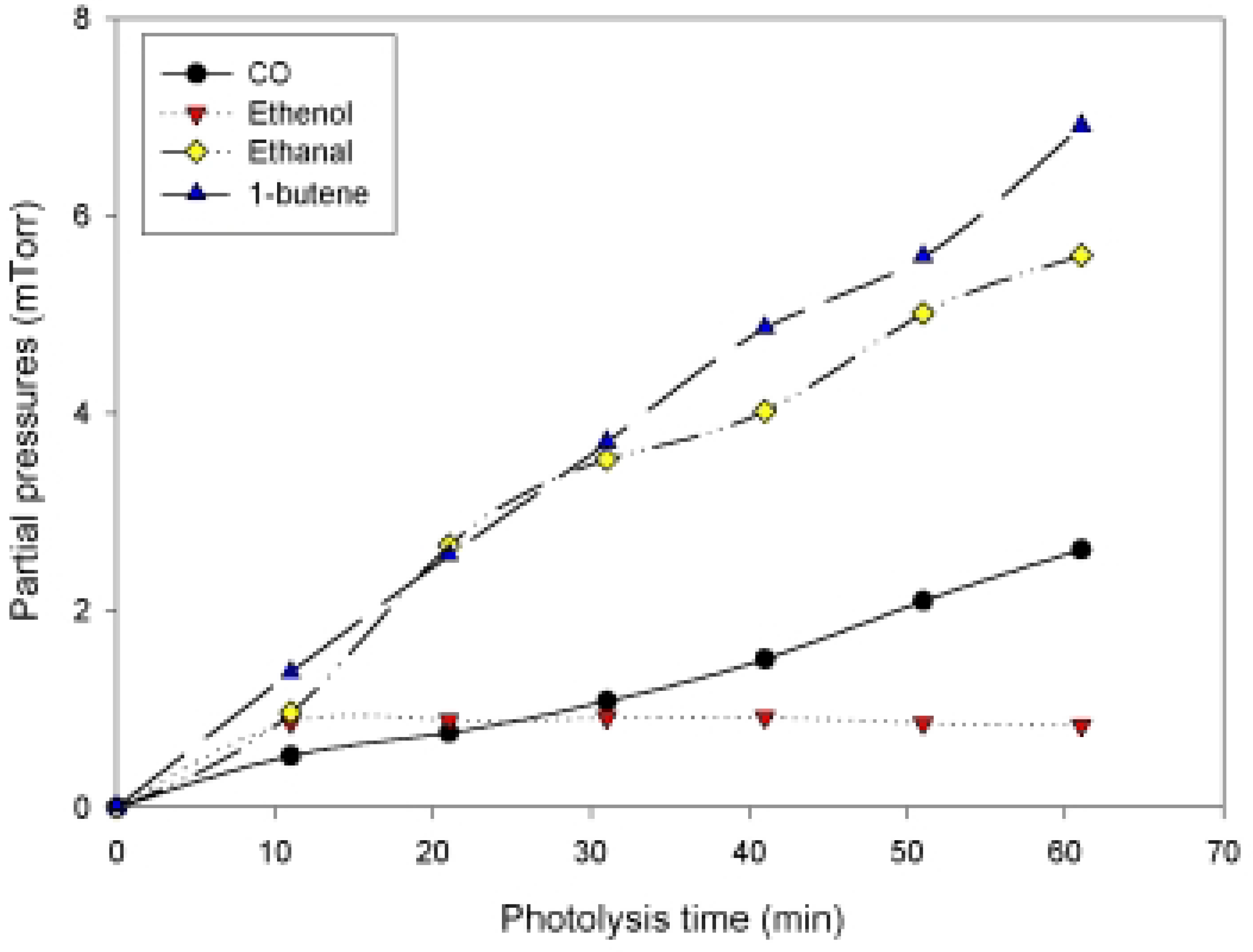


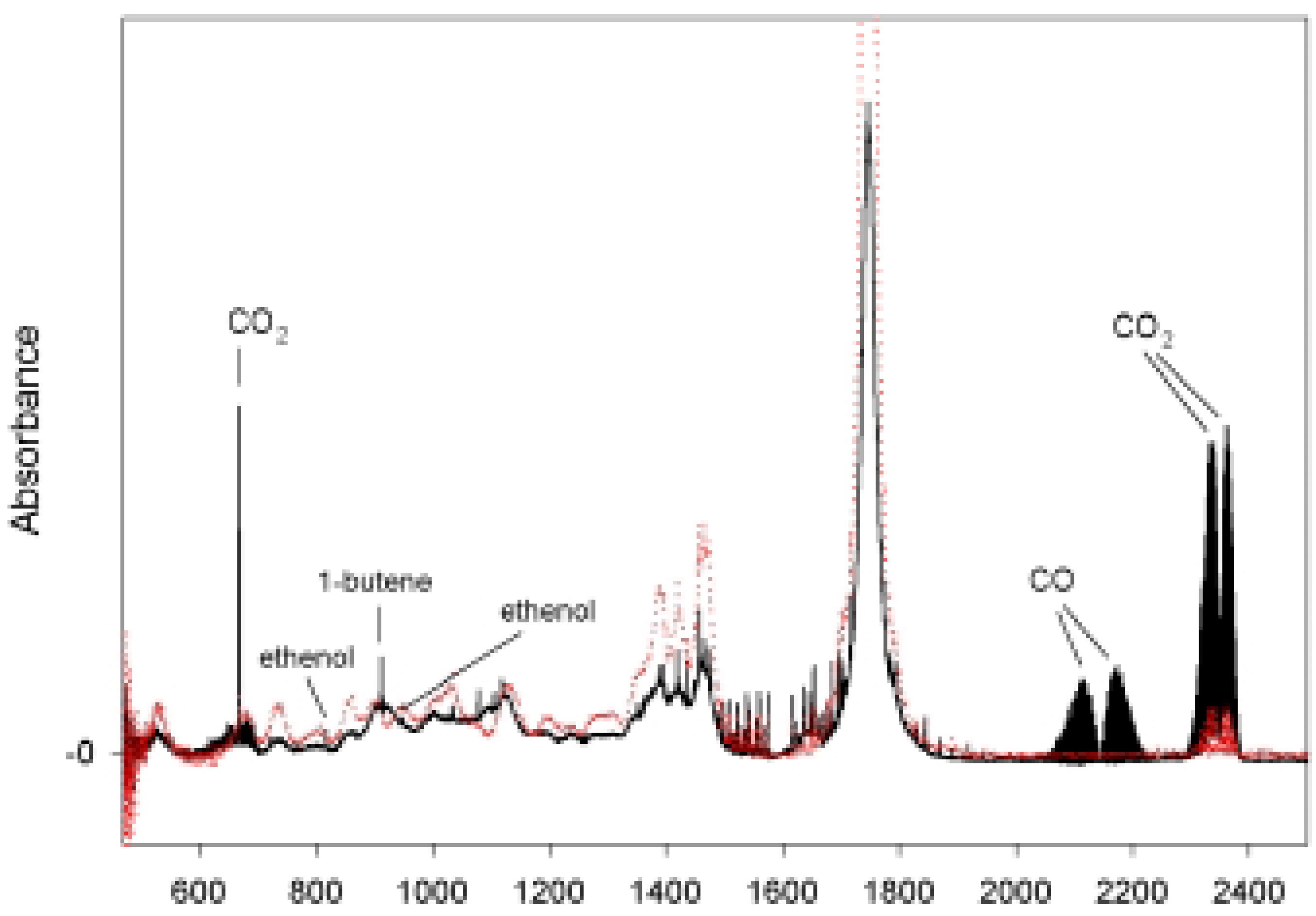{kind=link}
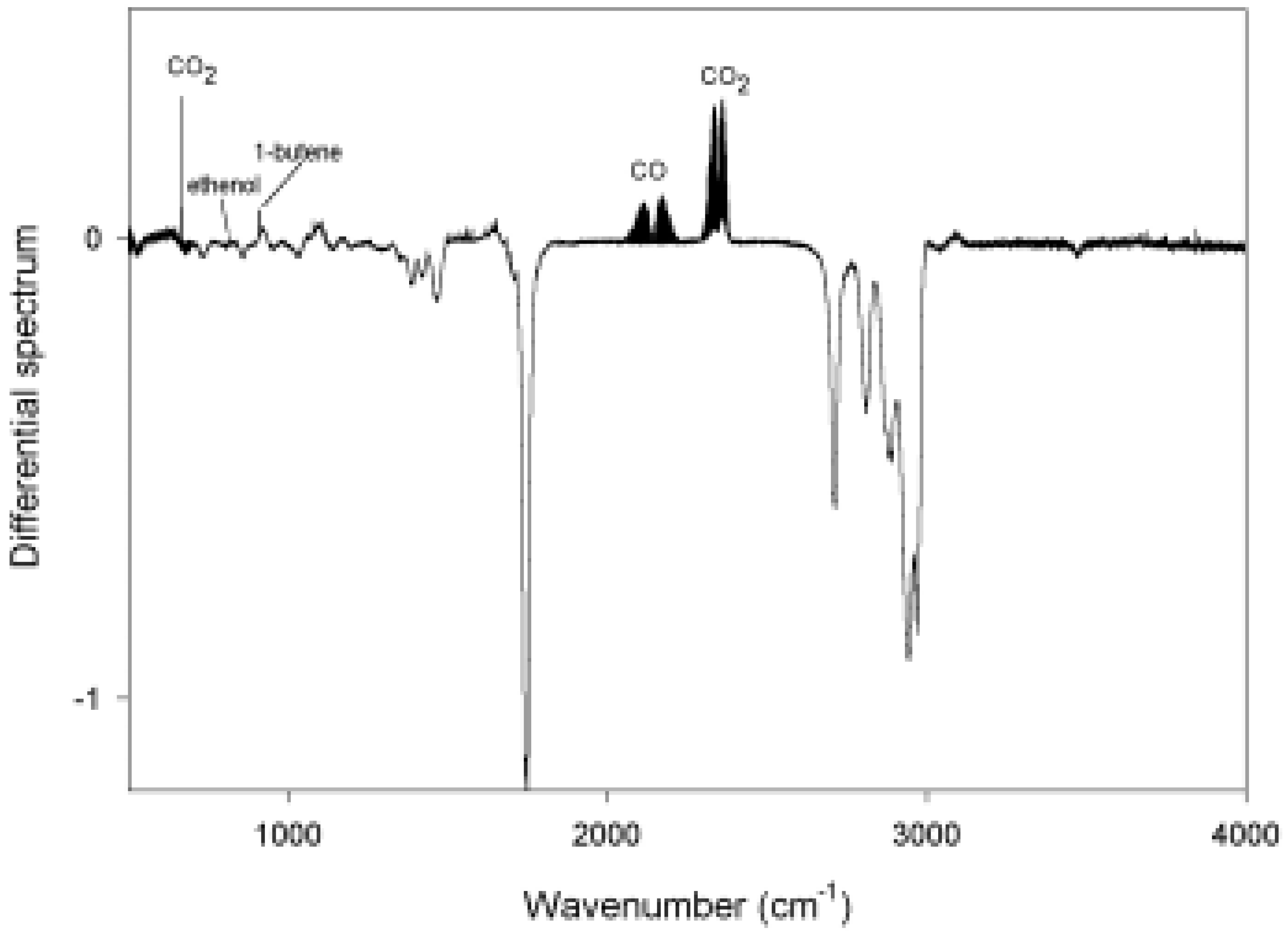{kind=link}
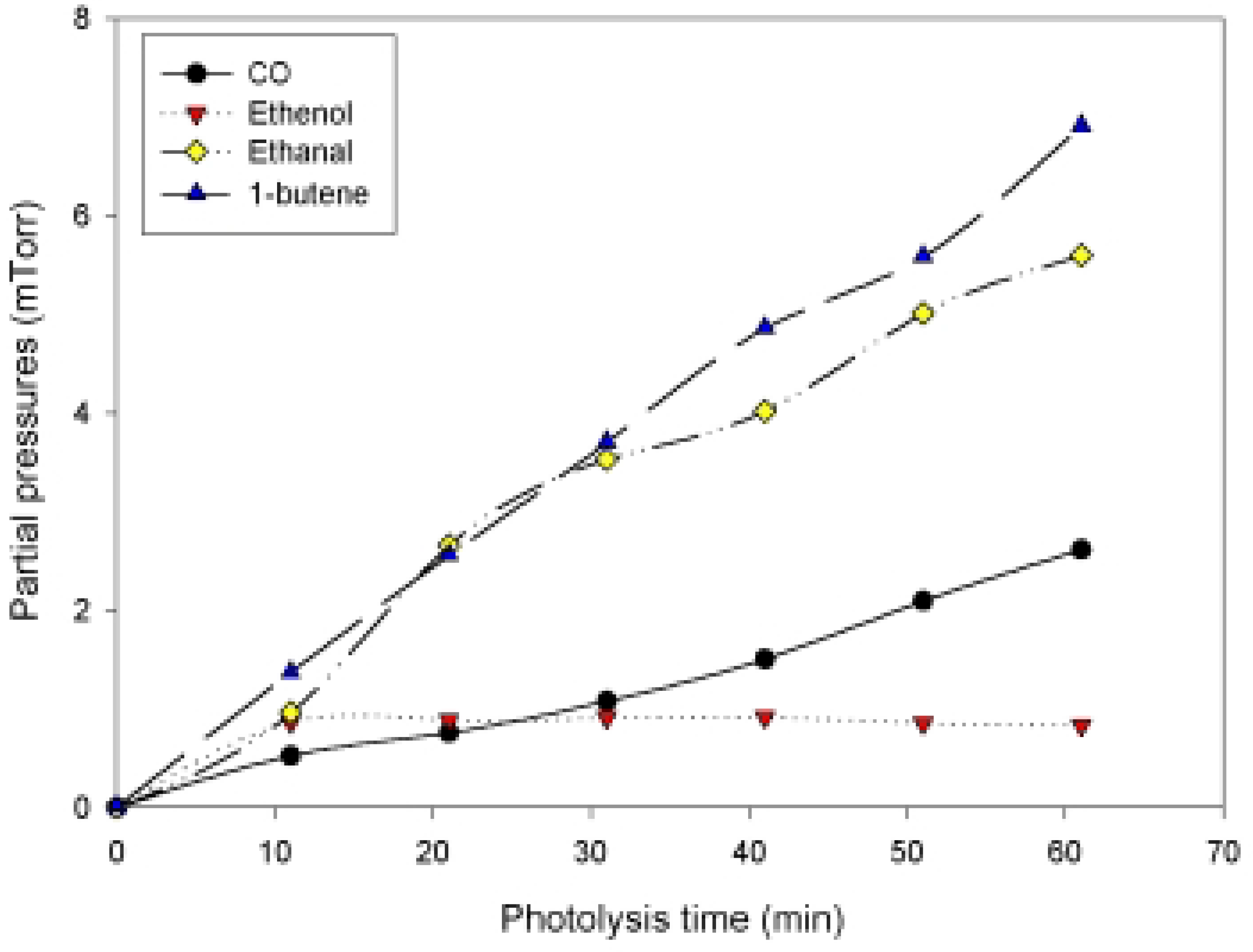{kind=link}
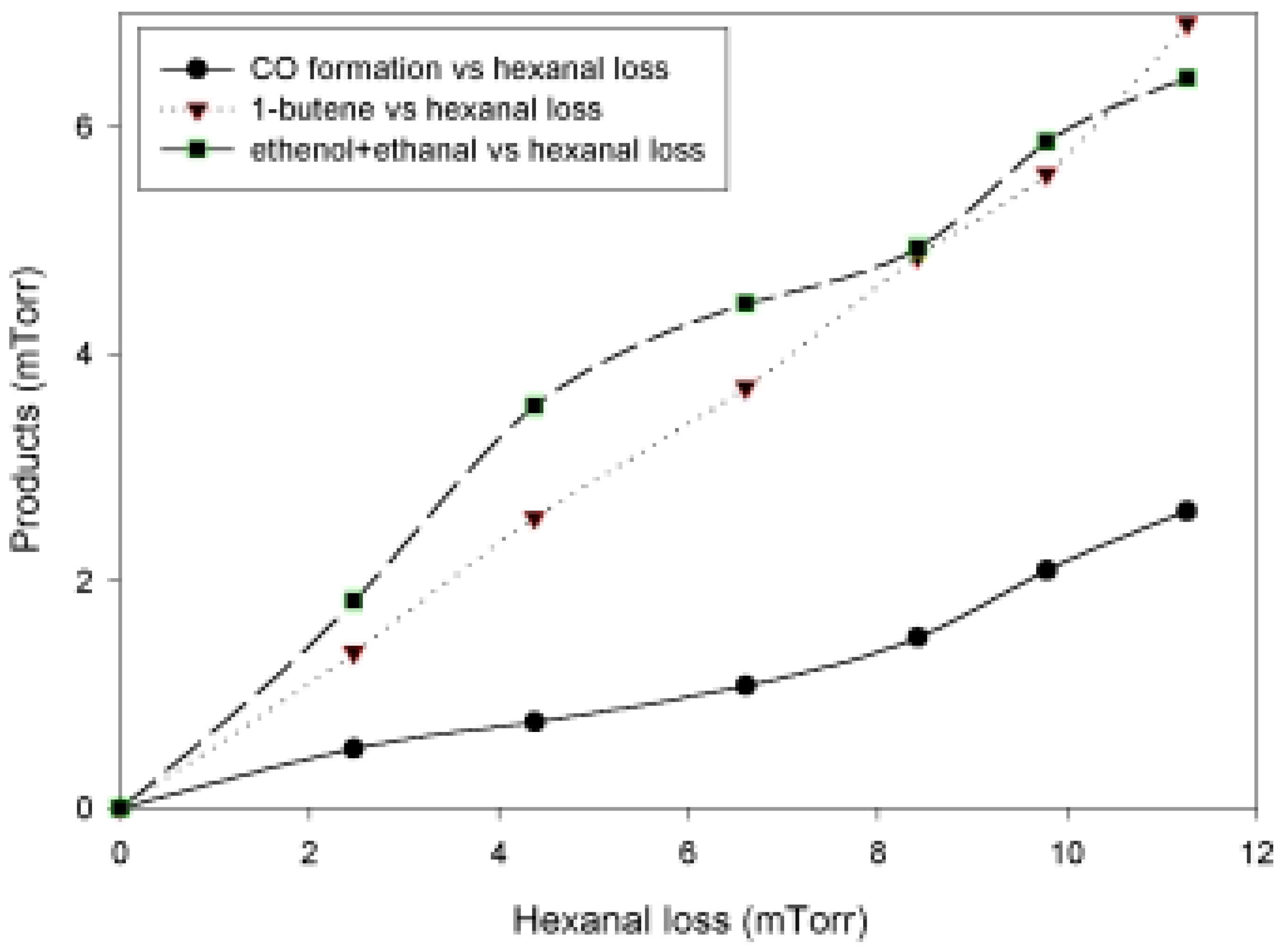{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total pressure (Torr) | Norrish type I | Norrish type II |
| 100.00 | 0.35 | 0.66 |
| 100.00 | 0.38 | 0.62 |
| 100.00 | 0.35 | 0.65 |
| 0.36±0.02 | 0.64±0.02 | |
| 300.00 | 0.28 | 0.72 |
| 300.00 | 0.26 | 0.74 |
| 300.00 | 0.28 | 0.72 |
| 0.27±0.01 | 0.73±0.01 | |
| 500.00 | 0.29 | 0.71 |
| 500.00 | 0.29 | 0.71 |
| 500.00 | 0.30 | 0.70 |
| 0.29±0.01 | 0.71±0.01 | |
| 700.00 | 0.27 | 0.73 |
| 700.00 | 0.28 | 0.72 |
| 700.00 | 0.27 | 0.73 |
| 0.27±0.01 | 0.73±0.01 |
| Δ CO/ -Δ hexanal | Δ 1-butene / -Δhexanal | Δ (ethanal+ethenol )/ -Δhexanal |
| 0.23 | 0.61 | 0.57 |
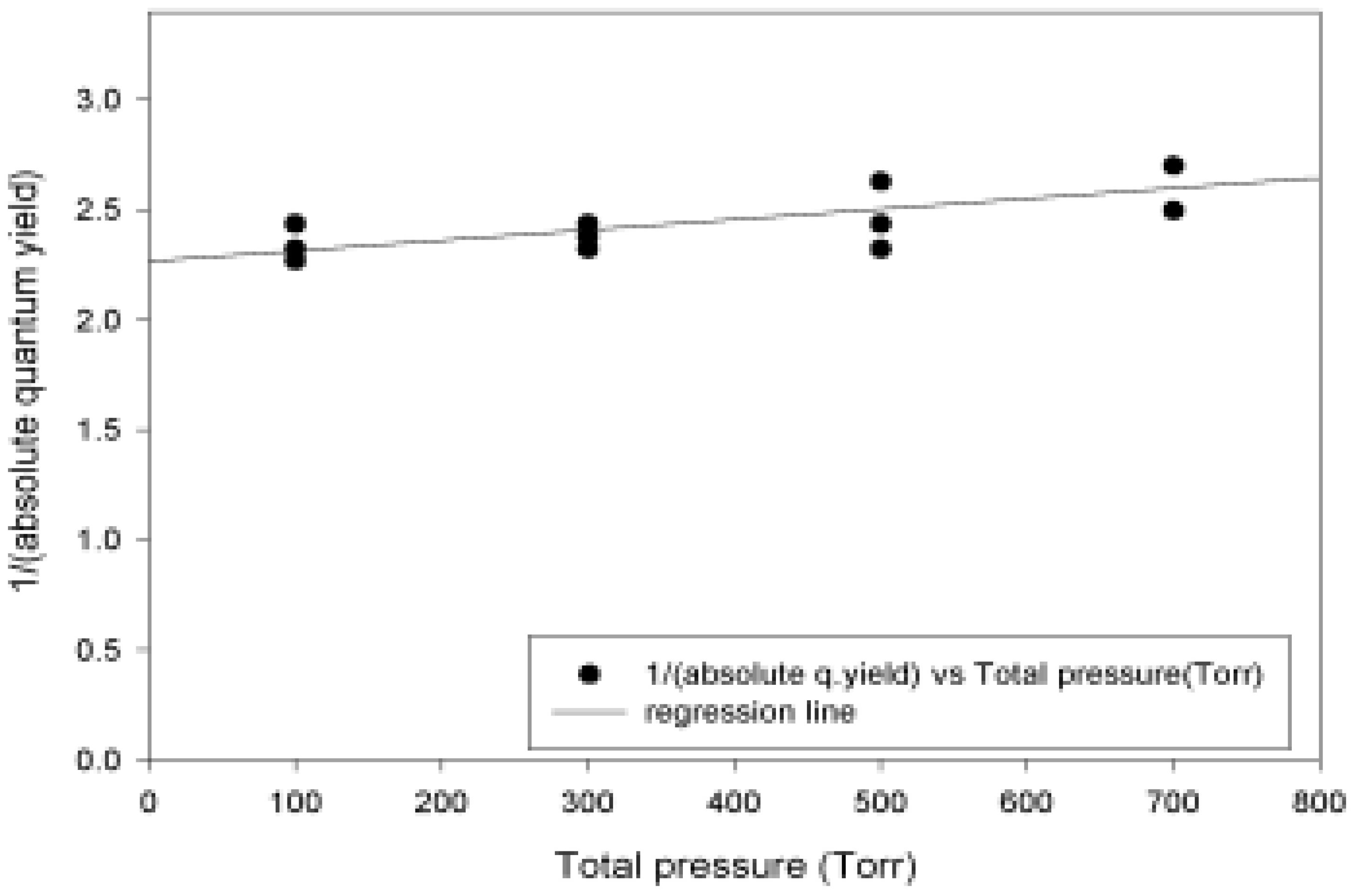
Absolute Quantum Yields
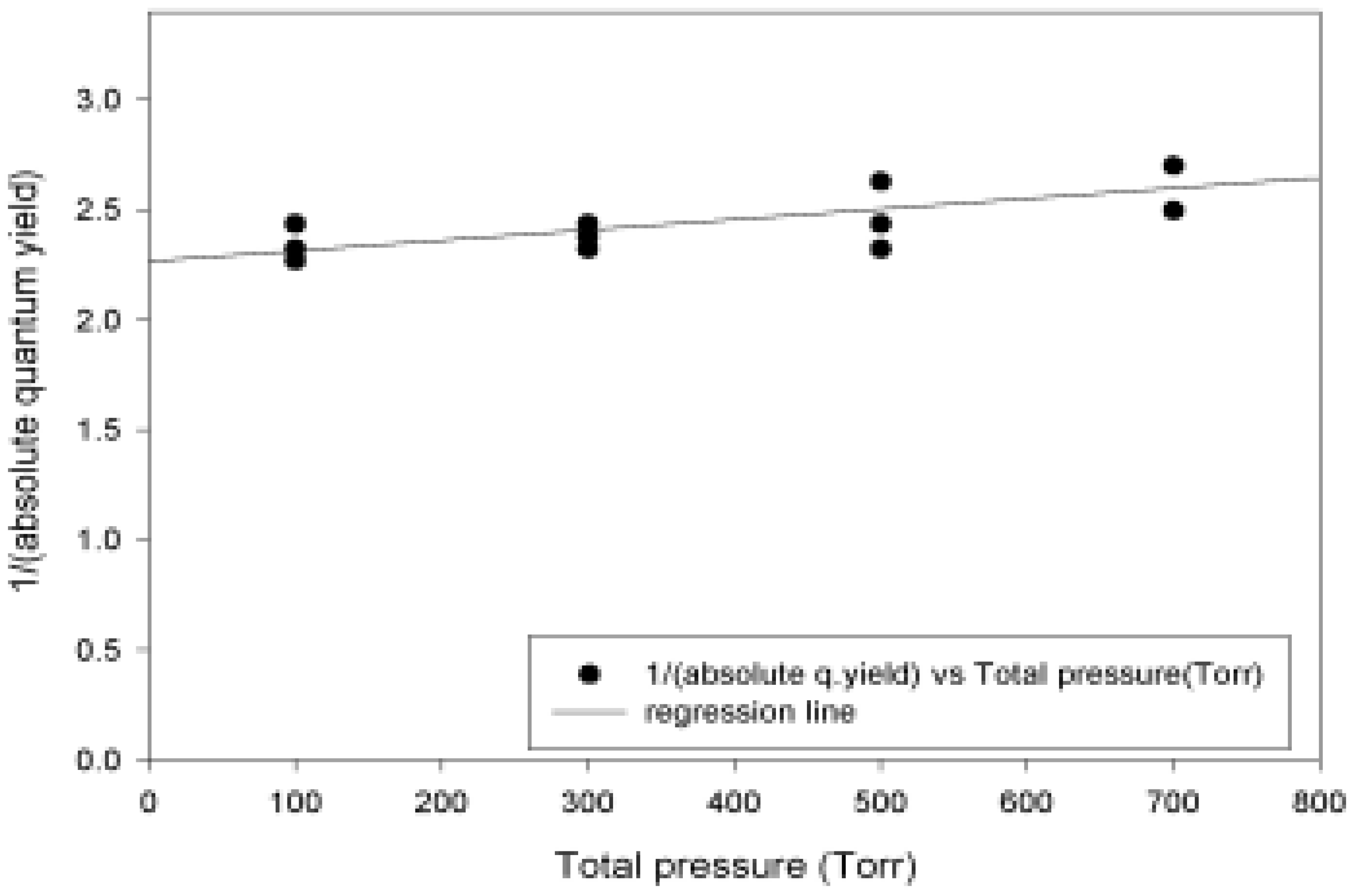
| Pressure (Torr) | Abs. Quantum yield |
| 100.00 | 0.43 |
| 100.00 | 0.41 |
| 100.00 | 0.44 |
| 0.43 ± 0.02 | |
| 300.00 | 0.41 |
| 300.00 | 0.42 |
| 300.00 | 0.43 |
| 0.42 ± 0.01 | |
| 500.00 | 0.43 |
| 500.00 | 0.38 |
| 500.00 | 0.41 |
| 0.40 ± 0.03 | |
| 700.00 | 0.40 |
| 700.00 | 0.37 |
| 700.00 | 0.37 |
| 0.38 ± 0.02 |
Conclusions
- ·
- Identification of the products of hexanal photolysis, detectable in IR region under applied conditions.
- ·
- Quantification of the products, and deduction of the photodecomposition pattern, particularly of primary step, exploiting the benefit of applied experimental system
- ·
- Determination of the absolute quantum yields at different pressures, providing values necessary for atmospheric modeling, at the same time examining the influence of the total pressure on the absolute quantum yield, evaluating importance of collisional deactivations, and indicating other relaxation channels
Experimental

References and Notes
- Graedel, T.E.; Farrow, L.A.; Weber, T.A. Atmos. Environ. 1976, 10, 1095–1099.
- Grosjean, D. Environ. Sci. Technol. 1982, 16, 254–259. [PubMed]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Atmospheric Chemistry; John Wiley: New York, 1986. [Google Scholar]
- Owen, S.; Boissard, R.; Street, R.A.; Duckam, S.C.; Csiky, O.; Hewitt, C.N. Atmos. Environ. 1997, 31 S1, 101–105.
- Kirstine, W.; Galbally, I. J. Geophys. Res. 1998, 103, 10603–10607.
- Calvert, J.G.; Pitts, J.N., Jr. Photochemistry; John Wiley: New York, 1966; pp. 372–375. [Google Scholar]
- Lee, E.K.C.; Lewis, R.S. Adv. Photochem. 1980, 12, 1–7.
- Moortgat, G K.; Seiler, W.; Warneck, P. J. Chem. Phys. 1983, 78, 1185–1191.
- Carmely, Y.; Horowitz, A. Int. J. Chem. Kin. 1984, 16, 1585–1589.
- Moore, C.B.; Weishaar, J.C. Annu. Rev. Phys. Chem. 1983, 34, 525–532.
- Ho, P.; Bamford, D.J.; Buss, R.J.; Lee, Y.T.; Moore, C.B. J. Chem. Phys. 1982, 76, 3630–3635.
- Horowitz, A.; Calvert, J.G. J. Phys. Chem. 1982, 86, 3105–3111.
- Meyrahn, H.; Moortgat, G.K.; Warneck, P. Presented at the 15th Informal Conference on Photochemistry, Stanford, CA, July, 1982.
- Shepson, P.B.; Heicklen, J. J. Photochem. 1982, 19, 215–220.
- Heicklen, J.; Desai, J.; Bahta, A.; Harper, C.; Simonaitis, R. J. Photochem. 1986, 34, 117–121.
- Terentis, A.C.; Knepp, P.T.; Kable, S.H. J. Phys. Chem. 1995, 99, 12704–12709.
- Forgeter, S.; Berces, T.; Dobe, S. Int. J. Chem. Kin. 1979, 11, 219–237.
- Cronin, J.T.; Zhu, L. J. Phys. Chem. A 1998, 102, 10274–10279.
- Moortgat, G.K.; Cox, R.A.; Schuster, G.; Burrows, J.P.; Tyndall, G.S. J. Chem. Soc. Farad. Trans. II 1989, 85, 809–818.
- Raber, W.H.; Moortgat, G.K. Adv. Ser. in Phys. Chem., 3, "Progress and Problems in Atmospheric Chemistry"; Barker, J.R., Ed.; Word Scientific Publ. Co.: Singapore, 1995; pp. 318–373. [Google Scholar]
- Crowley, J.N.; Moortgat, G.K. J. Chem. Soc. Farad. Trans. 1992, 88, 2437–2448.
- Meller, R.; Raber, W.; Crowley, J.N.; Jenkin, M.; Moortgat, G.T. J. Photochem. 1991, A62, 163–174.
- Plagens, H.; Bröske, R.; Splitter, M.; Ruppert, L.; Barnes, I.; Becker, K.H. Proceedings of the Second Workshop of the Eurotrac-2 Subproject CMD, ”Chemical Mechanism Development”, Karlsruhe, 23-25- September 1998.
- Martinez, R.D.; Buitrago, A.A.; Howell, N.W.; Hearn, C.H.; Joens, J. A. Atmos. Environ. 1992, 26, 785–794.
- Moortgat, G.K. Final report on EU project RADICAL: ”Evaluation of radical sources in atmospheric chemistry through chamber and laboratory studies” ENV4-CT97-0419. March 2000. [Google Scholar]
- Moortgat, G.K.; Seiler, W.; Warneck, P. J. Chem. Phys. 1983, 78, 1185–1193.
- Horowitz, A.; Calvert, J.G. Int. J. Chem. Kin. 1978, 10, 805–813.
- Horowitz, A.; Su, F.; Calvert, J.G. Int. J. Chem. Kin. 1978, 10, 1099–1108.
- Atkinson, R. J. Phys. Chem. Ref. Data 1994. Monograph 2.
- Lightfoot, P.D.; Cox, R.A.; Crowley, J.N.; Destriau, M.; Hayman, G.D.; Jenkin, M.E.; Moortgat, G.K.; Zabel, F. Atmos. Environ. 1992, 26A, 1805–1811.
- Tadic, J.; Moortgat, G.K. Manuscript in preparation.
- Zhu, L.; Cronin, J.T.; Narang, A. J. Phys.Chem.A 1999, 103: 36, 7248–7253. [PubMed]
- Bell, R.P.; Smith, P.W. J. Chem. Soc. (B) 1966, 241–248.
- Sample Availability: Not available
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
Tadić, J.; Juranić, I.; Moortgat, G.K. Photooxidation of n-Hexanal in Air. Molecules 2001, 6, 287-299. https://doi.org/10.3390/60400287
Tadić J, Juranić I, Moortgat GK. Photooxidation of n-Hexanal in Air. Molecules. 2001; 6(4):287-299. https://doi.org/10.3390/60400287
Chicago/Turabian StyleTadić, Jovan, Ivan Juranić, and Geert K. Moortgat. 2001. "Photooxidation of n-Hexanal in Air" Molecules 6, no. 4: 287-299. https://doi.org/10.3390/60400287
APA StyleTadić, J., Juranić, I., & Moortgat, G. K. (2001). Photooxidation of n-Hexanal in Air. Molecules, 6(4), 287-299. https://doi.org/10.3390/60400287