Bis-Porphyrin Racks with Space-Separated Co-Planar Porphyrin Rings

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
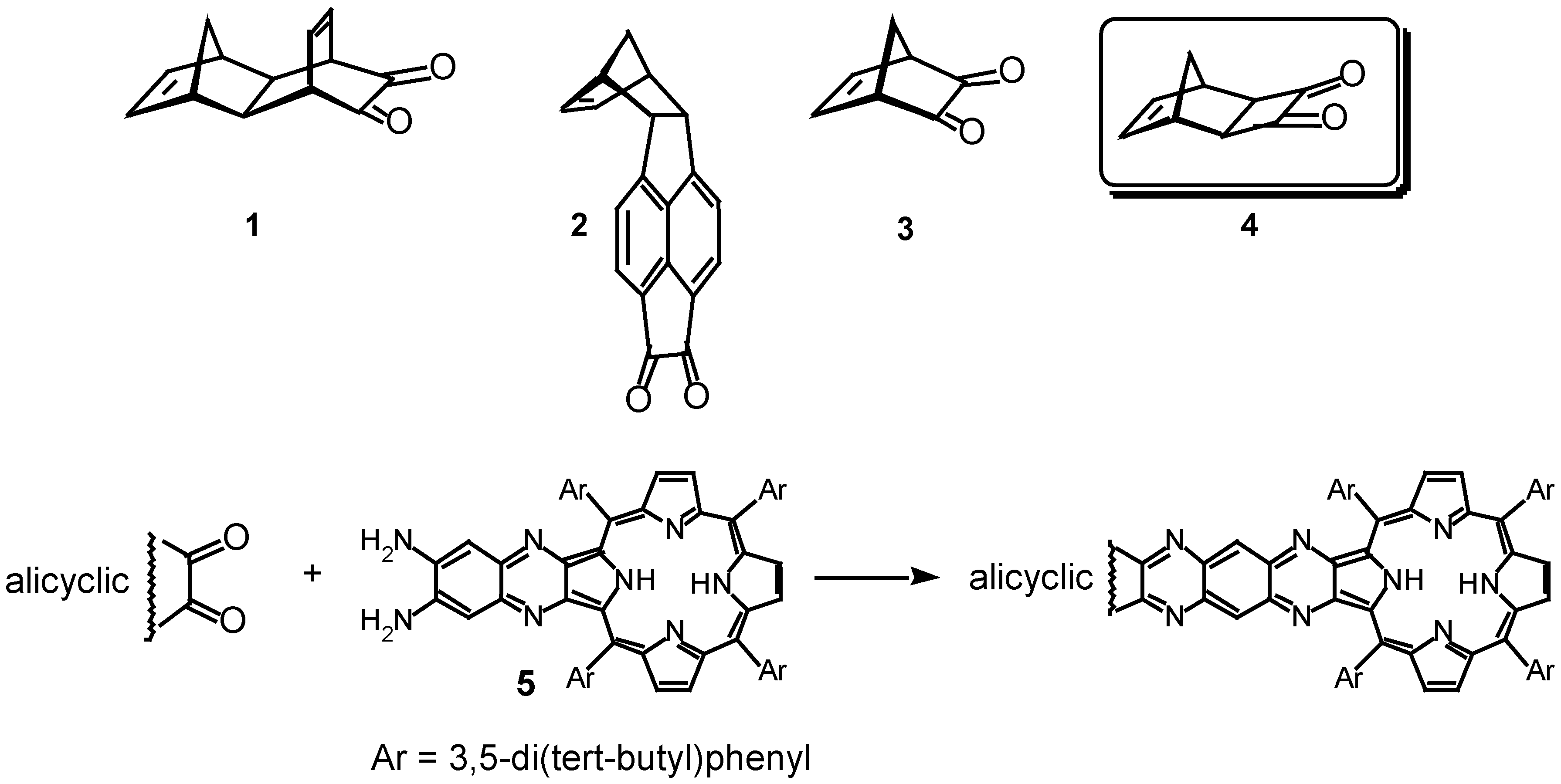
:Introduction
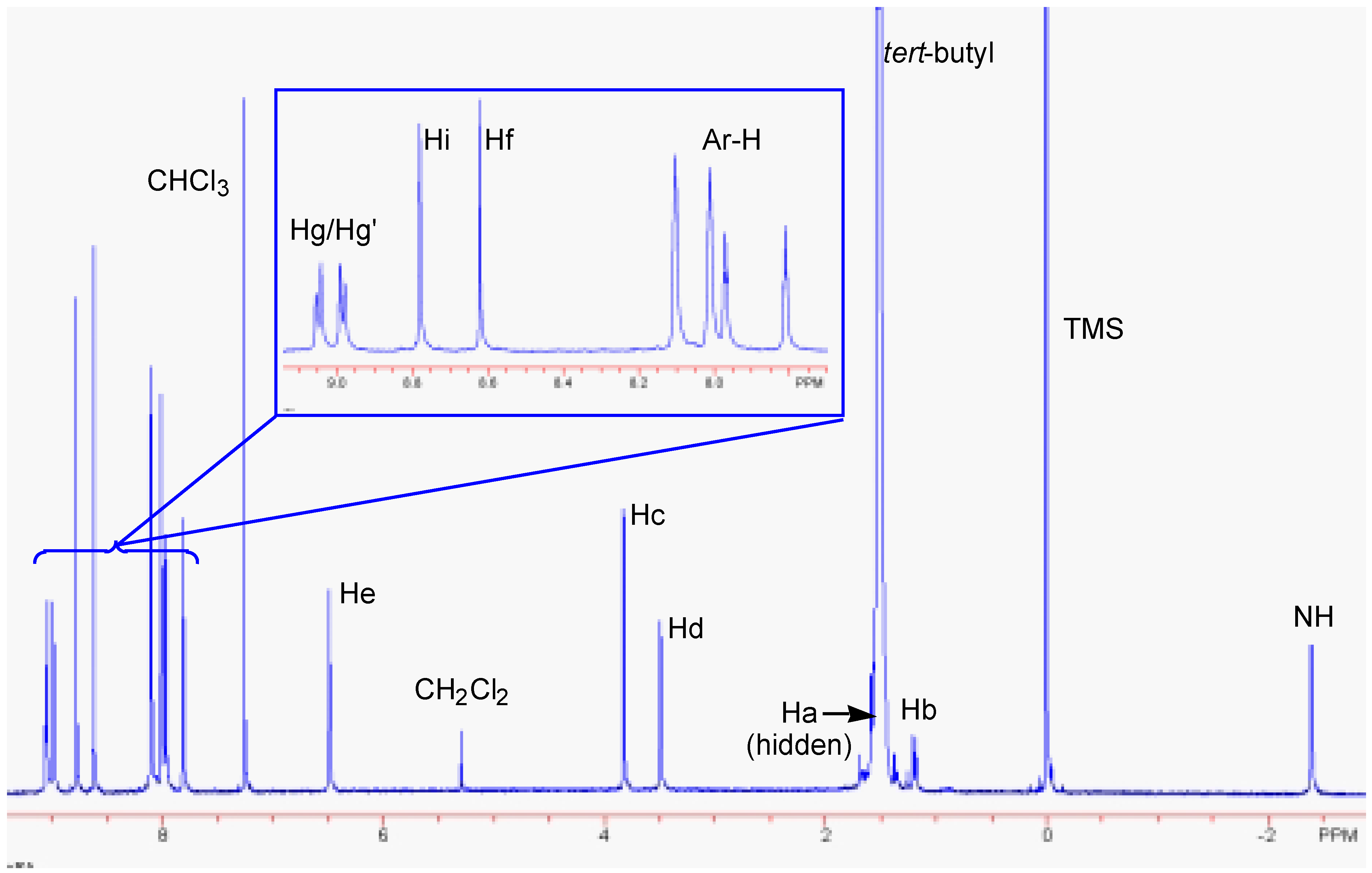
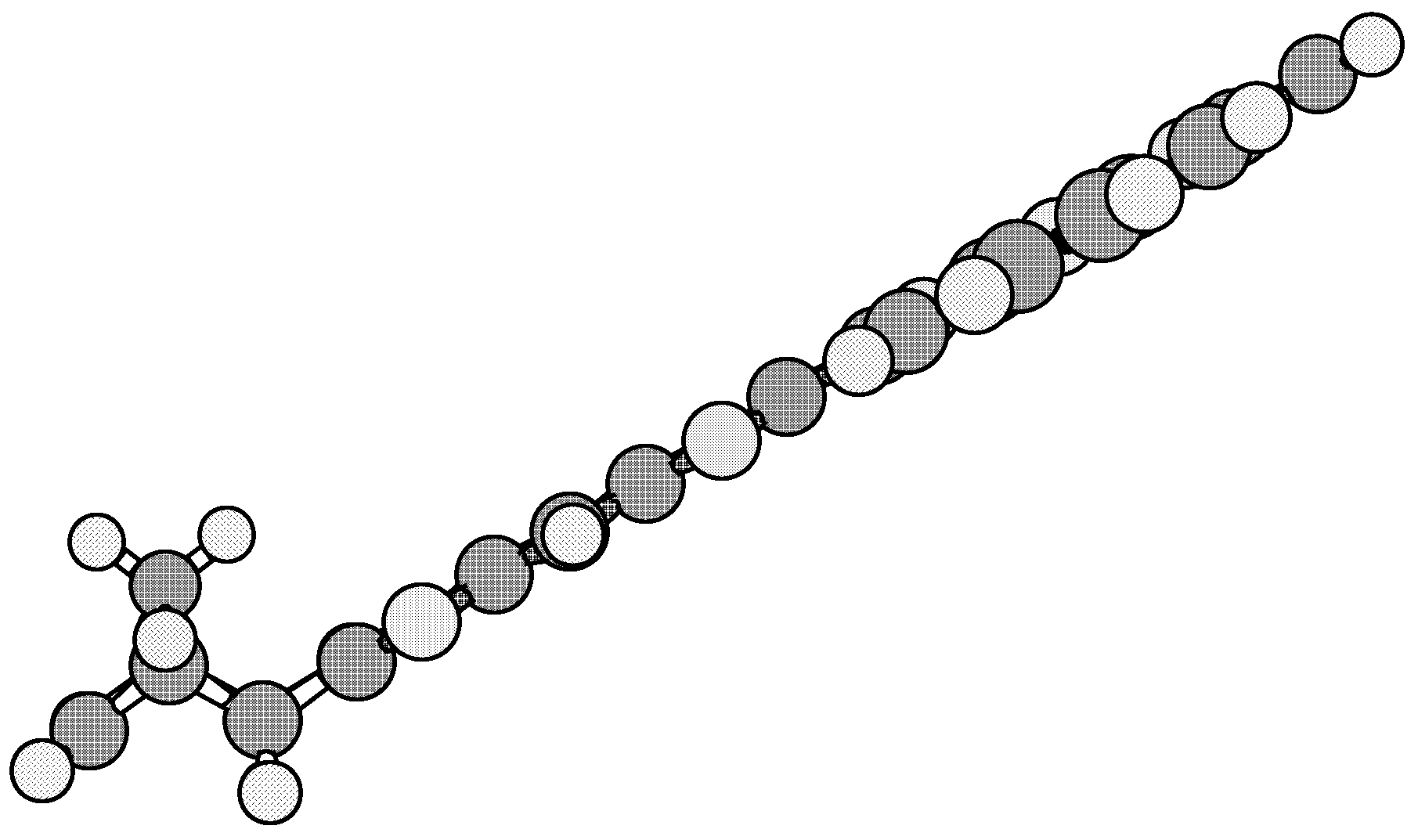
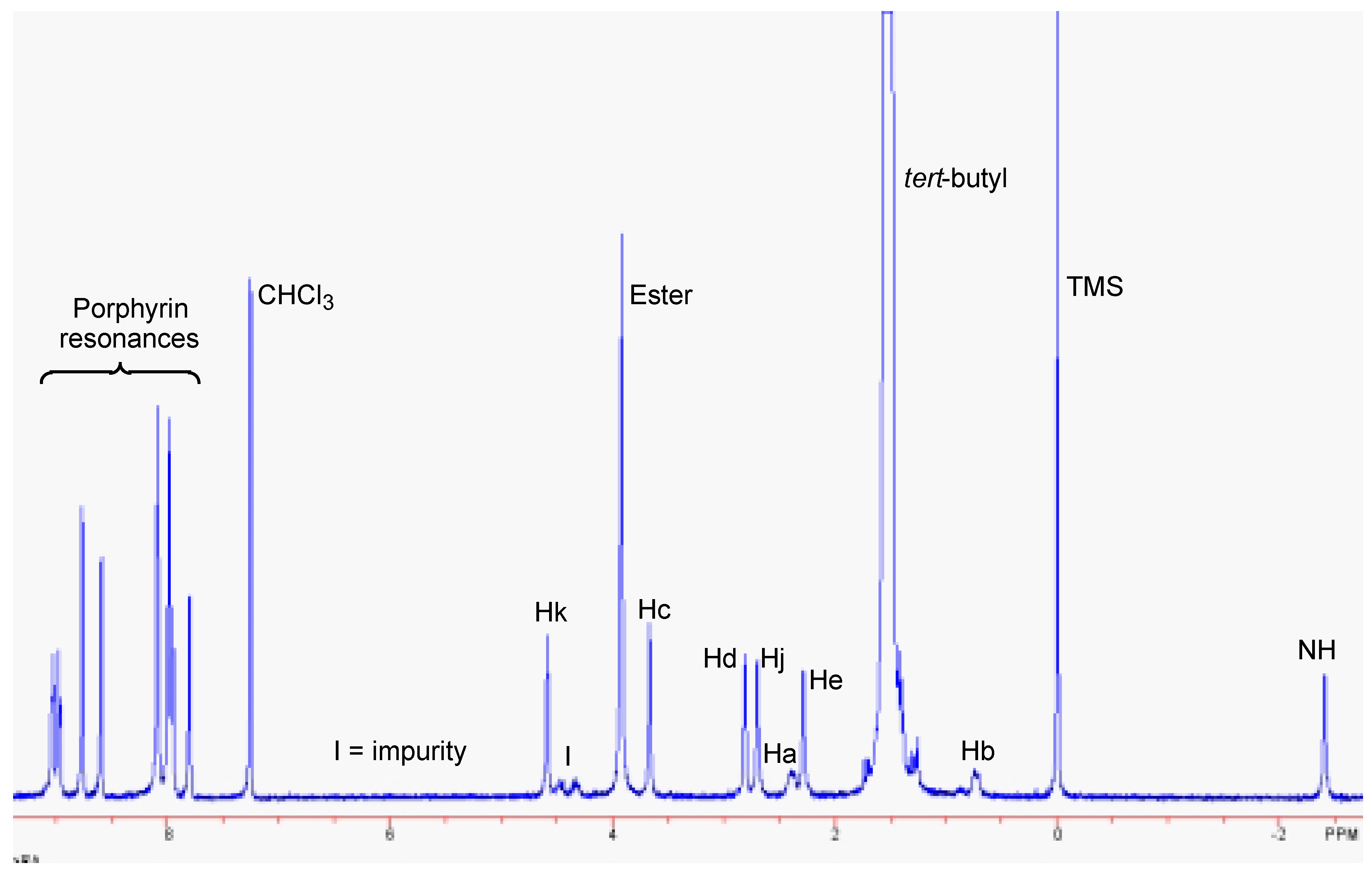
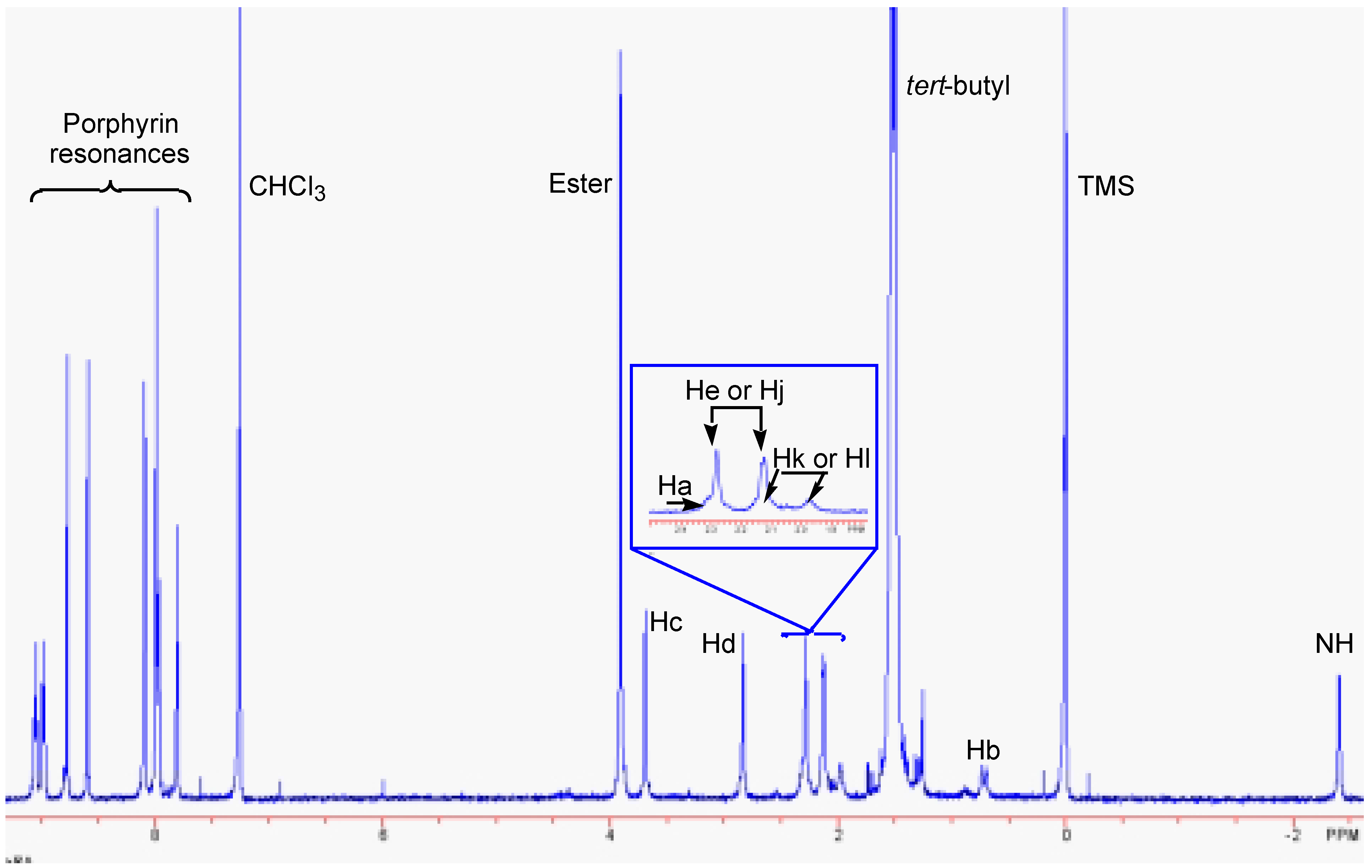
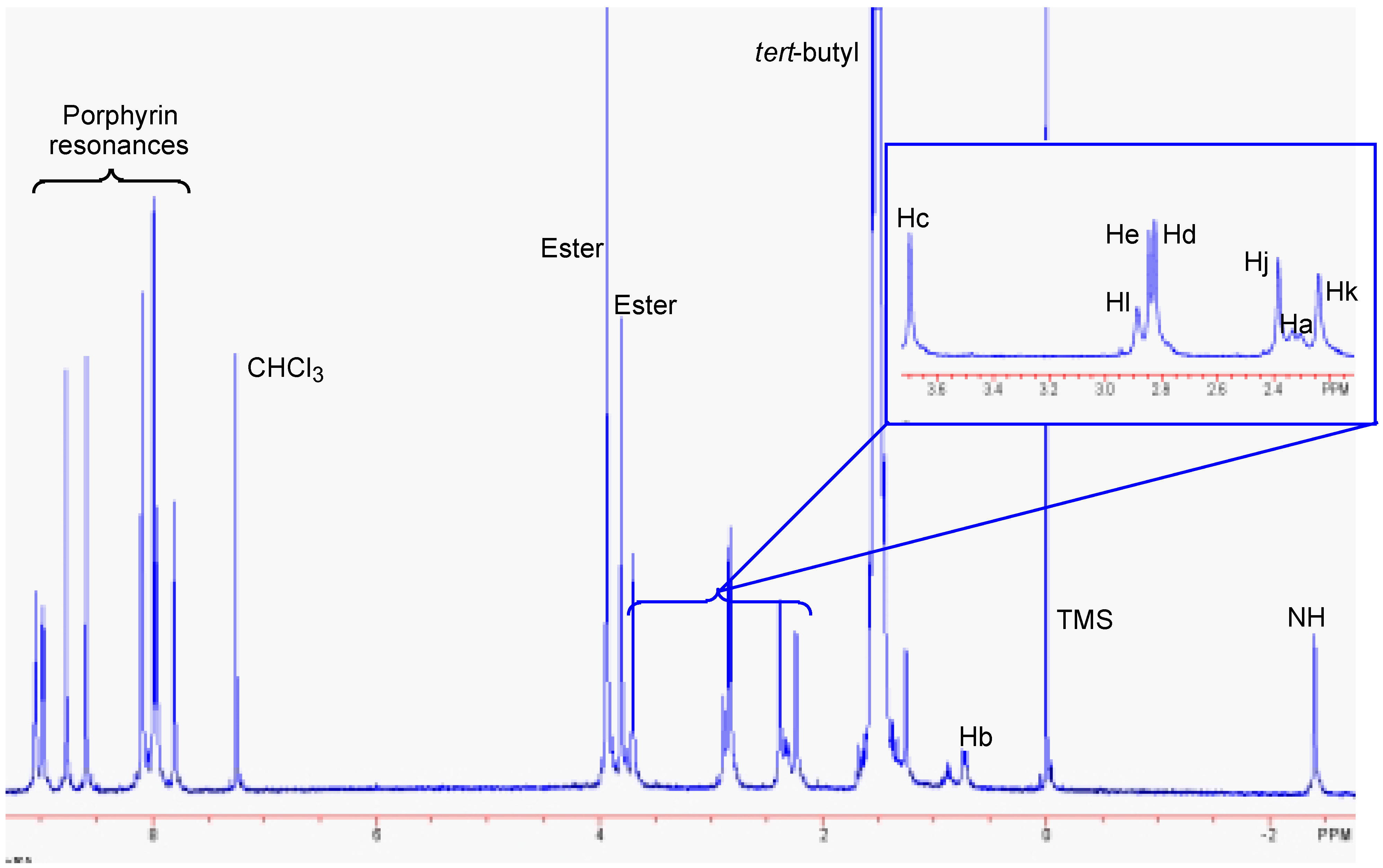
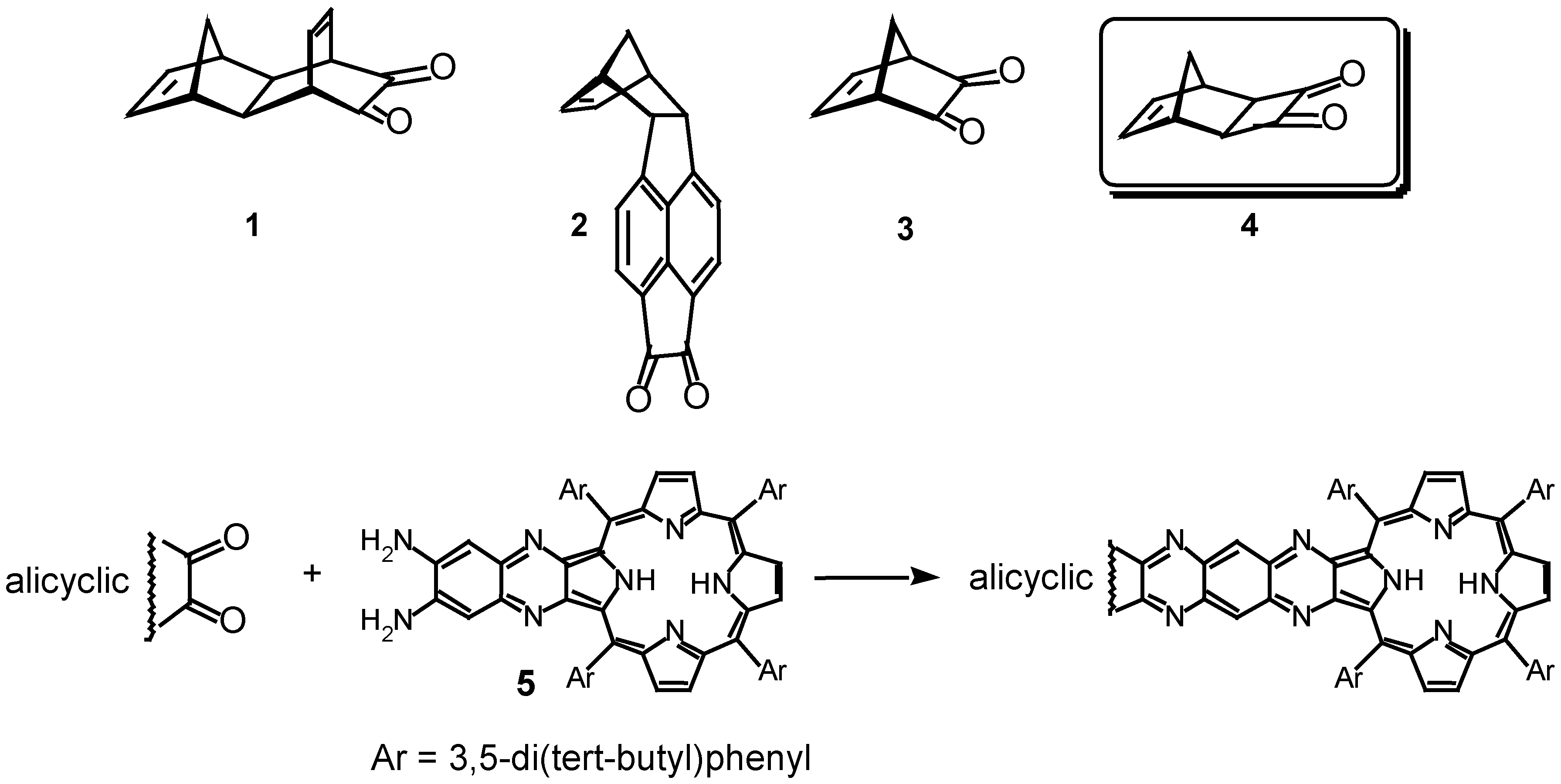
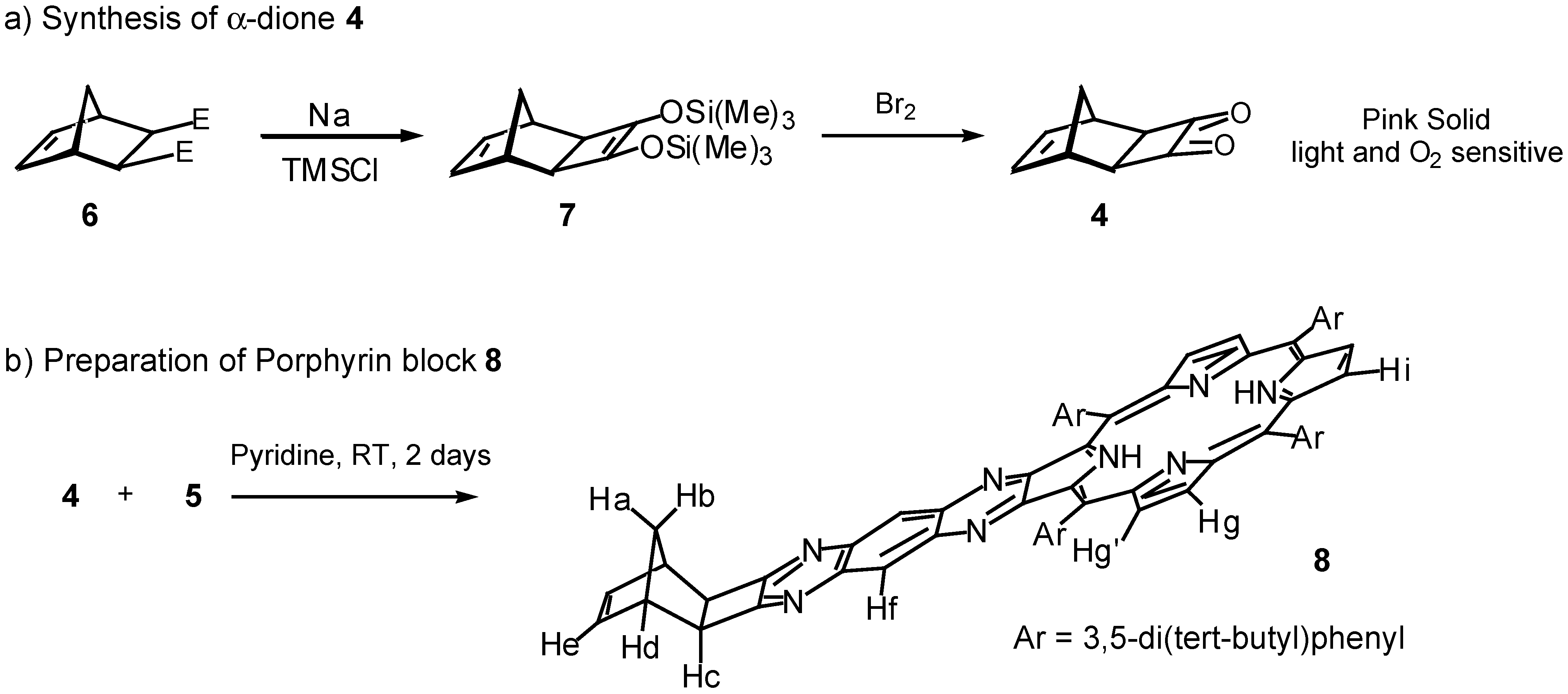
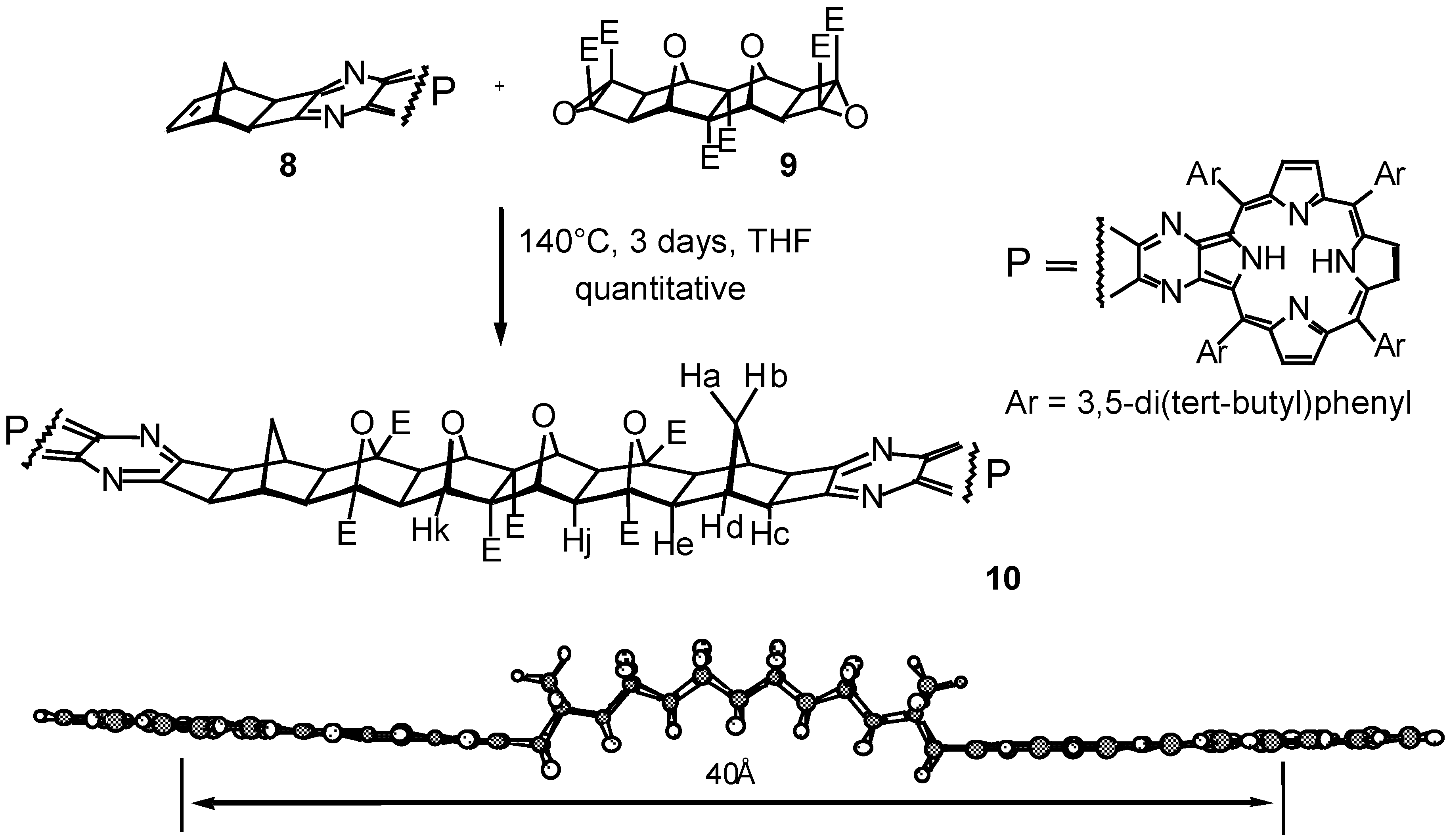
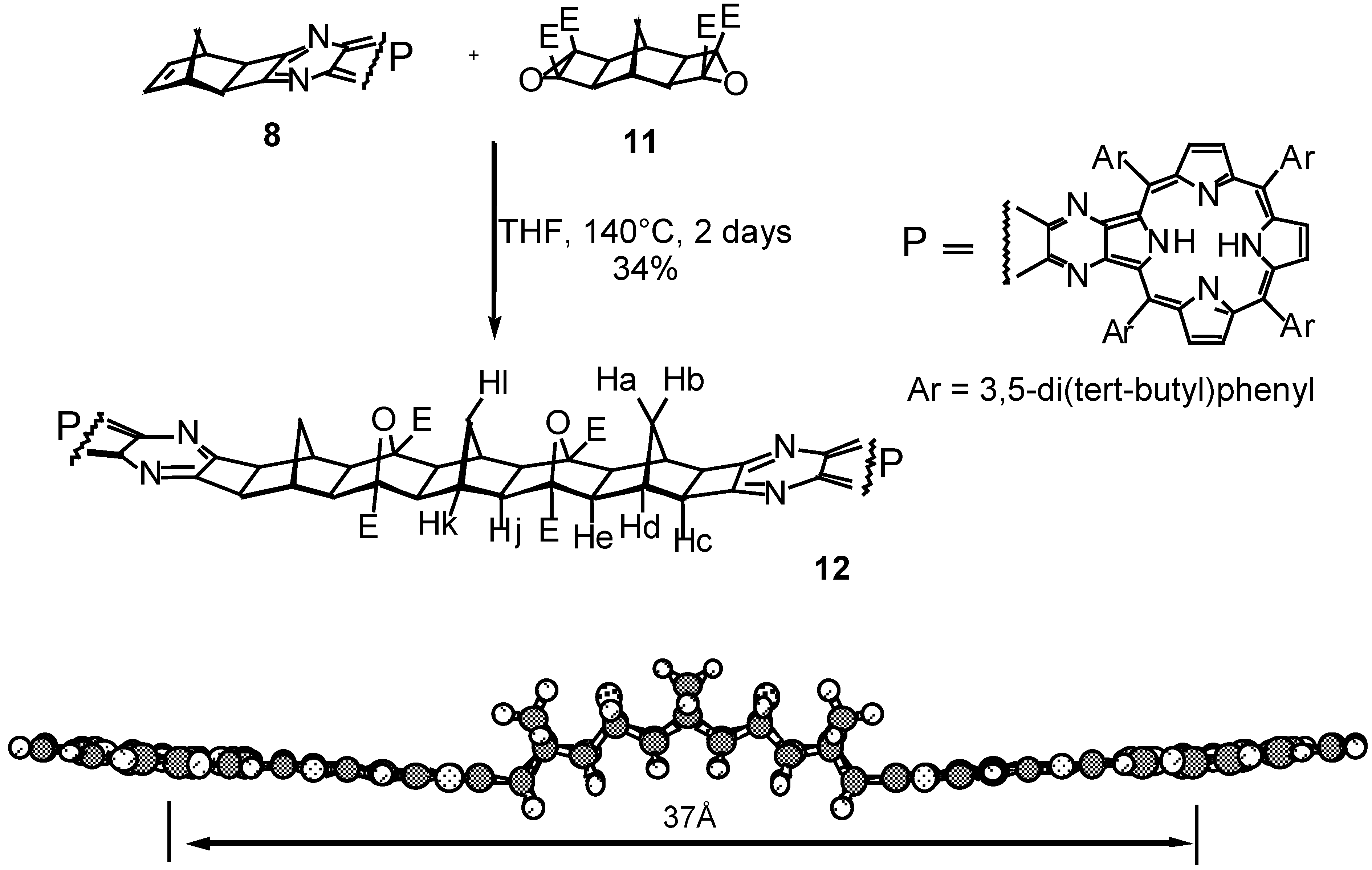
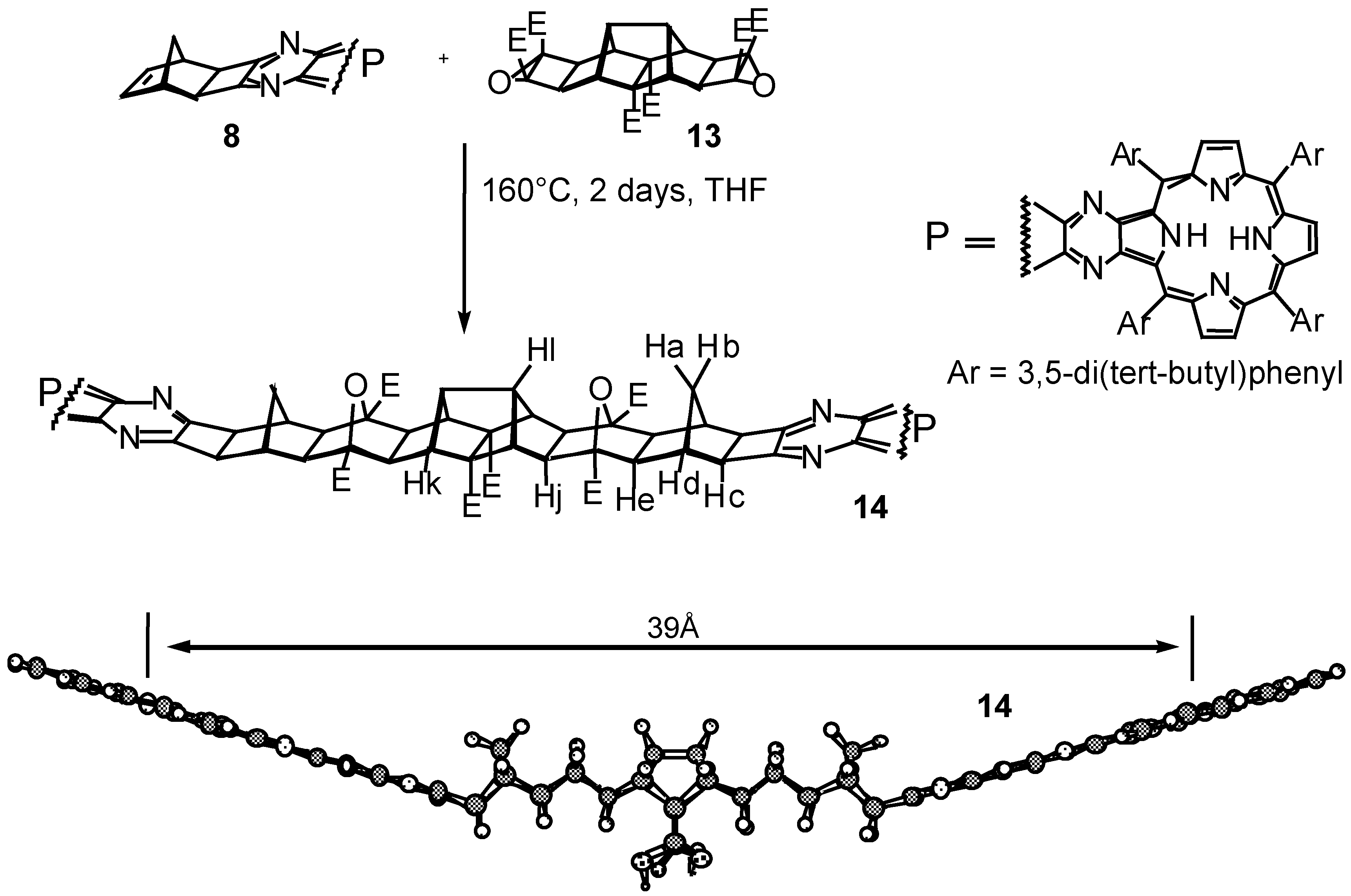
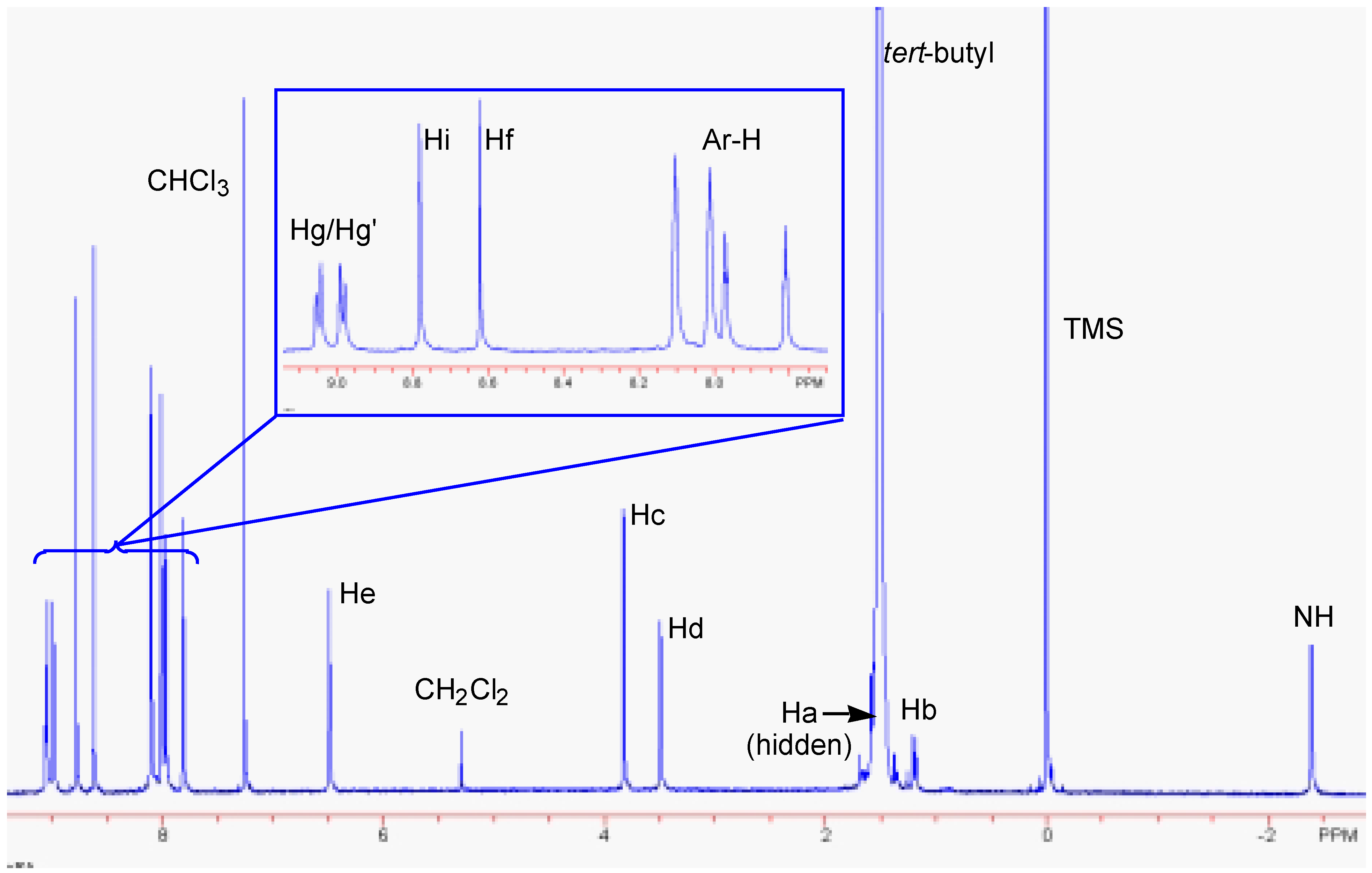
Results and Discussion

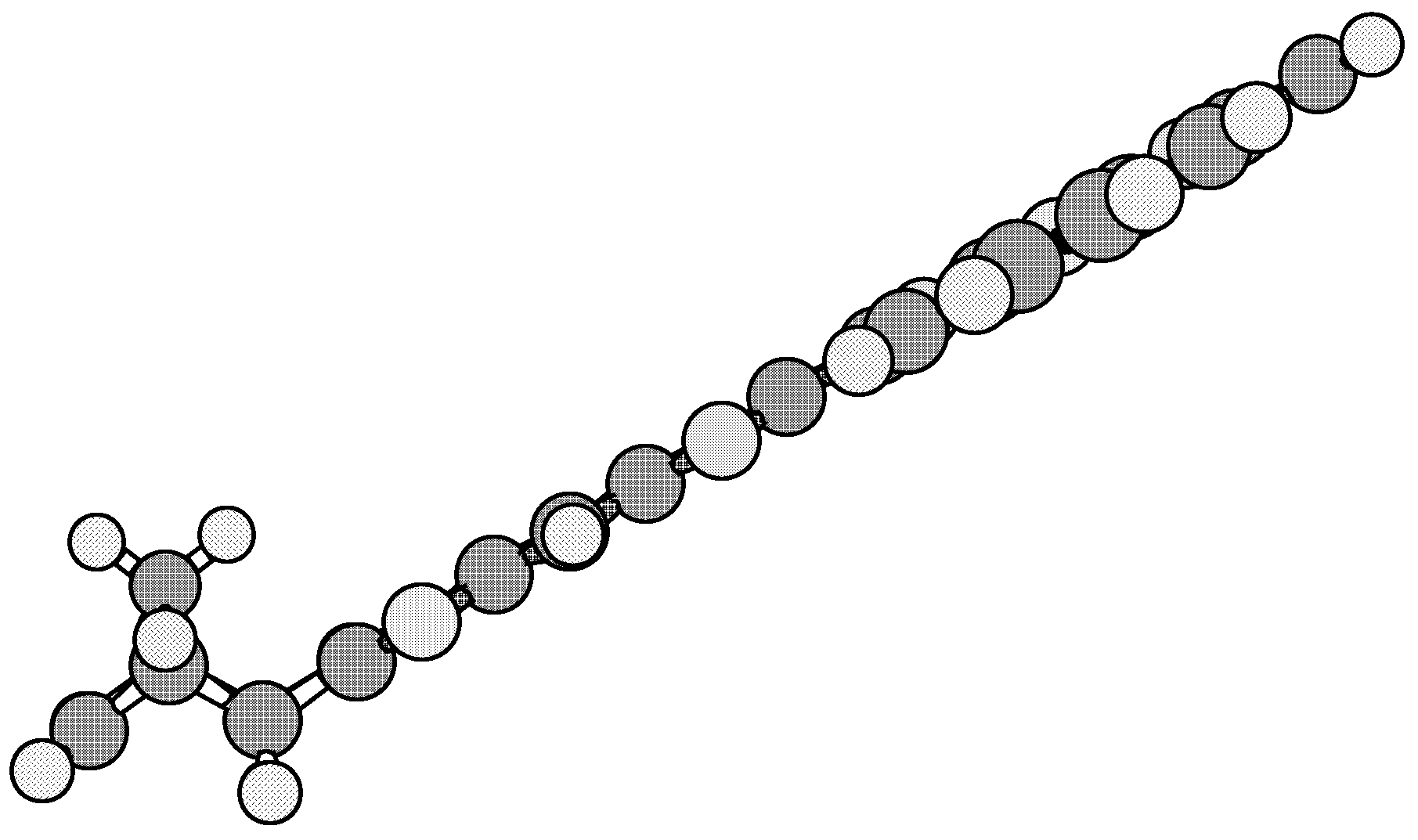
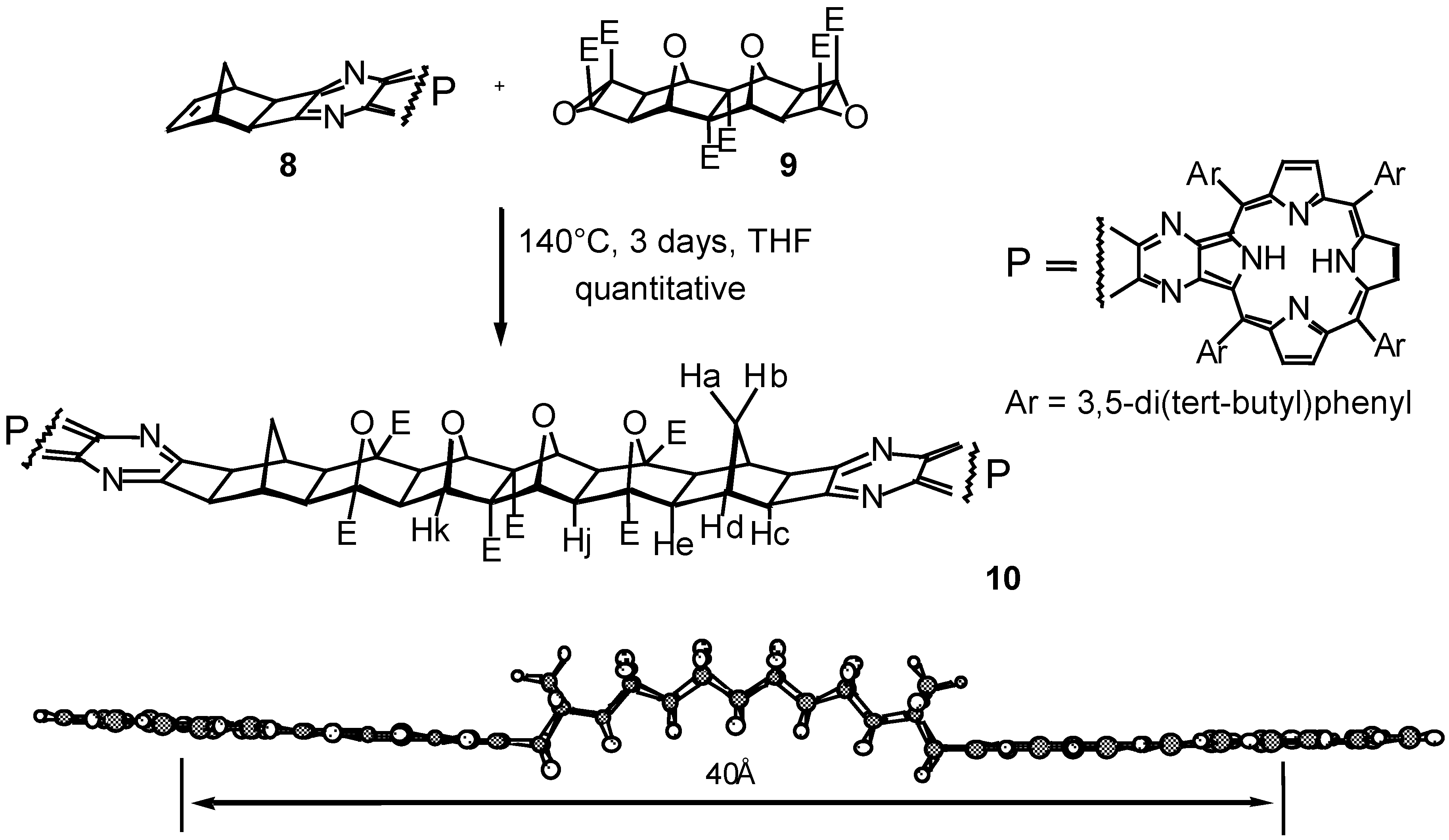
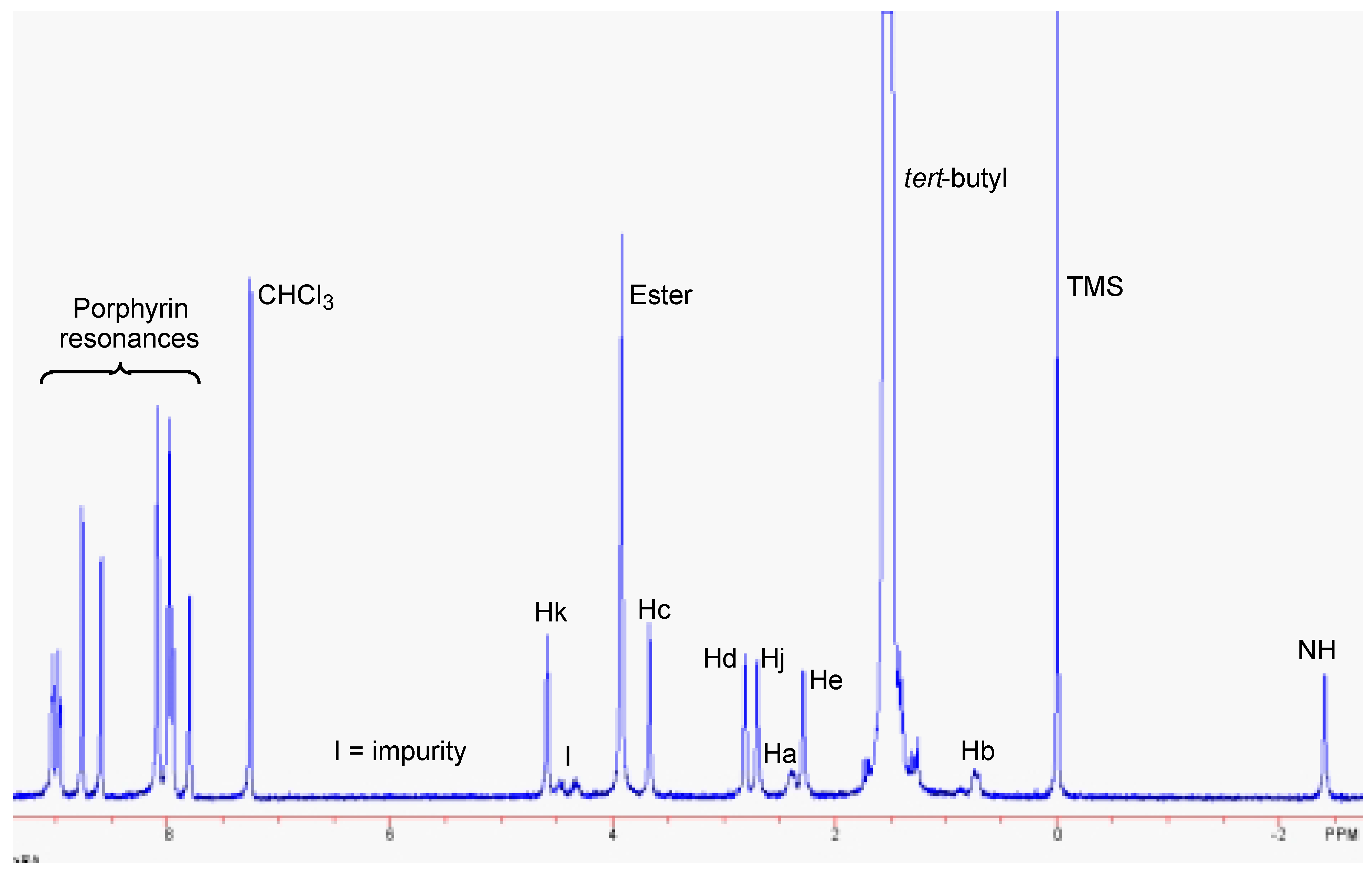
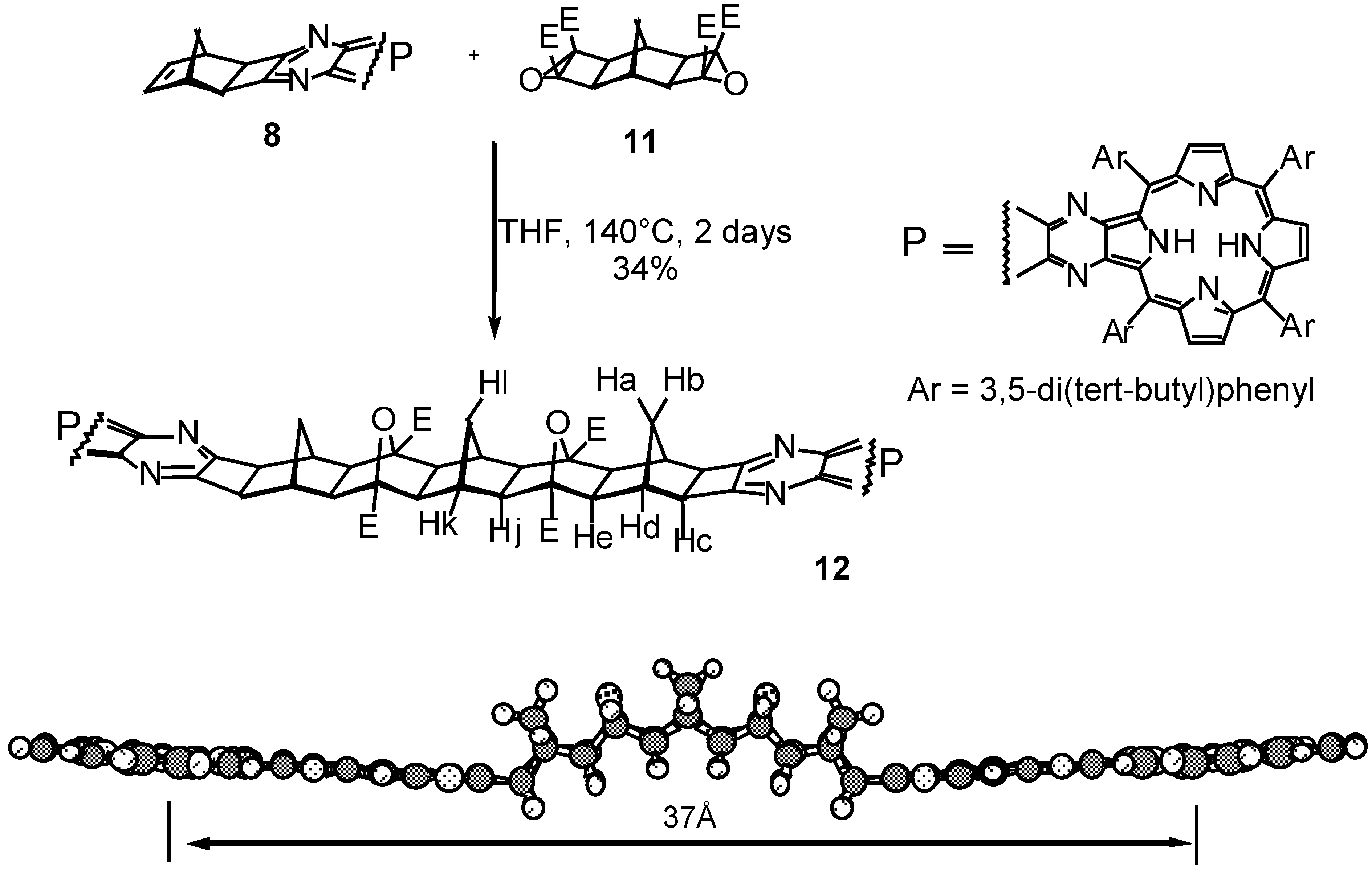
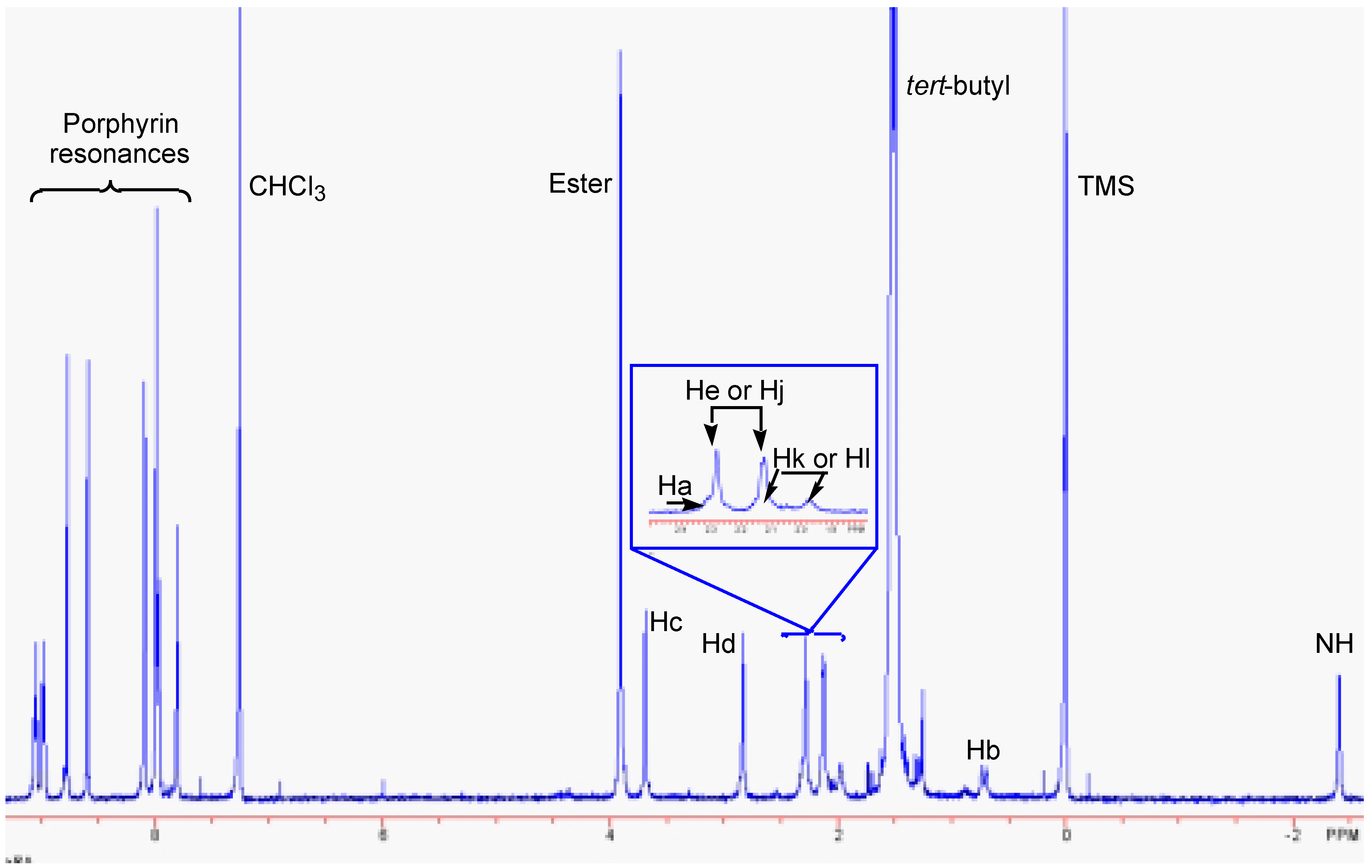
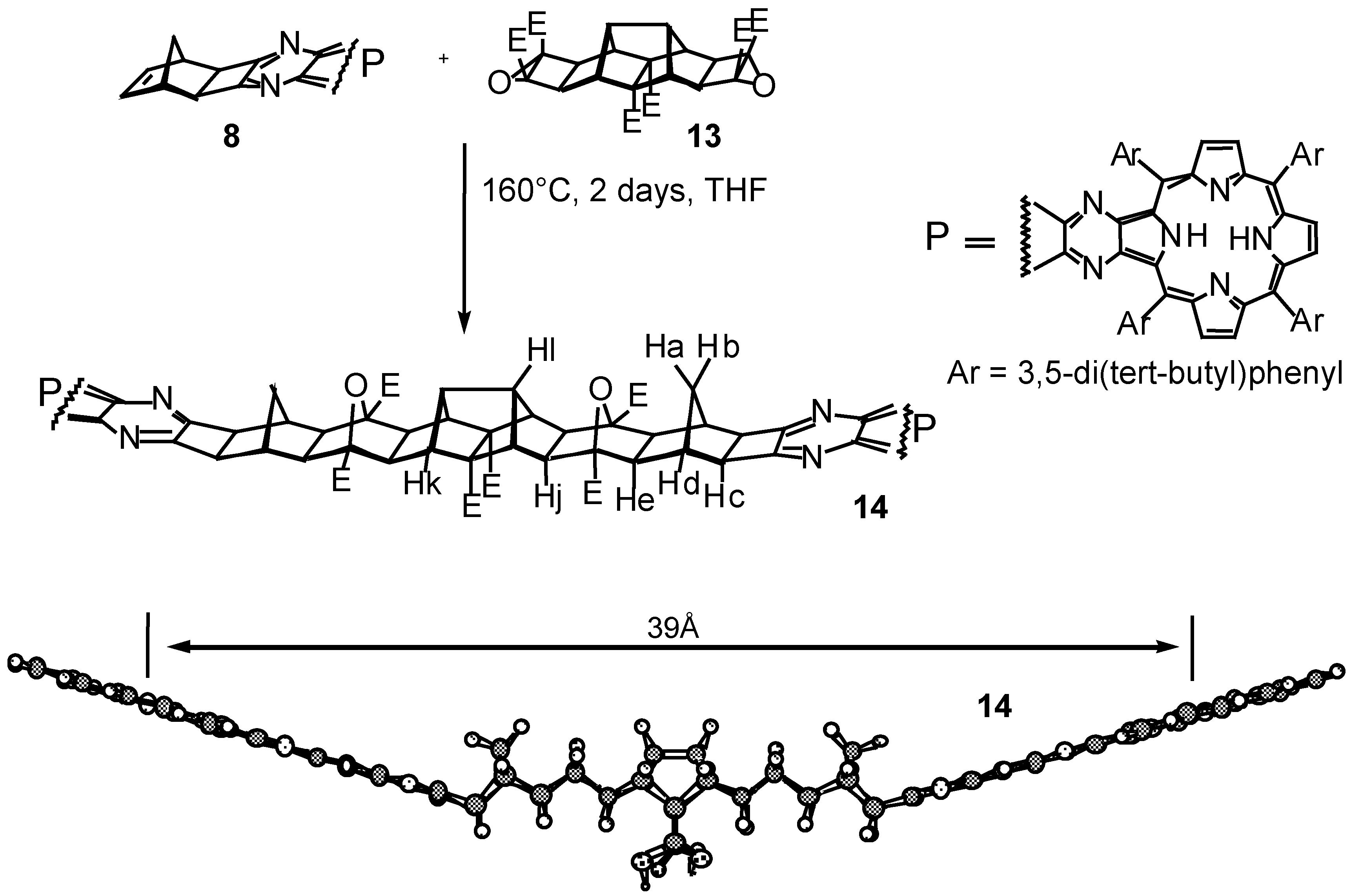
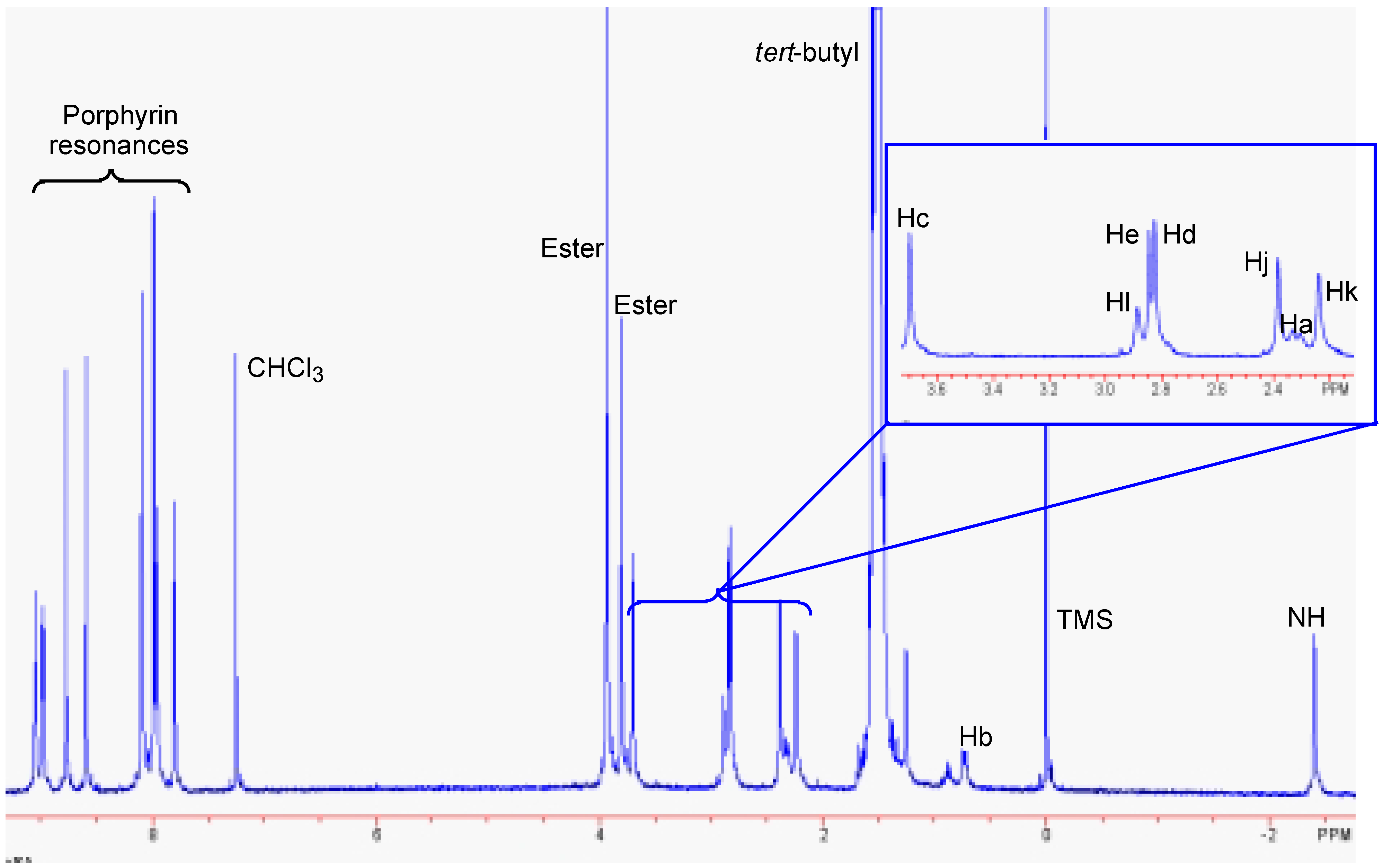
Conclusions
Experimental
General
Acknowledgements
References and Notes
- Pullerits, T.; Sundström, V. Photosynthetic Light-Harvesting Pigment-Protein Complexes: Toward Understanding How and Why. Acc. Chem. Res. 1996, 29, 381–389. [Google Scholar] McDermott, G.; Prince, S. M.; Freer, A. A.; Hawthornwaite-Lawless, A. M.; Papiz, M. Z.; Cogdell, R. J.; Isaacs, N. W. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 1995, 374, 517–521. [Google Scholar]
- For recent leading references see: Nakano, A.; Yamazaki, T.; Nishimura, Y.; Yamazaki, I.; Osuka, A. Three-Dimensionally Arranged Windmill and Grid Porphyrin Arrays by AgI-Promoted meso- meso Block Oligomerization. Chem. Eur. J. 2000, 6, 3254–3271. [Google Scholar] Vicente, M. G. H.; Jaquinod, L.; Smith, K. M. Oligomeric porphyrin arrays. Chem. Commun 1999, 1771–1782. [Google Scholar]
- For an excellent review see: Chambron, J-C.; Heitz, V.; Sauvage, J-P. Noncovalent multiporphyrin assemblies. In Porphyrin Handbook; pp. 1–42. vol. 6, 2000; Kadish, K. M., Smith, K. M., Guilard, R., Eds.; [Google Scholar]
- Warrener, R. N.; Johnston, M. R.; Gunter, M. J. Preparation of New Porphyrin BLOCKs and their Application to the Synthesis of Spacer and Cavity Ribbon Structures. Synlett 1998, 593–595. [Google Scholar] [CrossRef]
- Johnston, M. R.; Gunter, M. J.; Warrener, R. N. Templated formation of multiporphyrin assemblies resembling a molecular universal joint. Chem. Commun. 1998, 2739–2740. [Google Scholar] [CrossRef]
- Warrener, R. N.; Schultz, A. C.; Johnston, M. R.; Gunter, M. J. New Porphyrin 4π-Cycloaddition Reagents and their use in the Preparation of Porphyrin-(Rigid Spacer)-1,10-Phenanthrolines in which Geometric ‘Tuning’ of the Chromophores is a Feature. J. Org. Chem. 1999, 64, 4218–4219. [Google Scholar] [CrossRef]
- Warrener, R. N.; Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A. A New Building BlOCK Technique Based on Cycloaddition Chemistry for the Regiospecific Linking of Alicyclic Sub-units as a Route to Large, Custom-Functionalised Structures. Chem. Commun. 1997, 1023–1024. [Google Scholar] Margetic, D.; Russell, R. A.; Sun, G.; Warrener, R. N. A 1,3-Dipolar Cycloaddition Route to 7-Azanorbornanes: Application to the Synthesis of syn-Facial N-Bridged Polynorbornanes. Synlett 1998, 588–589. [Google Scholar]
- Crossley, M. J.; Govenlock, L. J.; Prashar, J. K. Synthesis of Porphyrin-2,3,12,13- and –2,3,7,8- tetrones: Building Blocks for the Synthesis of Extended Porphyrin Arrays. J. Chem. Soc., Chem. Commun. 1995, 2379–2380. [Google Scholar] Crossley, M. J.; Burn, P. L. An Approach to Porphyrin-based Molecular Wires: Synthesis of a Bis(porphyrin)tetraone and its Conversion to a Linearly Conjugated Tetrakisporphyrin System. J. Chem. Soc., Chem. Commun. 1991, 1569–1571. [Google Scholar]
- Johnston, M. R.; Warrener, R. N. manuscript in preparation.
- Warrener, R. N.; Margetic, D.; Amarasekara, A. S.; Russell, R. A. The synthesis of functionalised cavity structures via 1,3-dipolar cycloaddition of angle-shaped alkenes to curved norbornene framed dipoles. Org. Lett. 1999, 2, 203–206. [Google Scholar] [CrossRef]
- Martin, H-D.; Schiwek, H-J.; Spanget-Larsen, J.; Gleiter, R. Synthesis, Spectroscopic Properties and Transannular Interactions of Tricyclic 1,2-Cyclobutanediones. Chem. Ber. 1978, 111, 2557–2562. [Google Scholar] Albert, B.; Heller, C.; Iden, R.; Martin, G.; Martin, H-D.; Mayer, B.; Oftring, A. Design and Synthesis of α-Diketones. The Cyclobutane-1,2-dione Chromophore: Synthesis, Dienophilic Reactivity and Electronic Properties of Cyclobutenedione and Polycyclic Cyclobutanediones. Israel J. Chem. 1985, 25, 74–83. [Google Scholar]
- Molecular modeling was carried out using SPARTAN v5.0.3 (Wavefunction Inc.) on an Silicon Graphics Indigo2workstation. Minimisations were done without peripheral substituents such as aromatic rings or ester groups which have no bearing on the geometric outcome of the calculation.
- Margetic, D. M.; Warrener, R. N. unpublished results.
- Warrener, R. N.; Margetic, D. M.; Foley, P. J.; Butler, D. N.; Winling, A.; Beales, K., A.; Russell, R. A. syn-Facial hetero-bridged [n]polynorbornanes: a new class of polarofacial framework molecules composed of fused 7-oxa- and 7-azanorbornanes. Tetrahedron 2001. in press. [Google Scholar] [CrossRef]
- Margetic, D.; Johnston, M. R.; Tiekink, E. R. T.; Warrener, R. N. Synthesis and Modelling of Novel Rigid Rods derived from a Simple pentacyclic bis-norbornene. Tetrahedron Lett. 1998, 39, 5277–5280. [Google Scholar] [CrossRef]
- Sample availability: Available from the author.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for non-commercial purposes
Share and Cite
Johnston, M.R. Bis-Porphyrin Racks with Space-Separated Co-Planar Porphyrin Rings. Molecules 2001, 6, 406-416. https://doi.org/10.3390/60400406
Johnston MR. Bis-Porphyrin Racks with Space-Separated Co-Planar Porphyrin Rings. Molecules. 2001; 6(4):406-416. https://doi.org/10.3390/60400406
Chicago/Turabian StyleJohnston, Martin R. 2001. "Bis-Porphyrin Racks with Space-Separated Co-Planar Porphyrin Rings" Molecules 6, no. 4: 406-416. https://doi.org/10.3390/60400406
APA StyleJohnston, M. R. (2001). Bis-Porphyrin Racks with Space-Separated Co-Planar Porphyrin Rings. Molecules, 6(4), 406-416. https://doi.org/10.3390/60400406