Synthesis and X-ray Crystal Structure of Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene)

Abstract
:Introduction

Results and Discussion
where R = -CH2C10H7.


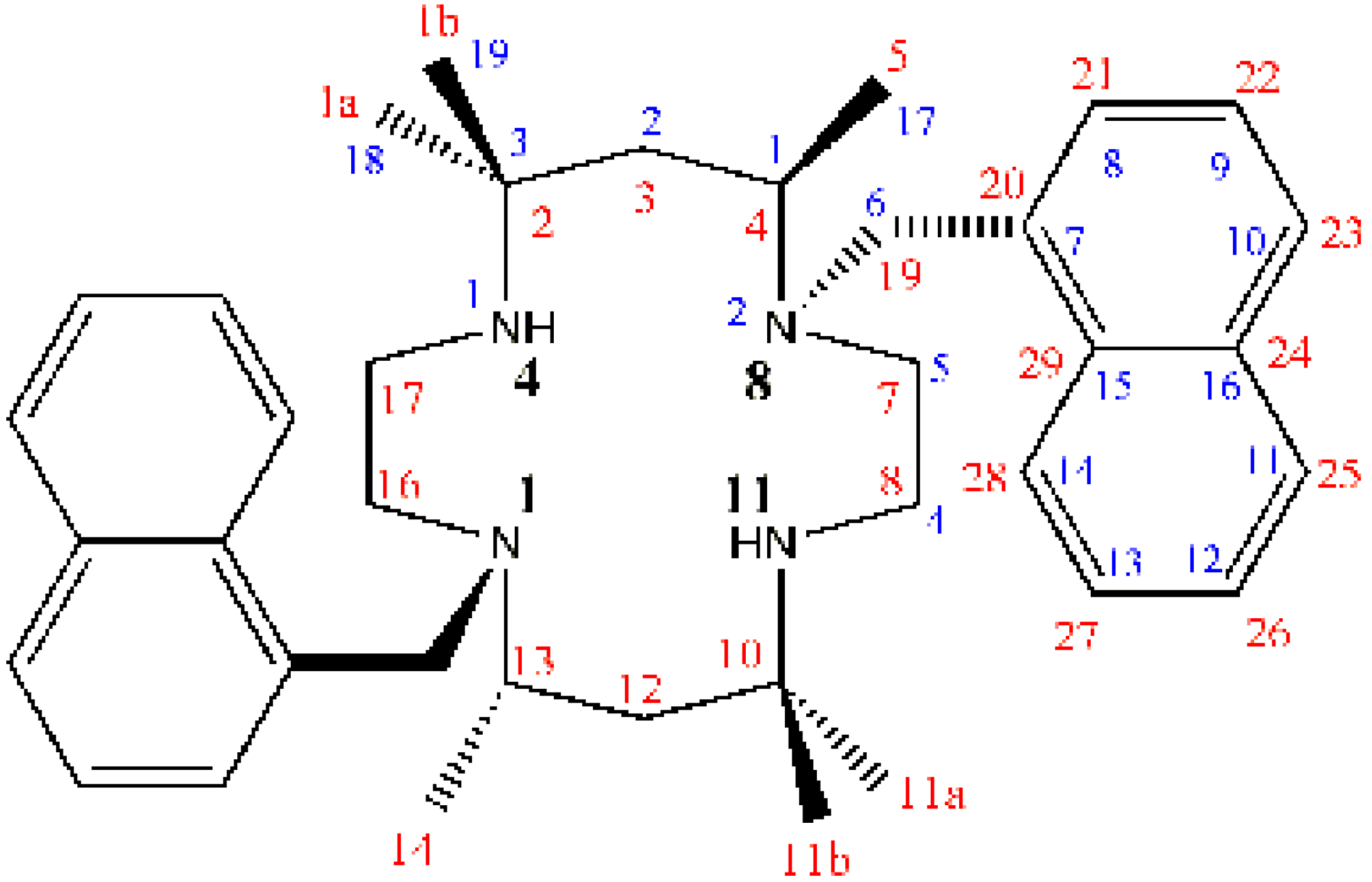{kind=link}
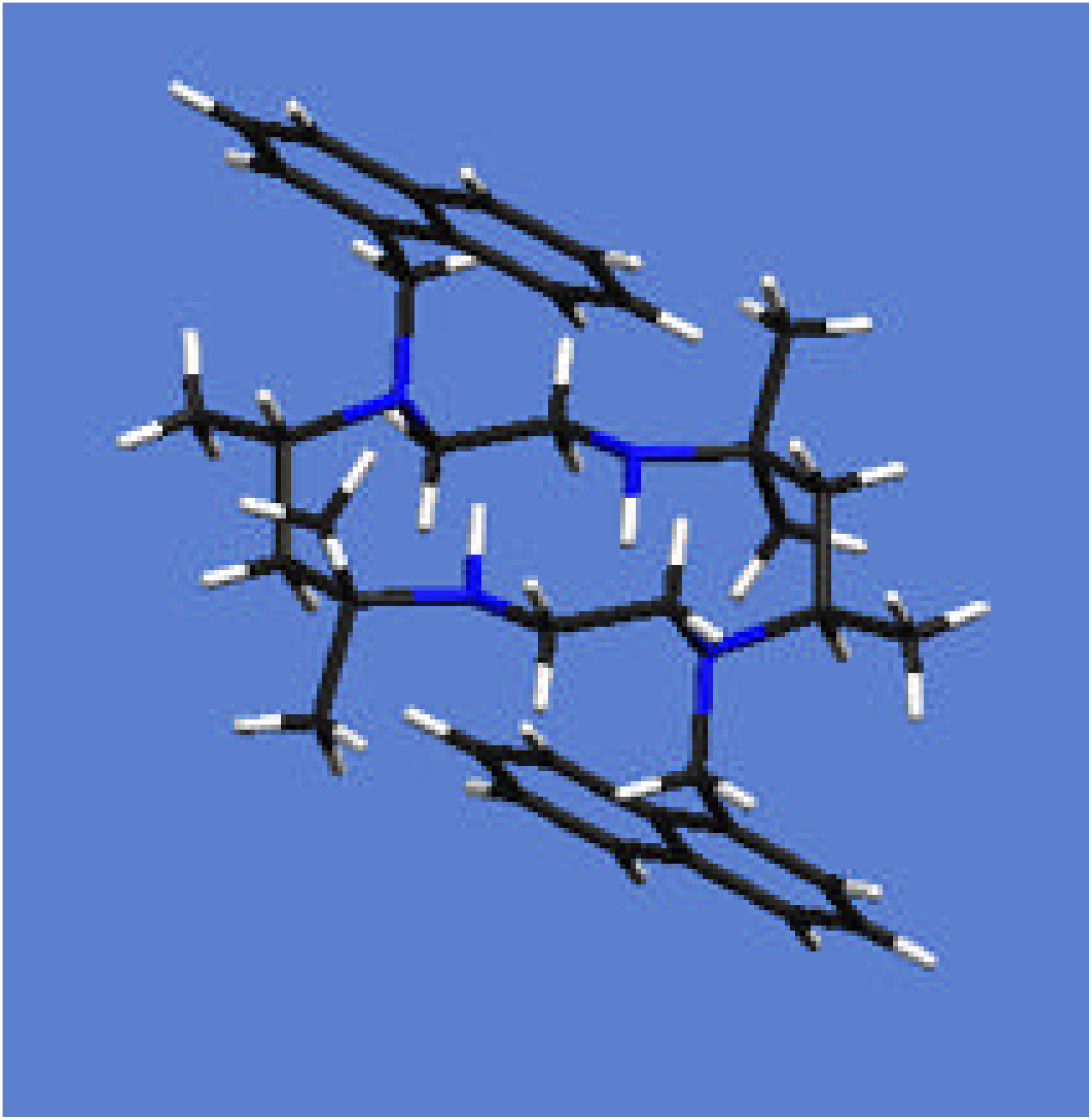{kind=link}
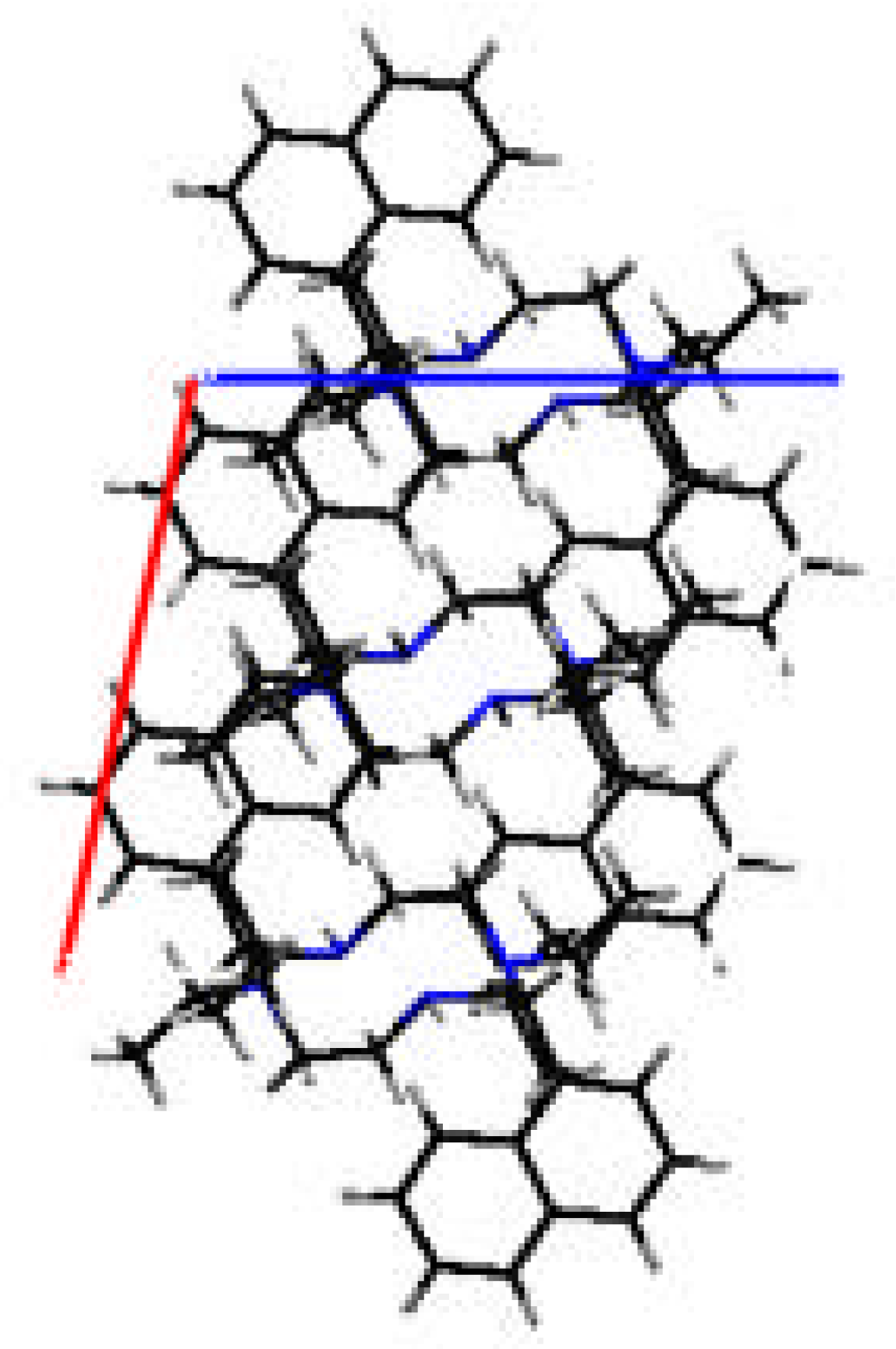{kind=link}
| atom | atom | Distance | Atom | atom | Distance |
| N1 | C3 | 1.49(1) | N1 | C4 | 1.46(1) |
| N2 | C1 | 1.50(1) | N2 | C5 | 1.47(1) |
| N2 | C6 | 1.46(1) | C1 | C2 | 1.51(1) |
| C1 | C17 | 1.53(1) | C2 | C3 | 1.52(1) |
| C3 | C18 | 1.55(1) | C3 | C19 | 1.54(1) |
| C4 | C5 | 1.51(1) | C6 | C7 | 1.52(1) |
| C7 | C8 | 1.37(1) | C7 | C15 | 1.41(1) |
| C8 | C9 | 1.41(1) | C9 | C10 | 1.35(1) |
| C10 | C16 | 1.41(1) | C11 | C12 | 1.34(1) |
| C11 | C16 | 1.40(1) | C12 | C13 | 1.40(1) |
| C13 | C14 | 1.38(1) | C14 | C15 | 1.42(1) |
| C15 | C16 | 1.41(1) |
| atom | atom | atom | Angle | atom | atom | atom | angle |
| C3 | N1 | C4 | 115.1(7) | C1 | N2 | C5 | 111.3(7) |
| C1 | N2 | C6 | 112.6(7) | C5 | N2 | C6 | 110.8(7) |
| N2 | C1 | C2 | 112.4(8) | N2 | C1 | C17 | 114.3(8) |
| C2 | C1 | C17 | 111.5(9) | C1 | C2 | C3 | 120.3(9) |
| N1 | C3 | C2 | 107.8(8) | N1 | C3 | C18 | 113.1(8) |
| N1 | C3 | C19 | 108.4(8) | C2 | C3 | C18 | 109.7(8) |
| C2 | C3 | C19 | 108.7(9) | C18 | C3 | C19 | 109.0(9) |
| N1 | C4 | C5 | 113.2(8) | N2 | C5 | C4 | 114.8(8) |
| N2 | C6 | C7 | 112.9(8) | C6 | C7 | C8 | 120.1(9) |
| C6 | C7 | C15 | 121.1(9) | C8 | C7 | C15 | 119(1) |
| C7 | C8 | C9 | 122(1) | C8 | C9 | C10 | 120(1) |
| C9 | C10 | C16 | 121(1) | C12 | C11 | C16 | 120(1) |
| C11 | C12 | C13 | 122(1) | C12 | C13 | C14 | 120(1) |
| C13 | C14 | C15 | 119.9(9) | C7 | C15 | C14 | 121.9(9) |
| C7 | C15 | C16 | 120(1) | C14 | C15 | C16 | 118.1(9) |
| C10 | C16 | C11 | 121(1) | C10 | C16 | C15 | 119(1) |
| C11 | C16 | C15 | 121(1) |

Experimental
General
| Empirical Formula | C38H52N4 |
| Formula Weight | 564.86 |
| Crystal Color, Habit | colourless, plate |
| Crystal Dimensions | 0.06 x 0.20 x 0.24 mm |
| Crystal System | monoclinic |
| Lattice Type | Primitive |
| Space Group | P21/a (#14) |
| Lattice Parameters | a = 10.778(3) Å b = 13.809(3) Å c = 11.420(2) Å β= 102.49(2)o |
| Volume | 1659.5(6) Å3 |
| No. of Reflections Used for Unit Cell Determination (2q range) | 25 ( 20.4 - 26.0o ) |
| Omega Scan Peak Width at Half-height | 0.25o |
| Z value | 2 |
| Dcalc | 1.130 g/cm3 |
| F000 | 616.00 |
| µ(MoK∀) | 0.66 cm-1 |
| Function Minimized | S w (|Fo| - |Fc|)2 |
| Least Squares Weights | 1/s2(Fo) = 4Fo2/s2(Fo2) |
| p-factor | 0.0142 |
| Anomalous Dispersion | All non-hydrogen atoms |
| No. Observations (I>3.00s(I)) | 588 |
| No. Variables | 101 |
| Reflection/Parameter Ratio | 5.82 |
| Residuals: R; Rw | 0.059 ; 0.056 |
| Goodness of Fit Indicator | 2.31 |
| Max Shift/Error in Final Cycle | 0.00 |
| Maximum peak in Final Diff. Map | 0.16 e-/Å3 |
| Minimum peak in Final Diff. Map | -0.18 e-/Å3 |
Syntheses
Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene)
Acknowledgements
References
- Collin, J.-P.; Sauvage, J.-P. J. Chem. Soc. Chem. Commun. 1987, 1075.
- Kimura, E.; Koike, T.; Takahashi, M. J. Chem. Soc. Chem. Commun. 1985, 385.
- Cox, J.P.L.; Jankowski, K.J.; Kataky, R.; Parker, D.; Beeley, N.R.A.; Boyce, B.A.; Eaton, M.A.W.; Miller, K.; Millian, A.T.; Harrison, A.; Walker, C. J. Chem. Soc. Chem. Commun. 1989, 797.Morphy, J.R.; Parker, D.; Kataky, R.; Harrison, A.; Eaton, M.A.W.; Millian, A.; Phipps, A.; Walker, C. J. Chem. Soc. Chem. Commun. 1989, 792.
- Costamagna, J.; Ferraudi, G.; Matsuhiro, B.; Campos-Vallette, M.; Canales, J.; Villagrán, M.; Vargas, J.; Aguirre, M.J. Coord. Chem. Rev. 2000, 196, 125.
- McAuley, A.; Subramanian, S. Coord. Chem. Rev. 2000, 200-202, 75.
- Wainwright, K.P. Coord. Chem. Rev. 1997, 166, 35.
- Haines, R.I. Rev. Inorg. Chem. 2001, 21, 165.
- Haines, R.I.; Baldwin, R. Transition Met. Chem. 2002, 27, 284.Bissessur, R.; Haines, R.I.; Hutchings, D.R.; Bruening, R. Chem. Commun. 2001, 1598.Haines, R.I.; Hutchings, D.R.; McCormack, T.M. J. Inorg. Biochem. 2001, 85, 1.Haines, R.I.; Hutchings, D.R.; Lucas, R.J.; Miller, D. Can. J. Chem. 2001, 79, 54.
- SIR92: Altomare, A.; Cascarano, M.; Giacovazzo, C.; Guagliardi, A. J. Appl. Cryst. 1994, 26, 343.
- DIRDIF94: Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; de Gelder, R.; Israel, R; Smits, J.M.M. The DIRDIF-94 program system, Technical Report of the Crystallography Laboratory. University of Nijmegen: The Netherlands, 1994. [Google Scholar]
- teXsan for Windows version 1.05: Crystal Structure Analysis Package, Molecular Structure Corporation, 1997-8
- CCDC 199958 contains the supplementary crystallographic data for this paper. These data can be obtained, free of charge, via www.ccdc.cam.ac.uk/conts/retreiving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; email: deposit@ccdc.cam.ac.uk).
- Douglas, B.E. Inorganic Syntheses; Vol. 18, John Wiley and Sons: New York, 1978; pp. 10–13. [Google Scholar]
- Curtis, N.F. J. Chem. Soc. 1964, 2644.
- Sample Availability: Samples of meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene) are available from MDPI
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
Haines, R.I.; Hutchings, D.R. Synthesis and X-ray Crystal Structure of Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene). Molecules 2003, 8, 243-250. https://doi.org/10.3390/80200243
Haines RI, Hutchings DR. Synthesis and X-ray Crystal Structure of Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene). Molecules. 2003; 8(2):243-250. https://doi.org/10.3390/80200243
Chicago/Turabian StyleHaines, Robert I., and Dean R. Hutchings. 2003. "Synthesis and X-ray Crystal Structure of Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene)" Molecules 8, no. 2: 243-250. https://doi.org/10.3390/80200243
APA StyleHaines, R. I., & Hutchings, D. R. (2003). Synthesis and X-ray Crystal Structure of Meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane-1,8-di-(1-methylnaphthalene). Molecules, 8(2), 243-250. https://doi.org/10.3390/80200243