Impact of Methylglyoxal and High Glucose Co-treatment on Human Mononuclear Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
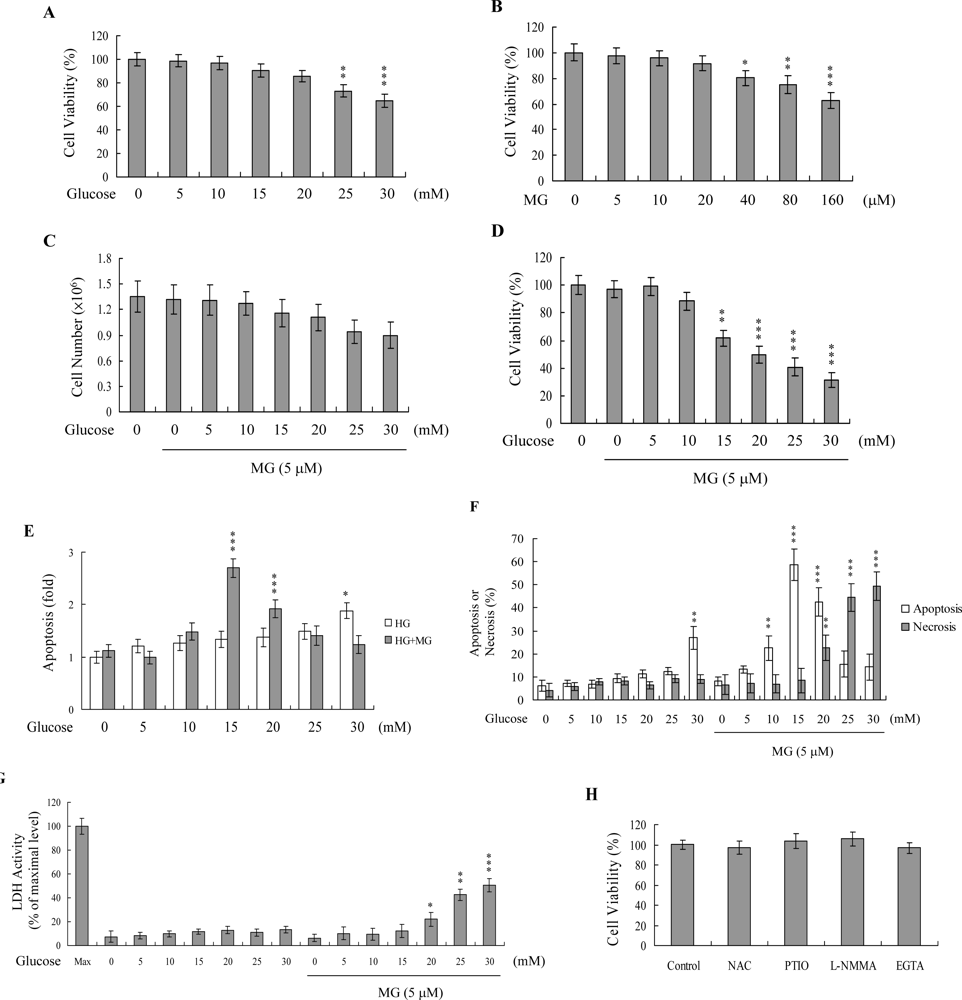
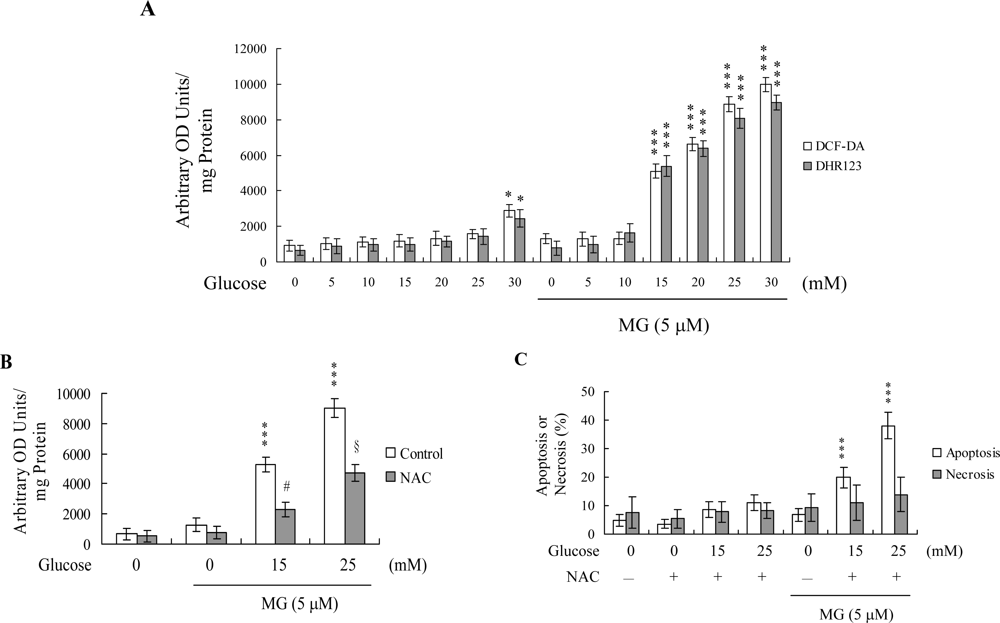
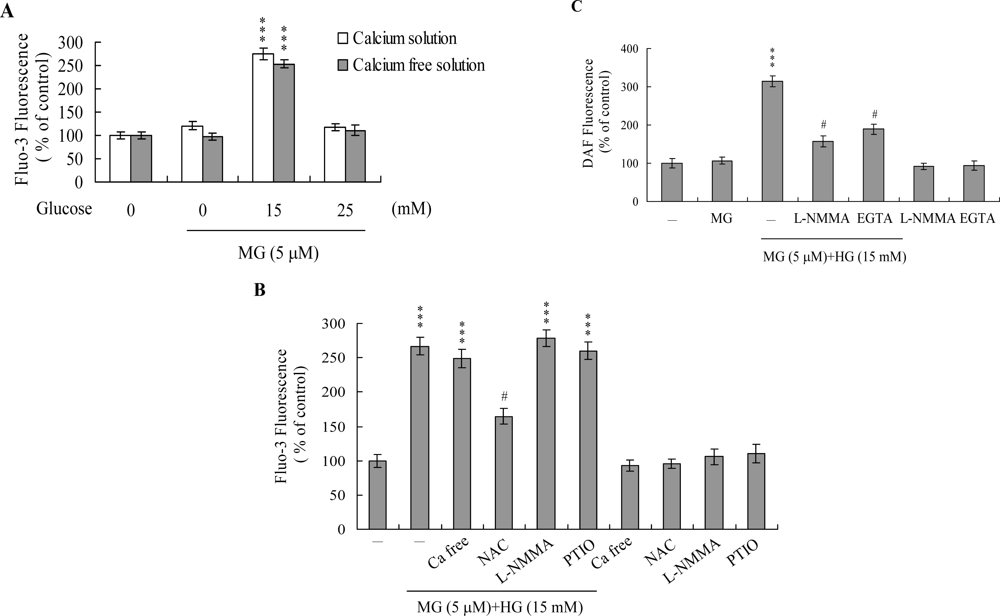
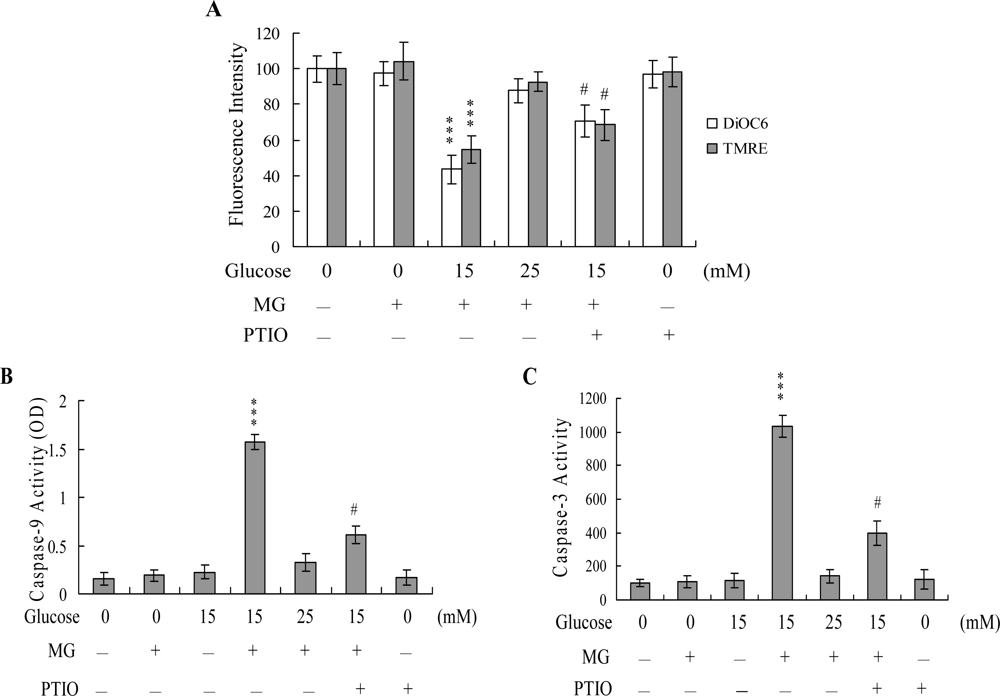
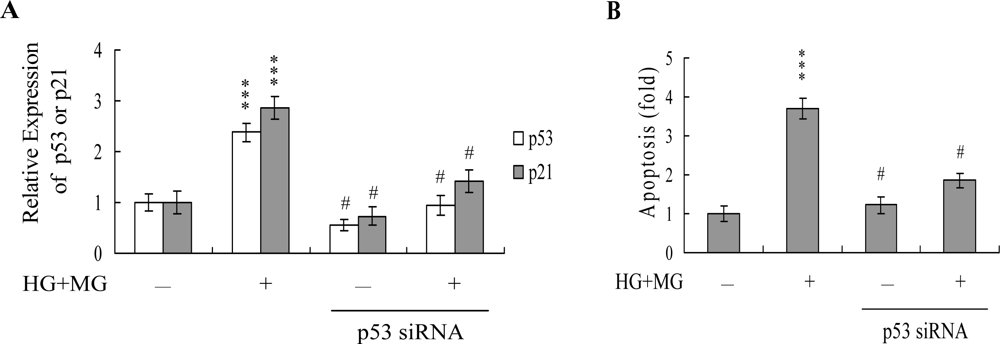
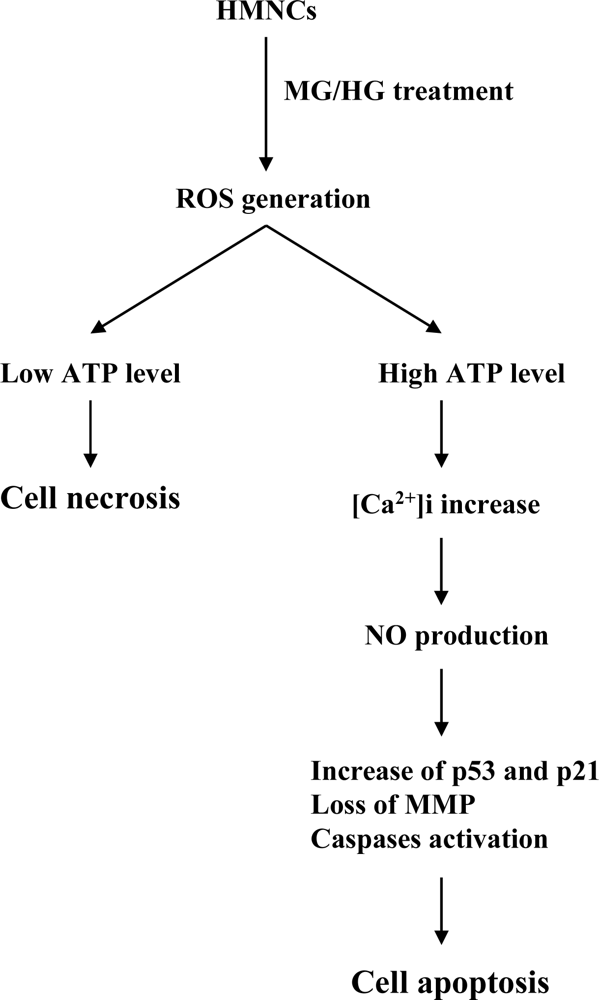
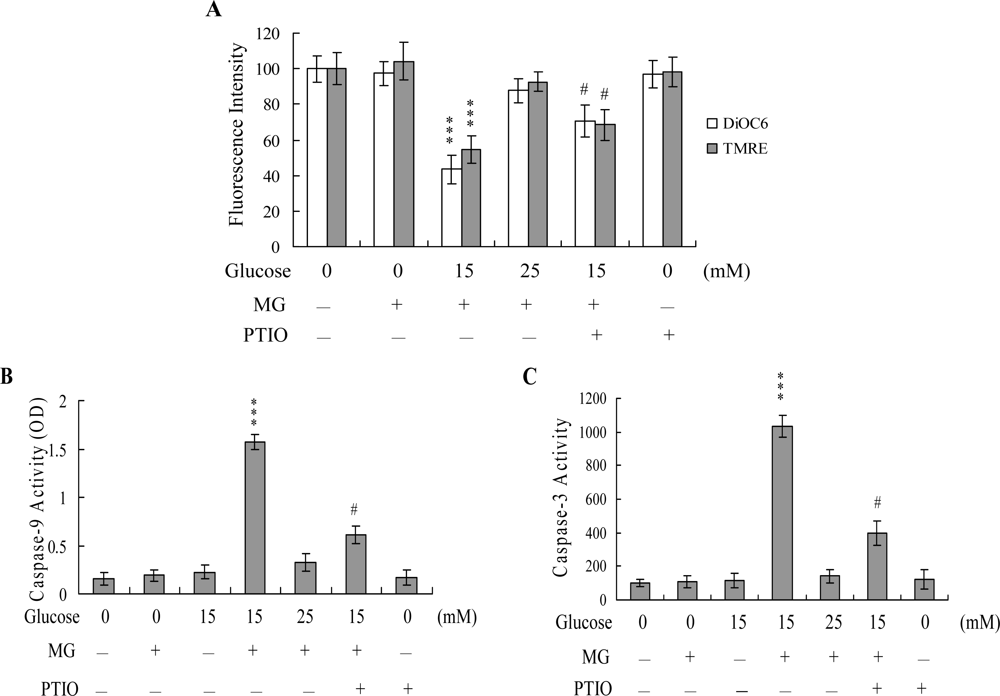
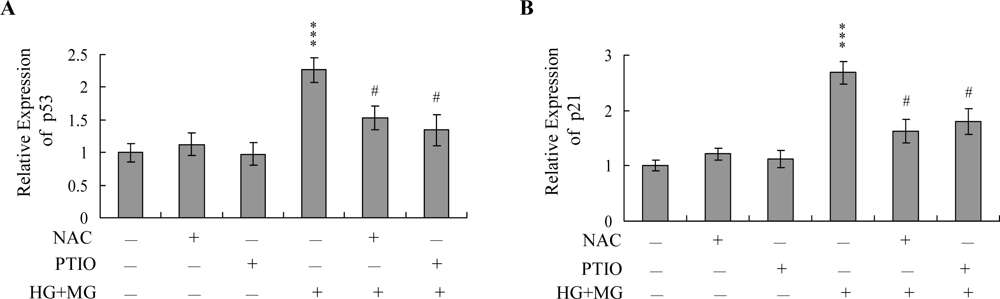
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. Isolation of human mononuclear cells (HMNCs) and treatment
4.3. MTT assay
4.4. Assessment of necrosis and apoptosis
4.5. ROS assay
4.6. ATP level analysis
4.7. Detection of intracellular calcium concentration ([Ca2+]i)
4.8. Detection of intracellular NO content
4.9. Caspase activity assays
4.10. Real-time RT-PCR assay
4.11. siRNA knockdown
4.12. Statistics
Acknowledgments
References and Notes
- Okado, A; Kawasaki, Y; Hasuike, Y; Takahashi, M; Teshima, T; Fujii, J; Taniguchi, N. Induction of apoptotic cell death by methylglyoxal and 3-deoxyglucosone in macrophagederived cell lines. Biochem. Biophys. Res. Commun 1996, 225, 219–224. [Google Scholar]
- Ho, FM; Liu, SH; Liau, CS; Huang, PJ; Lin-Shiau, SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation 2000, 101, 2618–2624. [Google Scholar]
- Chan, WH. Effect of resveratrol on high glucose-induced stress in human leukemia K562 cells. J. Cell. Biochem 2005, 94, 1267–1279. [Google Scholar]
- Hsuuw, YD; Chang, CK; Chan, WH; Yu, JS. Curcumin prevents methylglyoxal-induced oxidative stress and apoptosis in mouse embryonic stem cells and blastocysts. J. Cell. Physiol 2005, 205, 379–386. [Google Scholar]
- Chan, WH; Wu, HJ. Methylglyoxal and high glucose co-treatment induces apoptosis or necrosis in human umbilical vein endothelial cells. J. Cell. Biochem 2008, 103, 1144–1157. [Google Scholar]
- Qin, S; Minami, Y; Kurosaki, T; Yamamura, H. Distinctive functions of Syk and Lyn in mediating osmotic stress- and ultraviolet C irradiation-induced apoptosis in chicken B cells. J. Biol. Chem 1997, 272, 17994–17999. [Google Scholar]
- Chan, WH; Yu, JS; Yang, SD. PAK2 is cleaved and activated during hyperosmotic shockinduced apoptosis via a caspase-dependent mechanism: evidence for the involvement of oxidative stress. J. Cell. Physiol 1999, 178, 397–408. [Google Scholar]
- Kasai, H; Kumeno, K; Yamaizumi, Z; Nishimura, S; Nagao, M; Fujita, Y; Sugimura, T; Nukaya, H; Kosuge, T. Mutagenicity of methylglyoxal in coffee. Gann 1982, 73, 681–683. [Google Scholar]
- Baden, T; Yamawaki, H; Saito, K; Mukohda, M; Okada, M; Hara, Y. Telmisartan inhibits methylglyoxal-mediated cell death in human vascular endothelium. Biochem. Biophys. Res. Commun 2008, 373, 253–257. [Google Scholar]
- Phillips, SA; Thornalley, PJ. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem 1993, 212, 101–105. [Google Scholar]
- Ahmed, MU; Brinkmann Frye, E; Degenhardt, TP; Thorpe, SR; Baynes, JW. N-epsilon-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J 1997, 324, 565–570. [Google Scholar]
- Fosmark, DS; Torjesen, PA; Kilhovd, BK; Berg, TJ; Sandvik, L; Hanssen, KF; Agardh, CD; Agardh, E. Increased serum levels of the specific advanced glycation end product methylglyoxal-derived hydroimidazolone are associated with retinopathy in patients with type 2 diabetes mellitus. Metabolism 2006, 55, 232–236. [Google Scholar]
- Mostafa, AA; Randell, EW; Vasdev, SC; Gill, VD; Han, Y; Gadag, V; Raouf, AA; El Said, H. Plasma protein advanced glycation end products, carboxymethyl cysteine, and carboxyethyl cysteine, are elevated and related to nephropathy in patients with diabetes. Mol. Cell. Biochem 2007, 302, 35–42. [Google Scholar]
- Wu, L. Is methylglyoxal a causative factor for hypertension development? Can. J. Physiol. Pharmacol 2006, 84, 129–139. [Google Scholar]
- Shipanova, IN; Glomb, MA; Nagaraj, RH. Protein modification by methylglyoxal: chemical nature and synthetic mechanism of a major fluorescent adduct. Arch. Biochem. Biophys 1997, 344, 29–36. [Google Scholar]
- Uchida, K; Khor, OT; Oya, T; Osawa, T; Yasuda, Y; Miyata, T. Protein modification by a Maillard reaction intermediate methylglyoxal. Immunochemical detection of fluorescent 5-methylimidazolone derivatives in vivo. FEBS Lett 1997, 410, 313–318. [Google Scholar]
- Yim, HS; Kang, SO; Hah, YC; Chock, PB; Yim, MB. Free radicals generated during the glycation reaction of amino acids by methylglyoxal. A model study of protein-cross-linked free radicals. J. Biol. Chem 1995, 270, 28228–28233. [Google Scholar]
- Wu, HJ; Chan, WH. Genistein protects methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Toxicol. In Vitro 2007, 21, 335–342. [Google Scholar]
- Chan, WH; Wu, HJ. Protective effects of curcumin on methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Acta Pharmacol. Sin 2006, 27, 1192–1198. [Google Scholar]
- Messier, C; Gagnon, M. Glucose regulation and cognitive functions: relation to Alzheimer’s disease and diabetes. Behav. Brain Res 1996, 75, 1–11. [Google Scholar]
- Gerozissis, K. Brain insulin: regulation, mechanisms of action and functions. Cell. Mol. Neurobiol 2003, 23, 1–25. [Google Scholar]
- Ota, K; Nakamura, J; Li, W; Kozakae, M; Watarai, A; Nakamura, N; Yasuda, Y; Nakashima, E; Naruse, K; Watabe, K; Kato, K; Oiso, Y; Hamada, Y. Metformin prevents methylglyoxal-induced apoptosis of mouse Schwann cells. Biochem. Biophys. Res. Commun 2007, 357, 270–275. [Google Scholar]
- Okouchi, M; Okayama, N; Aw, TY. Hyperglycemia potentiates carbonyl stress-induced apoptosis in naive PC-12 cells: relationship to cellular redox and activator protease factor-1 expression. Curr. Neurovasc. Res 2005, 2, 375–386. [Google Scholar]
- Jan, CR; Chen, CH; Wang, SC; Kuo, SY. Effect of methylglyoxal on intracellular calcium levels and viability in renal tubular cells. Cell Signal 2005, 17, 847–855. [Google Scholar]
- Halliwell, B; Gutteridge, JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol 1990, 186, 1–85. [Google Scholar]
- Chan, WH. Ginkgolide B induces apoptosis and developmental injury in mouse embryonic stem cells and blastocysts. Hum. Reprod 2006, 21, 2985–2995. [Google Scholar]
- Chan, WH; Shiao, NH; Lu, PZ. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol. Lett 2006, 167, 191–200. [Google Scholar]
- Ekmekcioglu, S; Tang, CH; Grimm, EA. NO news is not necessarily good news in cancer. Curr. Cancer Drug Targets 2005, 5, 103–115. [Google Scholar]
- Zhou, J; Brune, B. NO and transcriptional regulation: from signaling to death. Toxicology 2005, 208, 223–233. [Google Scholar]
- Rao, CV. Nitric oxide signaling in colon cancer chemoprevention. Mutat. Res 2004, 555, 107–119. [Google Scholar]
- Lu, Z; Tao, Y; Zhou, Z; Zhang, J; Li, C; Ou, L; Zhao, B. Mitochondrial reactive oxygen species and nitric oxide-mediated cancer cell apoptosis in 2-butylamino-2-demethoxyhypocrellin B photodynamic treatment. Free Radic. Biol. Med 2006, 41, 1590–1605. [Google Scholar]
- Nazarewicz, RR; Zenebe, WJ; Parihar, A; Larson, SK; Alidema, E; Choi, J; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res 2007, 67, 1282–1290. [Google Scholar]
- Ghafourifar, P; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci 2005, 26, 190–195. [Google Scholar]
- Brookes, PS. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar]
- Dennis, J; Bennett, JP, Jr. Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J. Neurosci. Res 2003, 72, 76–88. [Google Scholar]
- Elfering, SL; Sarkela, TM; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem 2002, 277, 38079–38086. [Google Scholar]
- Dedkova, EN; Ji, X; Lipsius, SL; Blatter, LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am. J. Physiol. Cell. Physiol 2004, 286, C406–415. [Google Scholar]
- Majno, G; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol 1995, 146, 3–15. [Google Scholar]
- Schwartz, SM; Bennett, MR. Death by any other name. Am. J. Pathol 1995, 147, 229–234. [Google Scholar]
- Chan, WH; Yu, JS; Yang, SD. Apoptotic signalling cascade in photosensitized human epidermal carcinoma A431 cells: involvement of singlet oxygen, c-Jun N-terminal kinase, caspase-3 and p21-activated kinase 2. Biochem. J 2000, 351, 221–232. [Google Scholar]
- Chan, WH; Wu, HY; Chang, WH. Dosage effects of curcumin on cell death types in a human osteoblast cell line. Food Chem. Toxicol 2006, 44, 1362–1371. [Google Scholar]
- Chan, WH; Chang, YJ. Dosage effects of resveratrol on ethanol-induced cell death in the human K562 cell line. Toxicol. Lett 2006, 161, 1–9. [Google Scholar]
- Thompson, CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Alison, MR; Sarraf, CE. Liver cell death: patterns and mechanisms. Gut 1994, 35, 577–581. [Google Scholar]
- Bonfoco, E; Krainc, D; Ankarcrona, M; Nicotera, P; Lipton, SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar]
- Hampton, MB; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett 1997, 414, 552–556. [Google Scholar]
- Eguchi, Y; Shimizu, S; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997, 57, 1835–1840. [Google Scholar]
- Leist, M; Single, B; Castoldi, AF; Kuhnle, S; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med 1997, 185, 1481–1486. [Google Scholar]
- Richter, C; Schweizer, M; Cossarizza, A; Franceschi, C. Control of apoptosis by the cellular ATP level. FEBS Lett 1996, 378, 107–110. [Google Scholar]
- Cosentino, F; Hishikawa, K; Katusic, ZS; Luscher, TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 1997, 96, 25–28. [Google Scholar]
- Baumgartner-Parzer, SM; Wagner, L; Pettermann, M; Grillari, J; Gessl, A; Waldhausl, W. High-glucose–triggered apoptosis in cultured endothelial cells. Diabetes 1995, 44, 1323–1327. [Google Scholar]
- Lee, C; Yim, MB; Chock, PB; Yim, HS; Kang, SO. Oxidation-reduction properties of methylglyoxal-modified protein in relation to free radical generation. J. Biol. Chem 1998, 273, 25272–25278. [Google Scholar]
- Oya, T; Hattori, N; Mizuno, Y; Miyata, S; Maeda, S; Osawa, T; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxalarginine adducts. J. Biol. Chem 1999, 274, 18492–18502. [Google Scholar]
- Formigli, L; Papucci, L; Tani, A; Schiavone, N; Tempestini, A; Orlandini, GE; Capaccioli, S; Orlandini, SZ. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol 2000, 182, 41–49. [Google Scholar]
- Kass, GE; Eriksson, JE; Weis, M; Orrenius, S; Chow, SC. Chromatin condensation during apoptosis requires ATP. Biochem J 1996, 318, 749–752. [Google Scholar]
- Almeida, RD; Manadas, BJ; Carvalho, AP; Duarte, CB. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar]
- Inanami, O; Yoshito, A; Takahashi, K; Hiraoka, W; Kuwabara, M. Effects of BAPTA-AM and forskolin on apoptosis and cytochrome c release in photosensitized Chinese hamster V79 cells. Photochem. Photobiol 1999, 70, 650–655. [Google Scholar]
- Monteiro, HP; Silva, EF; Stern, A. Nitric oxide: a potential inducer of adhesion-related apoptosis–anoikis. Nitric Oxide 2004, 10, 1–10. [Google Scholar]
- Li, CQ; Wogan, GN. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005, 226, 1–15. [Google Scholar]
- Gomes, ER; Almeida, RD; Carvalho, AP; Duarte, CB. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem. Photobiol 2002, 76, 423–430. [Google Scholar]
- Li, CQ; Robles, AI; Hanigan, CL; Hofseth, LJ; Trudel, LJ; Harris, CC; Wogan, GN. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res 2004, 64, 3022–3029. [Google Scholar]
- Okada, H; Mak, TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar]
- Menon, RP; Pillai, S; Sudhakaran, PR. Binding of collagen causes intracellular mobilization of calcium in human mononuclear cells. Biochem. Mol. Biol. Int 1993, 31, 833–840. [Google Scholar]
- Wu, HJ; Chan, WH. Genistein protects methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Toxicol In Vitro 2006. [Google Scholar]
- Behl, C; Davis, JB; Lesley, R; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar]
- Aoshima, H; Satoh, T; Sakai, N; Yamada, M; Enokido, Y; Ikeuchi, T; Hatanaka, H. Generation of free radicals during lipid hydroperoxide-triggered apoptosis in PC12h cells. Biochim. Biophys. Acta 1997, 1345, 35–42. [Google Scholar]
- Nakatsubo, N; Kojima, H; Kikuchi, K; Nagoshi, H; Hirata, Y; Maeda, D; Imai, Y; Irimura, T; Nagano, T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 1998, 427, 263–266. [Google Scholar]
- Chan, WH; Wu, CC; Yu, JS. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. J. Cell. Biochem 2003, 90, 327–338. [Google Scholar]
- Hsieh, YJ; Wu, CC; Chang, CJ; Yu, JS. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell. Physiol 2003, 194, 363–375. [Google Scholar]






© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hsieh, M.-S.; Chan, W.-H. Impact of Methylglyoxal and High Glucose Co-treatment on Human Mononuclear Cells. Int. J. Mol. Sci. 2009, 10, 1445-1464. https://doi.org/10.3390/ijms10041445
Hsieh M-S, Chan W-H. Impact of Methylglyoxal and High Glucose Co-treatment on Human Mononuclear Cells. International Journal of Molecular Sciences. 2009; 10(4):1445-1464. https://doi.org/10.3390/ijms10041445
Chicago/Turabian StyleHsieh, Ming-Shu, and Wen-Hsiung Chan. 2009. "Impact of Methylglyoxal and High Glucose Co-treatment on Human Mononuclear Cells" International Journal of Molecular Sciences 10, no. 4: 1445-1464. https://doi.org/10.3390/ijms10041445