Cyclin D1 Expression and the Inhibitory Effect of Celecoxib on Ovarian Tumor Growth in Vivo

Abstract
:1. Introduction
2. Results and Discussion
2.1. Inhibition of Ovarian Cancer Growth
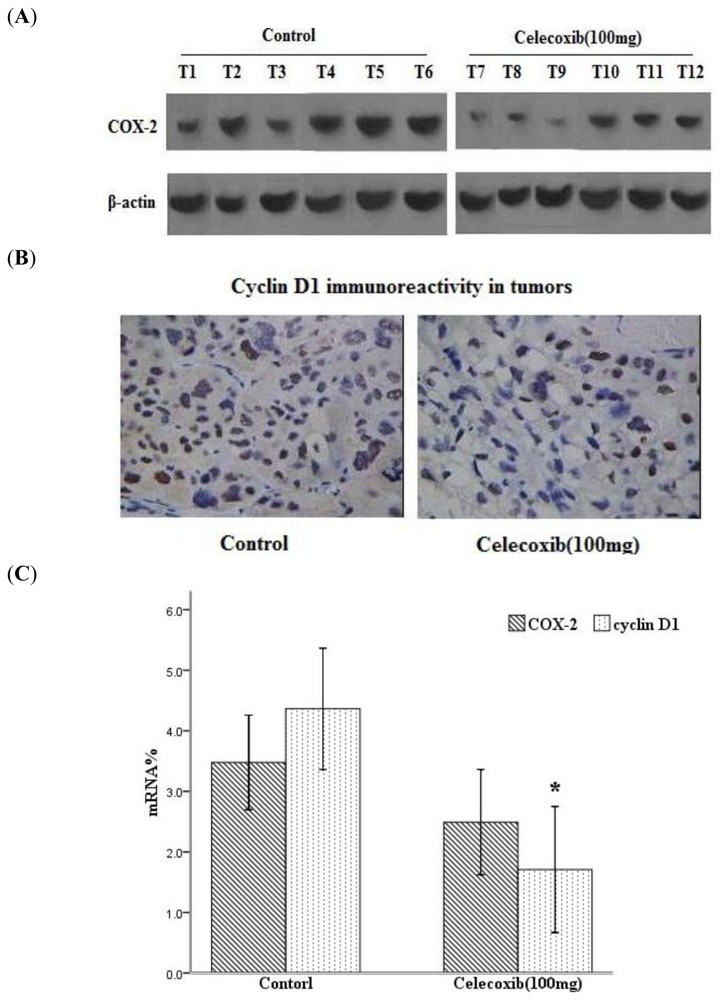
2.2. Celecoxib Decreases COX-2 and Cyclin D1 Expression in Tumors
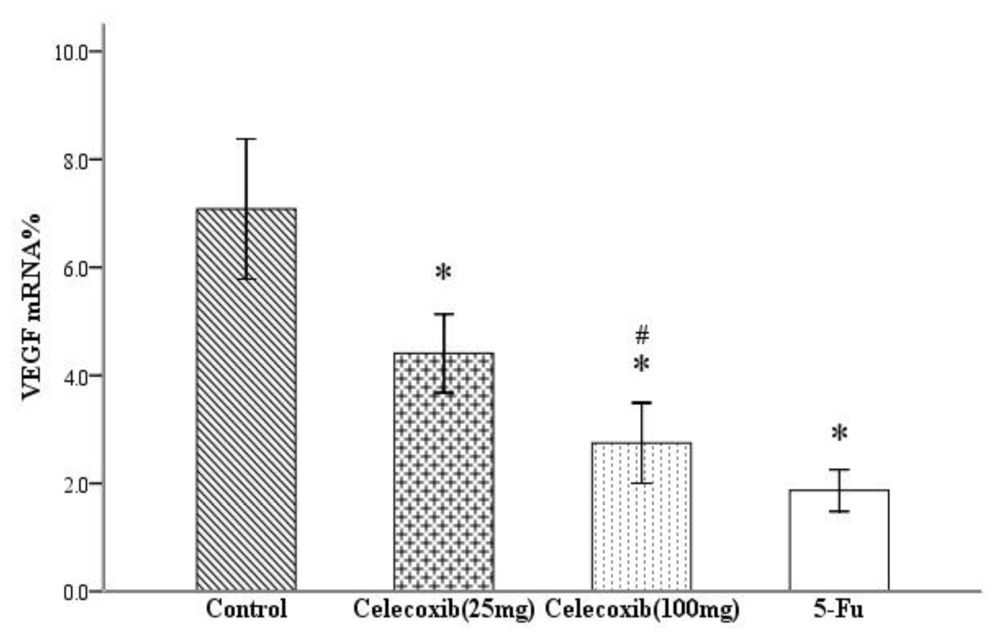
2.3. Effect on VEGF Production
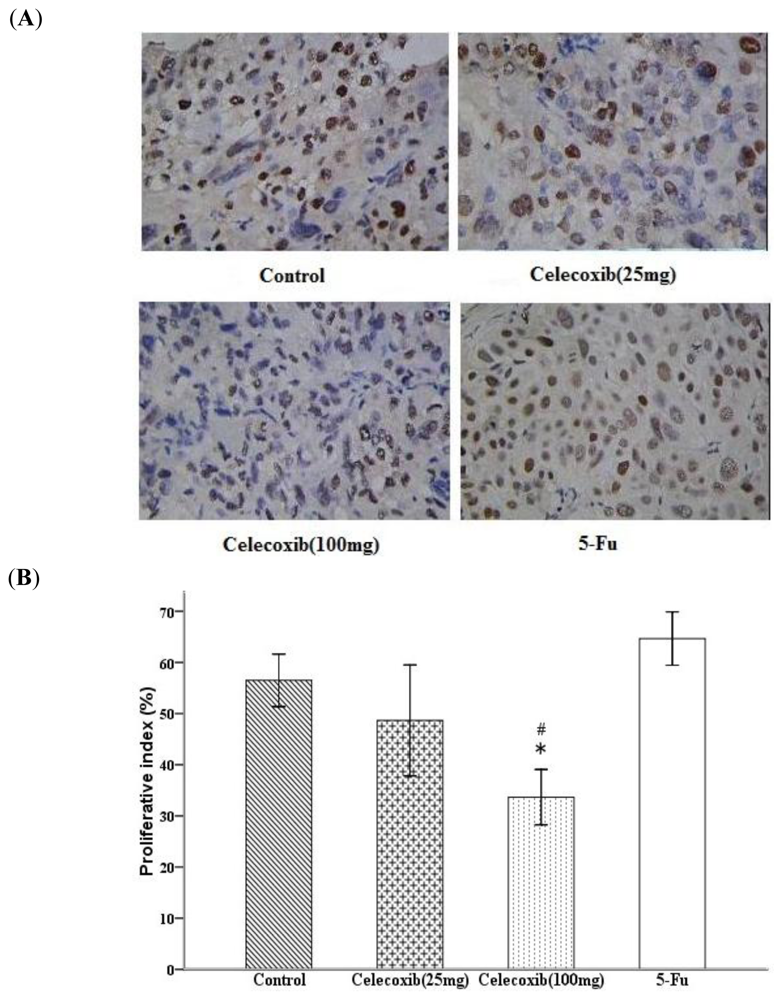
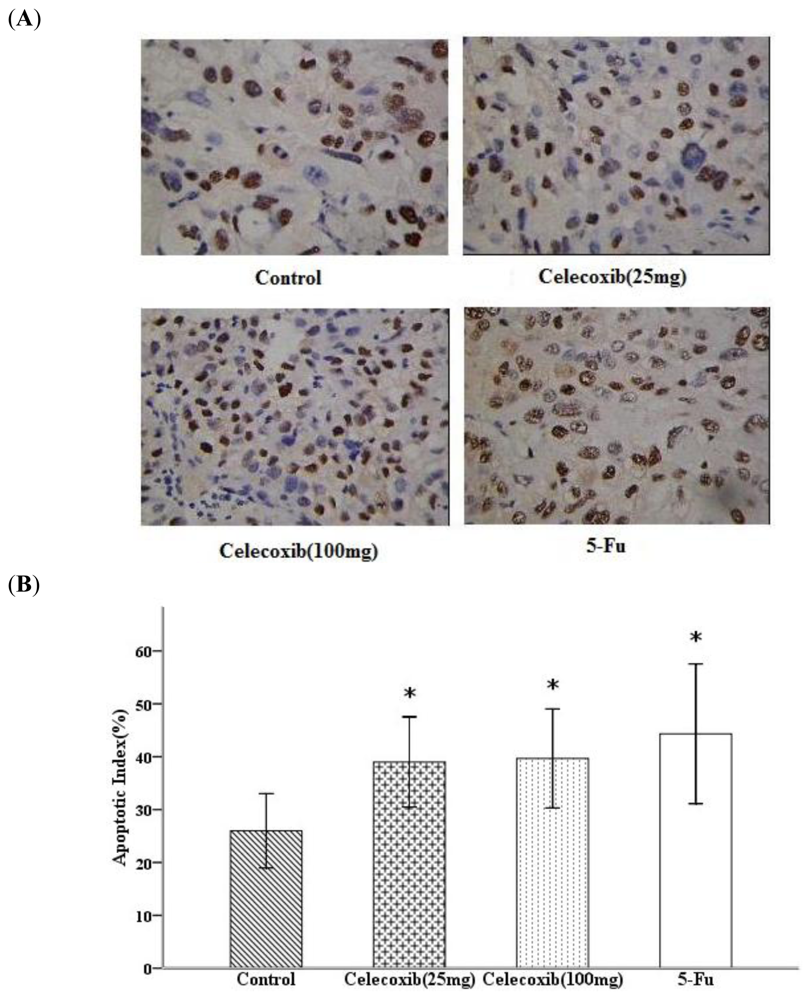
2.4. Celecoxib Inhibits Tumor Cell Proliferation and Induces Apoptosis
2.5. Discussion
3. Experimental Section
3.1. Human Ovarian Tumors in Nude Mice
3.2. Real-Time PCR
3.3. Western Blot
3.4. TUNEL
3.5. Immunohistochemistry
3.6. Statistical Analyses
4. Conclusions
Acknowledgements
References
- Yokoyama, Y; Sakamoto, T; Sato, S; Saito, Y. Evaluation of cytoreductive surgery with pelvic and paraaortic lymphadenectomy and intermittent cisplatin-based combination chemotherapy for improvement of long-term survival in ovarian cancer. Eur. J. Gynaecol. Oncol 1999, 20, 361–366. [Google Scholar]
- Williams, CS; Mann, M; DuBois, RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar]
- Dempke, W; Rie, C; Grothey, A; Schmoll, HJ. Cyclooxygenase-2: A novel target for cancer chemotherapy? J. Cancer Res. Clin. Oncol 2001, 127, 411–417. [Google Scholar]
- Li, S; Miner, K; Fannin, R; Barrett, JC; Davis, BJ. Cyclooxygenase-1 and 2 in normal and malignant human ovarian epithelium. Gynecol. Oncol 2004, 92, 622–627. [Google Scholar]
- Denkert, C; Köbel, M; Pest, S; Koch, I; Berger, S; Schwabe, M; Siegert, A; Reles, A; Klosterhalfen, B; Hauptmann, S. Expression of cyclooxygenase-2 is an independent prognostic factor in human ovarian carcinoma. Am. J. Pathol 2002, 160, 893–903. [Google Scholar]
- Erkinheimo, TL; Lassus, H; Finne, P; van Rees, BP; Leminen, A; Ylikorkala, O; Haglund, C; Butzow, R; Ristimäki, A. Elevated cyclooxygenase-2 expression is associated with altered expression of p53 and SMAD4, amplification of HER-2/neu, and poor outcome in serous ovarian carcinoma. Clin. Cancer Res 2004, 10, 538–545. [Google Scholar]
- Arico, S; Pattingre, S; Bauvy, C; Gane, P; Barbat, A; Codogno, P; Ogier-Denis, E. Celecoxib induces apoptosis by inhibiting 3-phosphoino-sitide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J. Biol. Chem 2002, 277, 27613–27621. [Google Scholar]
- Xin, B; Yokoyama, Y; Shigeto, T; Mizunuma, H. Anti-tumor effect of non-steroidal antiinflammatory drugs on human ovarian cancers. Pathol. Oncol. Res 2007, 13, 365–369. [Google Scholar]
- Xin, B; Yokoyama, Y; Shigeto, T; Futagami, M; Mizunuma, H. Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and Ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer 2007, 110, 791–800. [Google Scholar]
- Uddin, S; Ahmed, M; Hussain, A; Assad, L; Al-Dayel, F; Bavi, P; Al-Kuraya, KS; Munkarah, A. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int. J. Cancer 2010, 126, 386–394. [Google Scholar]
- Ragel, BT; Jensen, RL; Gillespie, DL; Prescott, SM; Couldwell, WT. Celecoxib inhibits meningioma tumor growth in a mouse xenograft model. Cancer 2007, 190, 588–597. [Google Scholar]
- Basu, GD; Pathangey, LB; Tinder, TL; Lagioia, M; Gendler, SJ; Mukherjee, P. Cyclooxygenase-2 inhibitor induces apoptosis in breast cancer cells in an in vivo model of spontaneous metastatic breast cancer. Mol. Cancer Res 2004, 2, 632–642. [Google Scholar]
- Wang, L; Chen, W; Xie, X; He, Y; Bai, X. Celecoxib inhibits tumor growth and angiogenesis in an orthotopic implantation tumor model of human colon cancer. Exp. Oncol 2008, 30, 42–51. [Google Scholar]
- Leahy, KM; Ornberg, RL; Wang, Y; Zweifel, BS; Koki, AT; Masferrer, JL. Cyclooxygenase-2 inhibition by Celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res 2002, 62, 625–631. [Google Scholar]
- Wu, GQ; Xie, D; Yang, GF; Liao, YJ; Mai, SJ; Deng, HX; Sze, J; Guan, XY; Zeng, YX; Lin, MC; Kung, HF. Cell cycle-related kinase supports ovarian carcinoma cell proliferation via regulation of cyclin D1 and is a predictor of outcome in patients with ovarian carcinoma. Int. J. Cancer 2009, 125, 2631–2642. [Google Scholar]
- Barbieri, F; Lorenzi, P; Ragni, N; Schettini, G; Bruzzo, C; Pedullà, F; Alama, A. Overexpression of cyclin D1 is associated with poor survival in epithelial ovarian cancer. Oncology 2004, 66, 310–315. [Google Scholar]
- Ferrara, N; Leung, DW; Cachianes, G; Winer, J; Henzel, WL. Purification and cloning of vascular endothelial growth factor secreted by pituitary folliculostellate cells. Methods Enzymol 1991, 198, 391–405. [Google Scholar]
- Denkert, C; Köbel, M; Pest, S; Koch, I; Berger, S; Schwabe, M; Siegert, A; Reles, A; Klosterhalfen, B; Hauptmann, S. Expression of cyclooxygenase 2 is an independent prognostic factor in human ovarian carcinoma. Am. J. Pathol 2002, 160, 893–903. [Google Scholar]
- Symowicz, J; Adley, BP; Woo, MM; Auersperg, N; Hudson, LG; Stack, MS. Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res 2005, 65, 2234–2242. [Google Scholar]
- Worsley, SD; Ponder, BA; Davies, BR. Overexpression of cyclin D1 in epithelial ovarian cancers. Gynecol. Oncol 1997, 64, 189–195. [Google Scholar]
- Masamha, CP; Benbrook, DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res 2009, 69, 6565–6572. [Google Scholar]
- Vital-Reyes, V; Rodríguez-Burford, C; Chhieng, DC; Oelschlager, DK; Reyes-Fuentes, A; Barnes, M; Grizzle, WE. Celecoxib inhibits cellular growth, decreases Ki-67 expression and modifies apoptosis in ovarian cancer cell lines. Arch. Med. Res 2006, 37, 689–695. [Google Scholar]
- Arnold, A; Papanikolaou, A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol 2005, 23, 4215–4224. [Google Scholar]
- Schiffmann, S; Maier, TJ; Wobst, I; Janssen, A; Corban-Wilhelm, H; Angioni, C; Geisslinger, G; Grösch, S. The anti-proliferative potency of celecoxib is not a class effect of coxibs. Biochem. Pharmacol 2008, 76, 179–187. [Google Scholar]
- Soh, JW; Kazi, JU; Li, H; Thompson, WJ; Weinstein, IB. Celecoxib-induced growth inhibition in SW480 colon cancer cells is associated with activation of protein kinase G. Mol. Carcinog 2008, 47, 519–525. [Google Scholar]
- D’Andrilli, G; Kumar, C; Scambia, G; Giordano, A. Cell cycle genes in ovarian cancer: steps toward earlier diagnosis and novel therapies. Clin. Cancer Res 2004, 10, 8132–8141. [Google Scholar]
- Macaluso, M; Paggi, MG; Giordano, A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene 2003, 22, 6472–6478. [Google Scholar]
- Narayanan, BA; Condon, MS; Bosland, MC; Narayanan, NK; Reddy, BS. Suppression of N-methyl-N-nitrosourea/testosterone-induced rat prostate cancer growth by celecoxib: effects on cyclooxygenase-2, cell cycle regulation, and apoptosis mechanism(s). Clin. Cancer Res 2003, 15, 3503–3513. [Google Scholar]
- Gastman, BR. Apoptosis and its clinical impact. Head Neck 2001, 23, 409–425. [Google Scholar]
- Song, YC; Kim, SH; Juhnn, YS; Song, YS. Apoptotic effect of celecoxib dependent upon p53 status in human ovarian cancer cells. Ann. N.Y. Acad. Sci 2007, 1095, 26–34. [Google Scholar]
- Olson, TA; Mohanraj, D; Carson, LF; Ramakrishnan, S. Vascular permeability factor gene expression in normal and neoplastic ovaries. Cancer Res 1994, 54, 276–280. [Google Scholar]
- Yoneda, J; Kuniyasu, H; Crispens, MA; Price, JE; Bucana, CD; Fidler, IJ. Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice. J. Natl. Cancer Inst 1998, 90, 447–454. [Google Scholar]
- Hartenbach, EM; Olson, TA; Goswitz, JJ; Mohanraj, D; Twiggs, LB; Carson, LF; Ramakrishnan, S. Vascular endothelial growth factor expression and survival in human epithelial ovarian carcinomas. Cancer Lett 1997, 121, 169–175. [Google Scholar]
- Paley, PJ; Staskus, KA; Gebhard, K; Mohanraj, D; Twiggs, LB; Carson, LF; Ramakrishnan, S. Vascular endothelial growth factor expression in early stage ovarian carcinomas. Cancer 1997, 80, 98–106. [Google Scholar]
- Yamamoto, S; Konishi, I; Mandai, M; Kuroda, H; Komatsu, T; Nanbu, K; Sakahara, H; Mori, T. Expression of vascular endothelial growth factor (VEGF) in epithelia ovarian neoplasms: correlation with clinicopathology and patient survival and analysis of serum VEGF levels. Br. J. Cancer 1997, 76, 1221–1227. [Google Scholar]
- Lee, JS; Choi, YD; Lee, JH; Nam, JH; Choi, C; Lee, MC; Park, CS; Juhng, SW; Min, KW. Expression of cyclooxygenase-2 in epithelial ovarian tumors and its relation to vascular endothelial growth factor and p53 expression. Int. J. Gynecol. Cancer 2006, 16, 247–253. [Google Scholar]
- Fujimoto, J; Toyoki, H; Sakaguchi, H; Jahan, I; Alam, SM; Tamaya, T. Clinical implications of expression of cyclooxygenase-2 related to angiogenesis in ovarian cancer. Oncol. Rep 2006, 15, 21–25. [Google Scholar]
- Gavrieli, Y; Sherman, Y; Ben-Sasson, SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol 1992, 119, 493–501. [Google Scholar]
- Del-Vecchio, MT; Leoncinel, L; Buerdi, K; Kraft, R; Megha, T; Barbini, P; Tosi, P; Cottier, H. Diffuse controcytic and/or centroblastic malignant non-Hodgkins lymphomas: comparison of mitotic and pyknotic (apoptotic) indices. Int. J. Cancer 1991, 47, 38–43. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Celecoxib (25 mg) | Celecoxib (100 mg) | 5-FU | |
|---|---|---|---|---|
| VEGF 121 | 3.55 ± 0.17 | 3.72 ± 0.16 | 4.91 ± 0.22 | 5.47 ± 0.19 |
| VEGF 165 | 3.28 ± 0.21 | 5.00 ± 0.19 | 5.13 ± 0.19 | 6.12 ± 0.24 |
| VEGF 189 | 4.72 ± 0.17 | 5.58 ± 0.12 | 5.70 ± 0.34 | 6.02 ± 0.29 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, W.; Jiang, H.-R.; Xu, X.-L.; Wang, J.; Zhang, J.; Liu, M.-L.; Zhai, L.-Y. Cyclin D1 Expression and the Inhibitory Effect of Celecoxib on Ovarian Tumor Growth in Vivo. Int. J. Mol. Sci. 2010, 11, 3999-4013. https://doi.org/10.3390/ijms11103999
Li W, Jiang H-R, Xu X-L, Wang J, Zhang J, Liu M-L, Zhai L-Y. Cyclin D1 Expression and the Inhibitory Effect of Celecoxib on Ovarian Tumor Growth in Vivo. International Journal of Molecular Sciences. 2010; 11(10):3999-4013. https://doi.org/10.3390/ijms11103999
Chicago/Turabian StyleLi, Wei, Hong-Ru Jiang, Xiao-Li Xu, Jie Wang, Jun Zhang, Mei-Lin Liu, and Ling-Yun Zhai. 2010. "Cyclin D1 Expression and the Inhibitory Effect of Celecoxib on Ovarian Tumor Growth in Vivo" International Journal of Molecular Sciences 11, no. 10: 3999-4013. https://doi.org/10.3390/ijms11103999