Combined 3D-QSAR and Docking Modelling Study on Indolocarbazole Series Compounds as Tie-2 Inhibitors

Abstract
:1. Introduction
2. Computational Methods
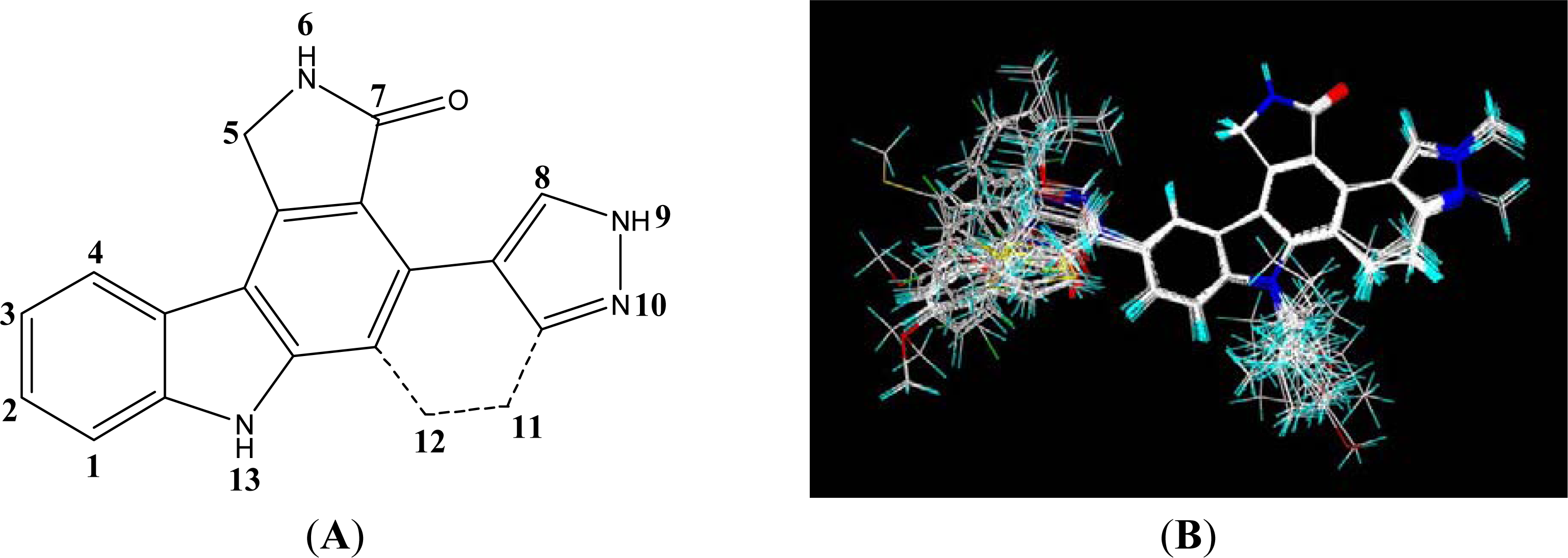
2.1. Data Set and Molecular Sketching
2.2. CoMFA and CoMSIA Study
2.3. Predictive Correlation Co-efficient (r2pred)
2.4. Molecular Docking
3. Results and Discussion
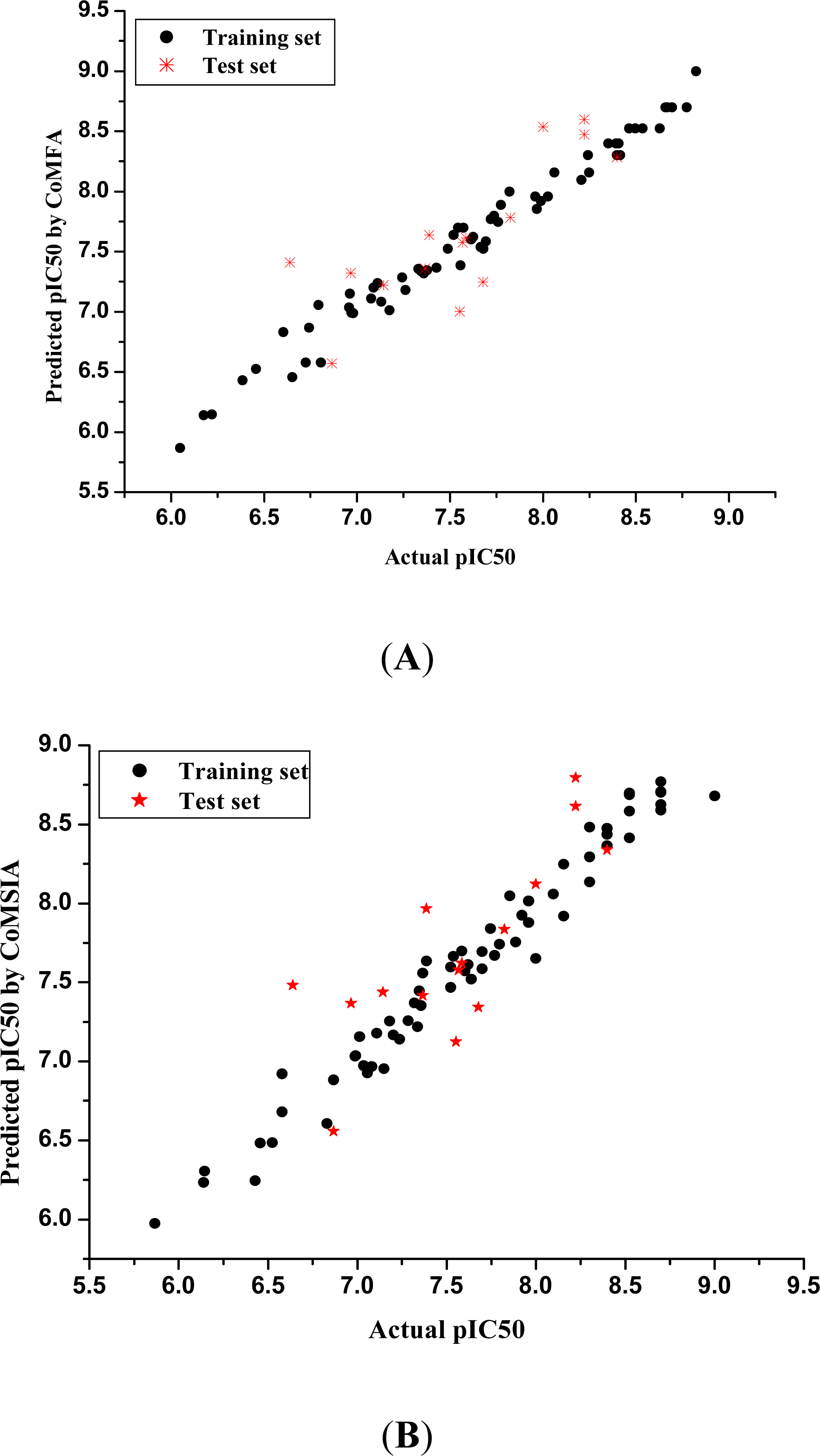
3.1. 3D-QSAR Models
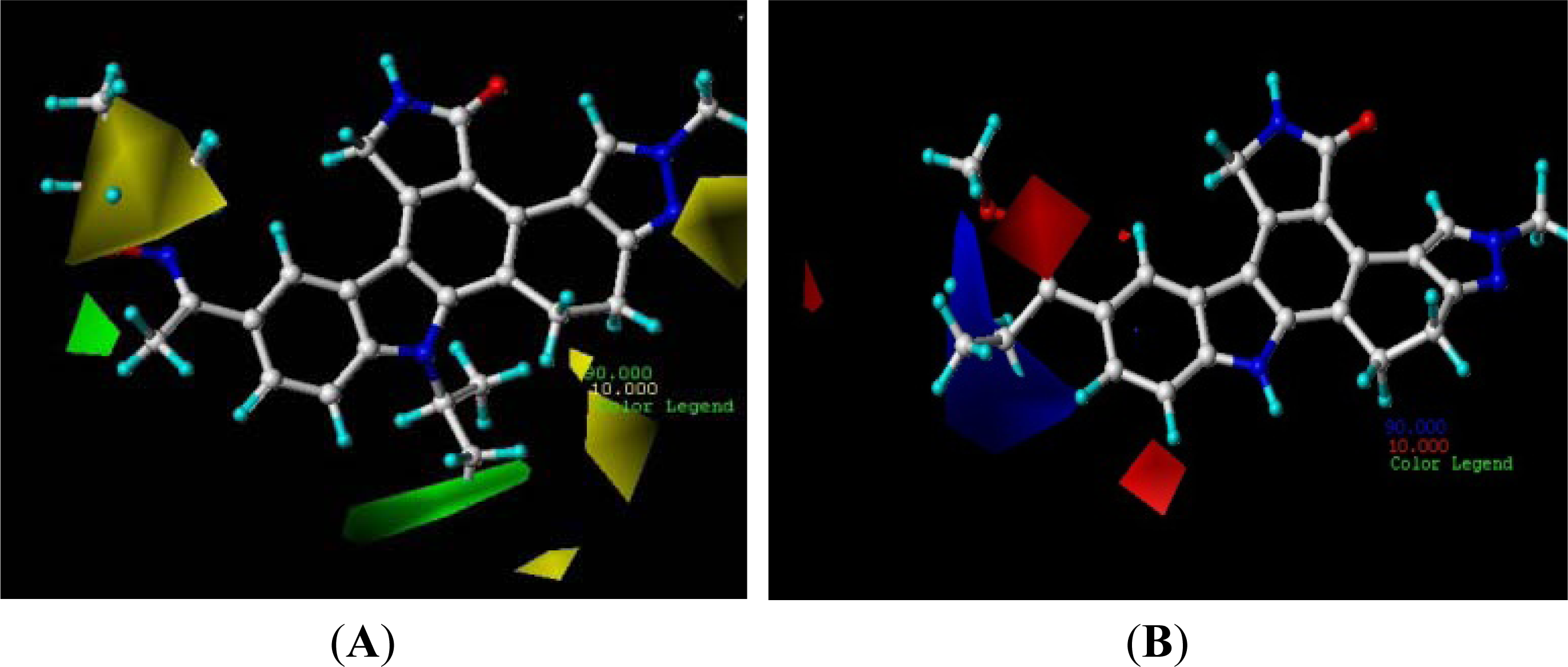
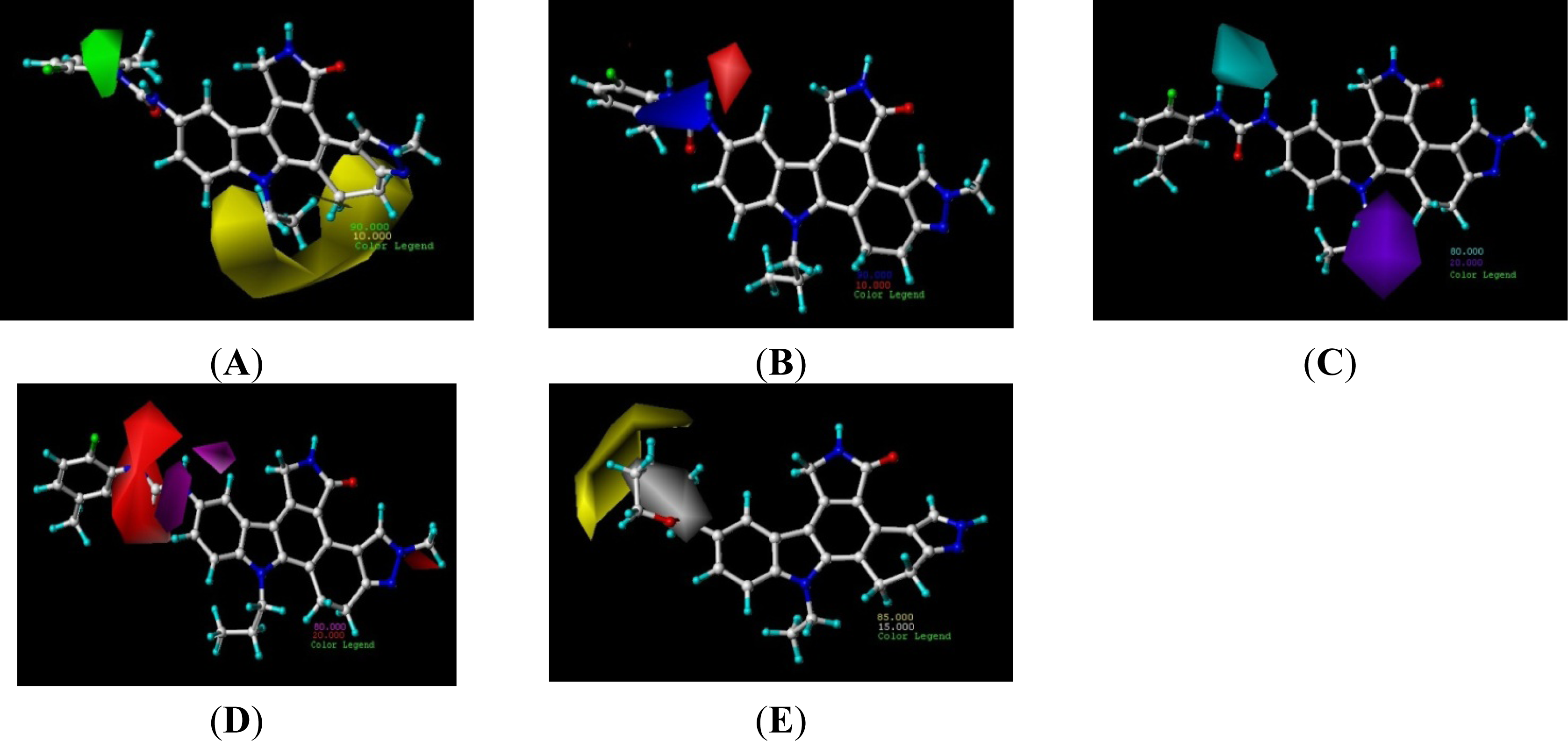
3.2. Contour Analysis
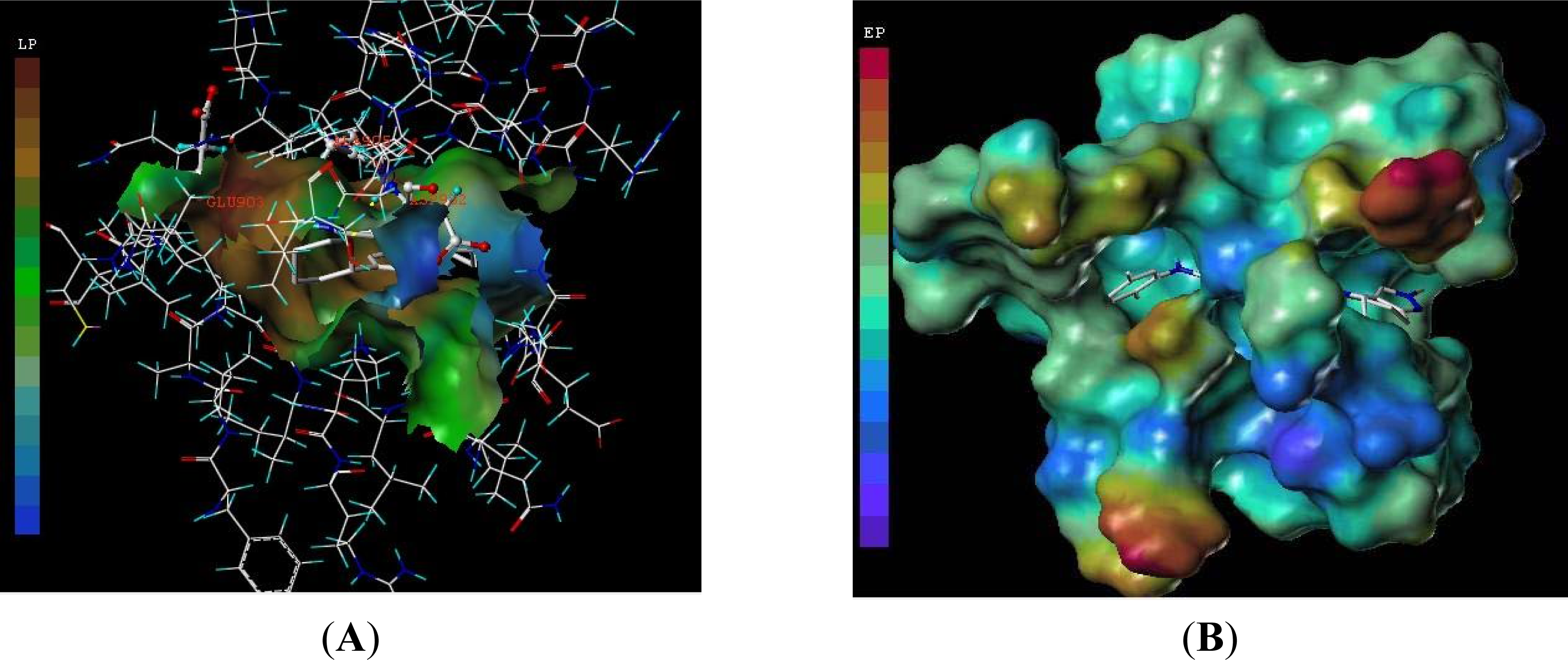
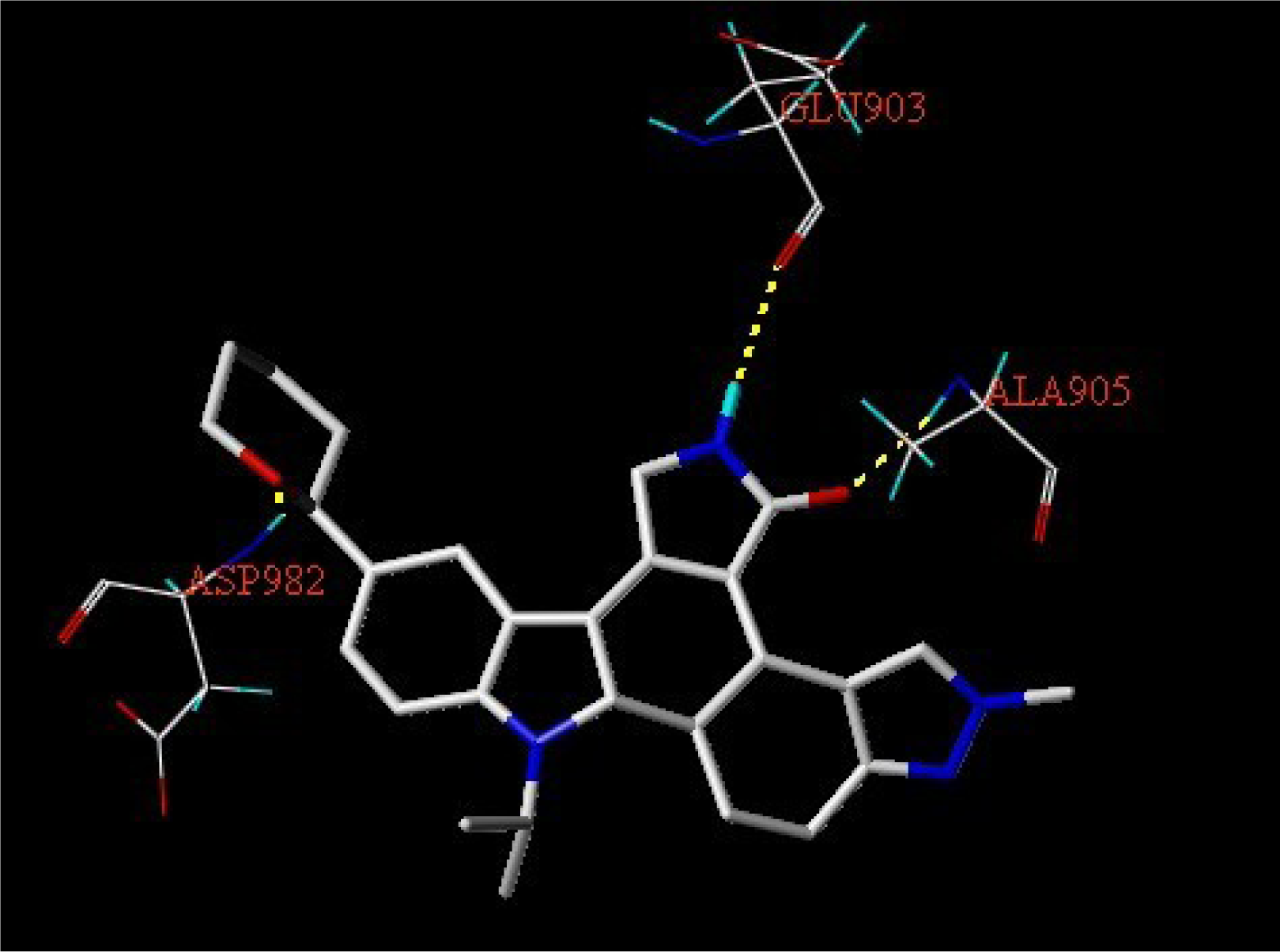
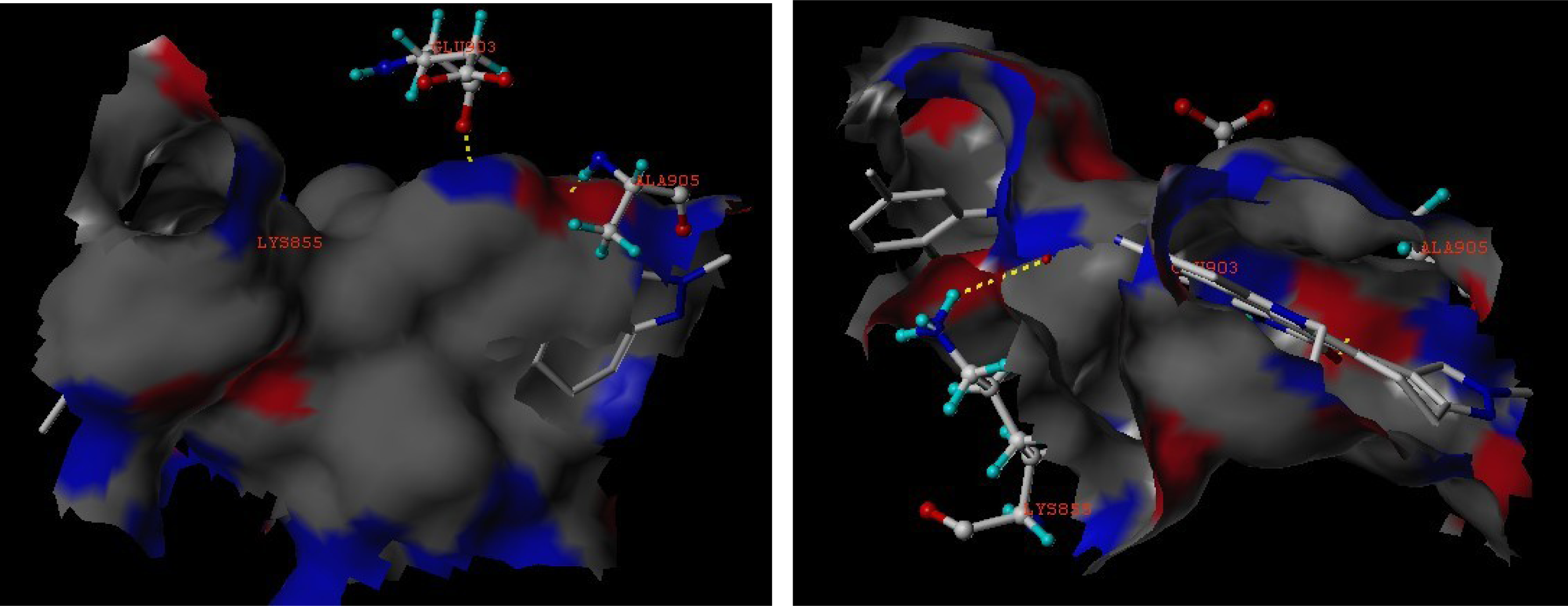
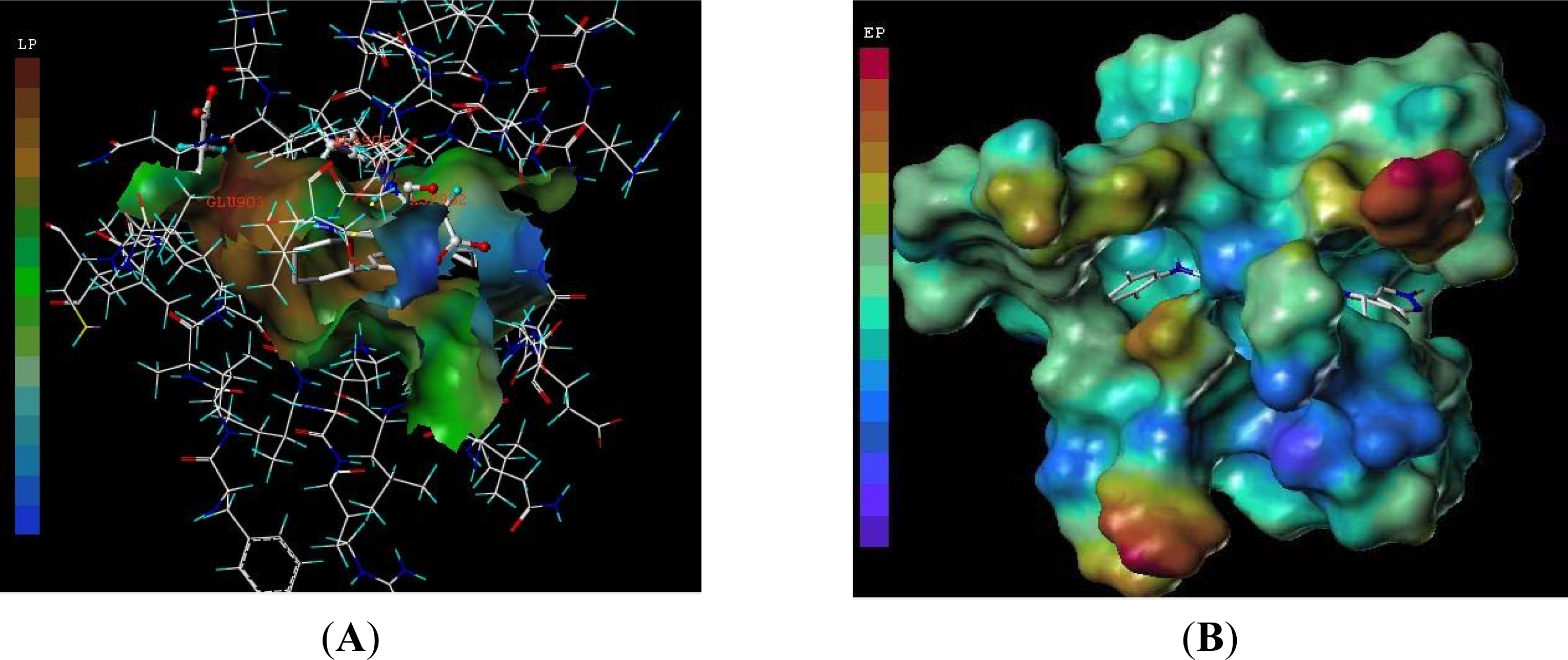
3.3. Docking Analysis
3.4. Design for New Inhibitors
4. Conclusion
Acknowledgments
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med 2003, 9, 653–660. [Google Scholar]
- Saharinen, P; Bry, M; Alitalo, K. How do angiopoietins Tie in with vascular endothelial growth factors? Curr. Opin. Hematol 2010, 17, 198–205. [Google Scholar]
- Zhang, J-M; Yang, PL; Gray, NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar]
- Ferrara, N; Kerbel, RS. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar]
- Nam, N-H; Parang, K. Current targets for anticancer drug discovery. Curr. Drug Targets 2003, 4, 159–179. [Google Scholar]
- Becknell, NC; Zulli, AL; Angeles, TS; Yang, S; Albom, MS; Aimone, LD; Robinson, C; Chang, H; Hudkins, RL. Novel C-3 N-urea, amide, and carbamate dihydroindazolo[5,4-a] pyrrolo[3,4-c]carbazole analogs as potent TIE-2 and VEGF-R2 dual inhibitors. Bioorg. Med. Chem. Lett 2006, 16, 5368–5372. [Google Scholar]
- Dandu, R; Zulli, AL; Bacon, ER; Underiner, T; Robinson, C; Chang, H; Miknyoczki, S; Grobelny, J; Ruggeri, BA; Yang, S; et al. Design and synthesis of dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole oximes as potent dual inhibitors of TIE-2 and VEGF-R2 receptor tyrosine kinases. Bioorg. Med. Chem. Lett 2008, 18, 1916–1921. [Google Scholar]
- Underiner, TL; Ruggeri, B; Aimone, L; Albom, M; Angeles, T; Chang, H; Hudkins, RL; Hunter, K; Josef, K; Robinson, C; et al. TIE-2/VEGF-R2 SAR and in vitro activity of C3-acyl dihydroindazolo[5,4-a] pyrrolo[3,4-c]carbazole analogs. Bioorg. Med. Chem. Lett 2008, 18, 2368–2372. [Google Scholar]
- Hudkins, RL; Zulli, AL; Underiner, TL; Angeles, TS; Aimone, LD; Meyer, SL; Pauletti, D; Chang, H; Fedorov, EV; Almo, SC; et al. 8-THP-DHI analogs as potent Type I dual TIE-2/VEGF-R2 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 3356–3360. [Google Scholar]
- Cramer, RD, III; Patterson, DE; Bunce, JD. Comparative molecular field analysis (CoMFA). Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Cramer, RD, III; Bunce, JD; Patterson, DE. Crossvalidation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant. Struct.-Act. Relat 1988, 7, 18–25. [Google Scholar]
- Lan, P; Chen, W-N; Chen, W-M. Molecular modeling studies on imidazo[4,5-b]pyridine derivatives as Aurora A Kinase inhibitors using 3D-QSAR and docking approaches. Eur. J. Med. Chem 2011, 46, 77–94. [Google Scholar]
- Sun, J; Cai, S; Yan, N; Mei, H. Docking and 3D-QSARstudies of influenza neuraminidase inhibitors using threedimensional holographic vector of atomic interaction field analysis. Eur. J. Med. Chem 2010, 45, 1008–1014. [Google Scholar]
- AbdulHameed, MD; Hamza, A; Liu, J-J; Zhan, C-G. Combined 3D-QSAR modeling and molecular docking study on indolinone derivatives as inhibitors of 3-Phosphoinositide-Dependent protein Kinase-1. J. Chem. Inf. Model 2008, 48, 1760–1772. [Google Scholar]
- Cao, H-Y; Zhang, H-B; Zheng, X-F; Gao, D-B. 3D QSAR studies on a series of potent and high selective inhibitors for three kinases of RTK family. J Mol Graph Model 2007, 236–245. [Google Scholar]
- Zhang, N; Zhong, R. Docking and 3D-QSAR studies of 7-hydroxycoumarin derivatives as CK2 inhibitors. Eur. J. Med. Chem 2010, 45, 292–297. [Google Scholar]
- Liao, S-Y; Qian, L; Mao, T-F; Lu, H-L; Zheng, K-C. CoMFA and docking studies of 2-phenylindole derivatives with anticancer activity. Eur. J. Med. Chem 2009, 44, 2822–2827. [Google Scholar]
- Ai, Y; Wang, S-T; Sun, P-H; Song, F-J. Molecular modeling studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline derivatives as potent CDK2/Cyclin a inhibitors using 3D-QSAR and docking. Int. J. Mol. Sci 2010, 11, 3705–3724. [Google Scholar]
- Zhang, B-D; Li, Y; Zhang, H-X; Ai, C-Z. 3D-QSAR and Molecular docking studies on derivatives of MK-0457, GSK1070916 and SNS-314 as inhibitors against Aurora B Kinase. Int. J. Mol. Sci 2010, 11, 4326–4347. [Google Scholar]
- Cao, R; Mi, N; Zhang, H-B. 3D-QSAR study of c-Src kinase inhibitor based on docking. J. Mol. Model 2010, 16, 361–375. [Google Scholar]
- Zeng, H-H; Zhang, H-B. Combined 3D-QSAR modeling and molecular docking study on 1,4-dihydroindenol[1,2-c] pyrazoles as VEGFR2 kinase inhibitors. J. Mol. Graph. Model 2010, 29, 54–71. [Google Scholar]
- Ai, Y; Wang, S-T; Sun, P-H; Song, F-J. Combined 3D-QSAR modeling and molecular docking studies on Pyrrole-Indolin-2-ones as Aurora A Kinase inhibitors. Int. J. Mol. Sci 2011, 12, 1605–1624. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
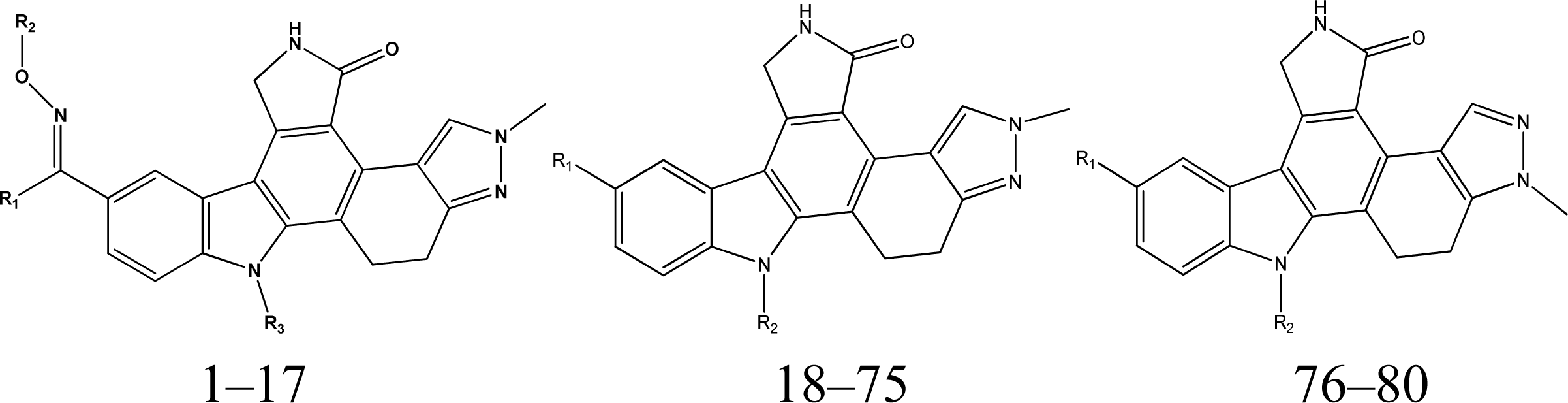 | ||||
|---|---|---|---|---|
| NO | R1 | R2 | R3 | pIC50(nM) |
| 1 | H | Me | H | 6.83 |
| 2 | H | Et | H | 6.58 |
| 3 | Me | Me | H | 7.15 |
| 4 | i-Pr | Me | H | 6.58 |
| 5 | i-Pr | Et | H | 7.04 |
| 6 | Me | i-Pr | H | 7.55 |
| 7 | Me | Me | Pr | 7.52 |
| 8 | Me | Me | i-Pr | 7.01 |
| 9 | Me | Me | i-Bu | 7.52 |
| 10 | Me | Me | n-Bu | 6.97 |
| 11 | Me | Et | i-Bu | 7.60 |
| 12 | Me | i-Pr | i-Bu | 7.28 |
| 13 | Me | i-Bu | i-Bu | 7.64 |
| 14 | Me | Et | i-Pr | 7.58 |
| 15 | Me | i-Pr | i-Pr | 7.18 |
| 16 | Me | i-Bu | i-Pr | 7.34 |
| 17 | Me | i-Bu | Et | 7.35 |
| NO | R1 | R2 | pIC50(nM) |
|---|---|---|---|
| 18 | NH2 | CH2CH(CH3)2 | 7.32 |
| 19 | NH2 | CH2CH2CH2CH3 | 6.64 |
| 20 | 4-OMe–phenyl NHCONH | CH2CH3 | 8.40 |
| 21 | 4-OMe–phenyl NHCONH | CH2CH2CH3 | 8.70 |
| 22 | 4-OMe–phenyl NHCONH | CH2CH(CH3)2 | 8.52 |
| 23 | 4-OMe–phenylNHCONH | CH2CH2CH2CH3 | 8.15 |
| 24 | 4-SMe–phenylNHCONH | CH2CH2CH3 | 8.52 |
| 25 | 4-NMe2–phenylNHCONH | CH2CH2CH3 | 8.22 |
| 26 | 4-Me–phenylNHCONH | CH2CH2CH3 | 8.52 |
| 27 | 2-F-5-Me-phenylNHCONH | CH2CH2CH3 | 9.00 |
| 28 | 2-F-5-CF3-phenylNHCONH | CH2CH2CH3 | 8.40 |
| 29 | PhenylNHCONH | CH2CH2CH3 | 8.30 |
| 30 | Phenyl(Me)NCONH | CH2CH2CH3 | 8.70 |
| 31 | 3-OMe–phenylNHCONH | CH2CH(CH3)2 | 8.40 |
| 32 | 2-OMe–phenylNHCONH | CH2CH(CH3)2 | 8.10 |
| 33 | 4-F-phenylNHCONH | CH2CH2CH3 | 8.70 |
| 34 | 3-F-phenylNHCONH | CH2CH2CH3 | 8.70 |
| 35 | 2-F-phenylNHCONH | CH2CH2CH3 | 8.70 |
| 36 | 4-Cl-phenylNHCONH | CH2CH2CH3 | 8.22 |
| 37 | 2-Cl-phenylNHCONH | CH2CH2CH3 | 8.52 |
| 38 | 2-Br-phenylNHCONH | CH2CH2CH3 | 8.40 |
| 39 | 2-ThienylCONH | CH3 | 7.92 |
| 40 | 2-ThienylCONH | CH2CH(CH3)2 | 8.00 |
| 41 | 2-FuranylCONH | CH2CH2CH3 | 7.54 |
| 42 | 2-FuranylCONH | CH2CH(CH3)2 | 7.70 |
| 43 | 4-OMe–phenylOCONH | CH2CH2CH3 | 8.30 |
| 44 | 4-F-phenylOCONH | CH2CH2CH3 | 7.96 |
| 45 | i-PrOCONH | CH2CH2CH3 | 7.96 |
| 46 | EtOCONH | CH2CH2CH3 | 7.74 |
| 47 | PrOCONH | CH2CH2CH3 | 8.15 |
| 48 | H | H | 5.87 |
| 49 | H | nPr | 6.87 |
| 50 | H | i-Bu | 6.43 |
| 51 | Ac | i-Pr | 6.14 |
| 52 | Ac | i-Bu | 7.08 |
| 53 | 2-Thiophene-CO | H | 6.87 |
| 54 | 2-Thiophene-CO | Et | 7.37 |
| 55 | 2-Thiophene-CO | Pr | 7.59 |
| 56 | 2-Thiophene-CO | i-Pr | 7.62 |
| 57 | 2-Thiophene-CO | i-Bu | 7.70 |
| 58 | 3-Thiophene-CO | i-Bu | 7.20 |
| 59 | 2-Furan-CO | i-Bu | 7.14 |
| 60 | 3-Cl-Thiophene-2-CO | CH2CH(CH3)2 | 7.89 |
| 61 | 3-Br-Thiophene-2-CO | CH2CH(CH3)2 | 7.82 |
| 62 | 3-Me-Thiophene-2-CO | CH2CH(CH3)2 | 8.30 |
| 63 | 4-Me-Thiophene-2-CO | CH2CH(CH3)2 | 7.37 |
| 64 | 3-Furan-CO | i-Bu | 7.24 |
| 65 | H | 7.36 | |
| 66 | c-Pentyl | 7.39 | |
| 67 | c-Hexyl | 6.99 | |
| 68 |  | CH2CH2OEt | 7.77 |
| 69 | (CH2)2OH | 7.57 | |
| 70 | (CH2)3OH | 7.80 | |
| 71 | Me | 7.39 | |
| 72 | Et | 7.85 | |
| 73 | i-Pr | 8.00 | |
| 74 | i-Pent | 7.68 | |
| 75 | n-Pent | 7.11 | |
| 76 | H | 6.15 | |
| 77 | Et | 7.06 | |
| 78 | i-Pr | 6.99 | |
| 79 | c-Pentyl | 6.52 | |
| 80 | (CH2)2OH | 6.46 |
| PLS Statistics | CoMFA | CoMSIA |
|---|---|---|
| q2a | 0.823 | 0.804 |
| r2b | 0.979 | 0.967 |
| ONC c | 8 | 8 |
| SEE d | 0.114 | 0.141 |
| F valuee | 319.429 | 207.935 |
| r2pred f | 0.948 | 0.935 |
| Field Contribution(%) | ||
| Steric | 0.527 | 0.121 |
| Electrostatic | 0.473 | 0.236 |
| Hydrophobic | – | 0.160 |
| H-bond Donor | – | 0.225 |
| H-bond Acceptor | – | 0.259 |
| Compd. No. | Experimental | CoMFA | CoMSIA | ||
| Pred. | Res. | Pred. | Res. | ||
| 1 | 6.830 | 6.604 | 0.226 | 6.606 | 0.224 |
| 2 | 6.578 | 6.726 | −0.148 | 6.679 | −0.101 |
| 3 | 7.149 | 6.961 | 0.188 | 6.953 | 0.196 |
| 4 | 6.578 | 6.807 | −0.229 | 6.921 | −0.343 |
| 5 | 7.036 | 6.958 | 0.078 | 6.973 | 0.063 |
| 6 * | 7.553 | 7.001 | 0.552 | 7.122 | 0.431 |
| 7 | 7.523 | 7.490 | 0.033 | 7.467 | 0.056 |
| 8 | 7.013 | 7.175 | −0.162 | 7.157 | −0.144 |
| 9 | 7.523 | 7.679 | −0.156 | 7.597 | −0.074 |
| 10 * | 6.967 | 7.321 | −0.354 | 7.366 | −0.399 |
| 11 | 7.602 | 7.615 | −0.013 | 7.571 | 0.031 |
| 12 | 7.284 | 7.243 | 0.041 | 7.258 | 0.026 |
| 13 | 7.638 | 7.520 | 0.118 | 7.519 | 0.109 |
| 14 * | 7.585 | 7.615 | −0.030 | 7.620 | −0.035 |
| 15 | 7.180 | 7.261 | −0.081 | 7.254 | −0.074 |
| 16 | 7.337 | 7.345 | −0.008 | 7.220 | 0.117 |
| 17 | 7.347 | 7.376 | −0.029 | 7.447 | −0.100 |
| 18 | 7.319 | 7.359 | −0.040 | 7.370 | −0.051 |
| 19 * | 6.638 | 7.410 | −0.771 | 7.481 | −0.842 |
| 20 | 8.398 | 8.394 | 0.004 | 8.366 | 0.032 |
| 21 | 8.699 | 8.774 | −0.075 | 8.589 | 0.110 |
| 22 | 8.523 | 8.496 | 0.027 | 8.415 | 0.108 |
| 23 | 8.155 | 8.249 | −0.094 | 8.249 | −0.094 |
| 24 | 8.523 | 8.537 | −0.014 | 8.583 | −0.060 |
| 25 * | 8.223 | 8.472 | −0.251 | 8.613 | −0.390 |
| 26 | 8.523 | 8.628 | −0.105 | 8.698 | −0.175 |
| 27 | 9.000 | 8.823 | 0.177 | 8.680 | 0.320 |
| 28 | 8.398 | 8.351 | 0.047 | 8.475 | −0.077 |
| 29 | 8.301 | 8.416 | −0.115 | 8.484 | −0.183 |
| 30 | 8.699 | 8.662 | 0.037 | 8.626 | 0.073 |
| 31 * | 8.398 | 8.286 | 0.112 | 8.338 | 0.060 |
| 32 | 8.097 | 8.208 | −0.111 | 8.059 | −0.038 |
| 33 | 8.699 | 8.659 | 0.040 | 8.701 | −0.002 |
| 34 | 8.699 | 8.694 | 0.005 | 8.707 | −0.008 |
| 35 | 8.699 | 8.668 | 0.031 | 8.771 | −0.072 |
| 36 * | 8.223 | 8.596 | −0.374 | 8.794 | −0.572 |
| 37 | 8.523 | 8.465 | 0.058 | 8.689 | −0.166 |
| Compd. No. | Experimental | CoMFA | CoMSIA | Compd. No. | Experimental |
| Pred. | Res. | ||||
| 38 | 8.398 | 8.406 | −0.008 | 8.436 | −0.038 |
| 39 | 7.921 | 7.989 | −0.068 | 7.925 | −0.004 |
| 40 * | 8.000 | 8.537 | −0.537 | 8.121 | −0.121 |
| 41 | 7.538 | 7.665 | −0.127 | 7.664 | −0.126 |
| 42 | 7.699 | 7.545 | 0.154 | 7.587 | 0.112 |
| 43 | 8.301 | 8.243 | 0.058 | 8.136 | 0.165 |
| 44 | 7.959 | 8.027 | −0.068 | 8.014 | −0.005 |
| 45 | 7.959 | 7.960 | −0.001 | 7.878 | 0.081 |
| 46 | 7.745 | 7.758 | −0.014 | 7.839 | −0.094 |
| 47 | 8.155 | 8.063 | 0.092 | 7.919 | 0.236 |
| 48 | 5.867 | 6.049 | −0.182 | 5.975 | −0.108 |
| 49 * | 6.867 | 6.571 | 0.296 | 6.557 | 0.310 |
| 50 | 6.429 | 6.384 | 0.045 | 6.244 | 0.185 |
| 51 | 6.139 | 6.177 | −0.038 | 6.233 | –0.094 |
| 52 | 7.081 | 7.132 | −0.051 | 6.968 | 0.113 |
| 53 | 6.866 | 6.742 | 0.124 | 6.882 | −0.016 |
| 54 * | 7.366 | 7.360 | 0.006 | 7.416 | −0.050 |
| 55 | 7.585 | 7.694 | −0.109 | 7.669 | −0.114 |
| 56 | 7.620 | 7.625 | −0.005 | 7.614 | 0.006 |
| 57 | 7.699 | 7.572 | 0.127 | 7.694 | 0.005 |
| 58 | 7.201 | 7.089 | 0.112 | 7.168 | 0.032 |
| 59 * | 7.143 | 7.219 | −0.076 | 7.437 | −0.294 |
| 60 | 7.886 | 7.775 | 0.111 | 7.755 | 0.131 |
| 61 * | 7.824 | 7.782 | 0.042 | 7.834 | −0.010 |
| 62 | 8.301 | 8.397 | −0.096 | 8.293 | 0.008 |
| 63 | 7.366 | 7.428 | −0.062 | 7.559 | −0.193 |
| 64 | 7.237 | 7.111 | 0.126 | 7.139 | 0.098 |
| 65 | 7.356 | 7.330 | 0.026 | 7.354 | 0.002 |
| 66 | 7.387 | 7.557 | −0.170 | 7.634 | −0.267 |
| 67 | 6.991 | 6.971 | 0.002 | 7.037 | −0.044 |
| 68 | 7.770 | 7.719 | 0.051 | 7.671 | 0.099 |
| 69 * | 7.569 | 7.574 | −0.005 | 7.577 | −0.009 |
| 70 | 7.796 | 7.738 | 0.058 | 7.741 | 0.055 |
| 71 * | 7.387 | 7.639 | −0.252 | 7.966 | −0.579 |
| 72 | 7.854 | 7.968 | −0.114 | 8.049 | −0.195 |
| 73 | 8.000 | 7.821 | 0.179 | 7.650 | 0.350 |
| 74 * | 7.678 | 7.247 | 0.431 | 7.343 | 0.335 |
| 75 | 7.108 | 7.077 | 0.031 | 7.179 | −0.071 |
| 76 | 6.146 | 6.219 | −0.073 | 6.305 | −0.159 |
| 77 | 7.056 | 6.794 | 0.262 | 6.926 | 0.130 |
| 78 | 6.987 | 6.979 | 0.008 | 7.032 | −0.045 |
| 79 | 6.522 | 6.456 | 0.066 | 6.485 | 0.037 |
| 80 | 6.456 | 6.652 | −0.196 | 6.484 | 0.028 |
 | ||||||
|---|---|---|---|---|---|---|
| No. | Substituent | Predicted pIC _50(nM) | Total-Score | |||
| R | R1 | R2 | CoMFA | CoMSIA | ||
| D1 | 2-F-5-Me-phenyl | CH3 | H | 9.024 | 8.380 | 13.35 |
| D2 | 2-F-5-Me-phenyl | CH2CH3 | H | 8.966 | 8.508 | 14.02 |
| D3 | 5-Me-phenyl | CH(CH3)2 | H | 8.906 | 8.519 | 11.88 |
| D4 | 2-F-5-Me-phenyl | CH(CH3)2 | H | 8.951 | 8.515 | 11.18 |
| D5 | 2-F-5-Me-phenyl | H | OH | 8.953 | 8.896 | 11.54 |
| D6 | 2-F-5-Me-phenyl | OH | OH | 8.971 | 8.588 | 11.43 |
| D7 | 2-F-5-Me-phenyl | OCH3 | OH | 8.947 | 8.572 | 11.31 |
| D8 | 2-F-5-Me-phenyl | OH | CH3 | 8.786 | 8.250 | 12.36 |
| D9 | 3-F-6-Me-2-pyridyl | H | H | 8.769 | 8.655 | 9.69 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, Y.; Xu, J.; Li, Z.; Zhu, Z.; Zhang, J.; Wu, S. Combined 3D-QSAR and Docking Modelling Study on Indolocarbazole Series Compounds as Tie-2 Inhibitors. Int. J. Mol. Sci. 2011, 12, 5080-5097. https://doi.org/10.3390/ijms12085080
Tian Y, Xu J, Li Z, Zhu Z, Zhang J, Wu S. Combined 3D-QSAR and Docking Modelling Study on Indolocarbazole Series Compounds as Tie-2 Inhibitors. International Journal of Molecular Sciences. 2011; 12(8):5080-5097. https://doi.org/10.3390/ijms12085080
Chicago/Turabian StyleTian, Yuanxin, Jian Xu, Zhonghuang Li, Zhengguang Zhu, Jiajie Zhang, and Shuguang Wu. 2011. "Combined 3D-QSAR and Docking Modelling Study on Indolocarbazole Series Compounds as Tie-2 Inhibitors" International Journal of Molecular Sciences 12, no. 8: 5080-5097. https://doi.org/10.3390/ijms12085080