A Tumor Surveillance Model: A Non-Coding RNA Senses Neoplastic Cells and Its Protein Partner Signals Cell Death

{kind=link}
Abstract
:1. Human Body Complexity and Cancer Incident Rates
2. Classical Tumor Surveillance Mechanisms
3. Facts about nc886 and Protein Kinase R (PKR) in Cancer
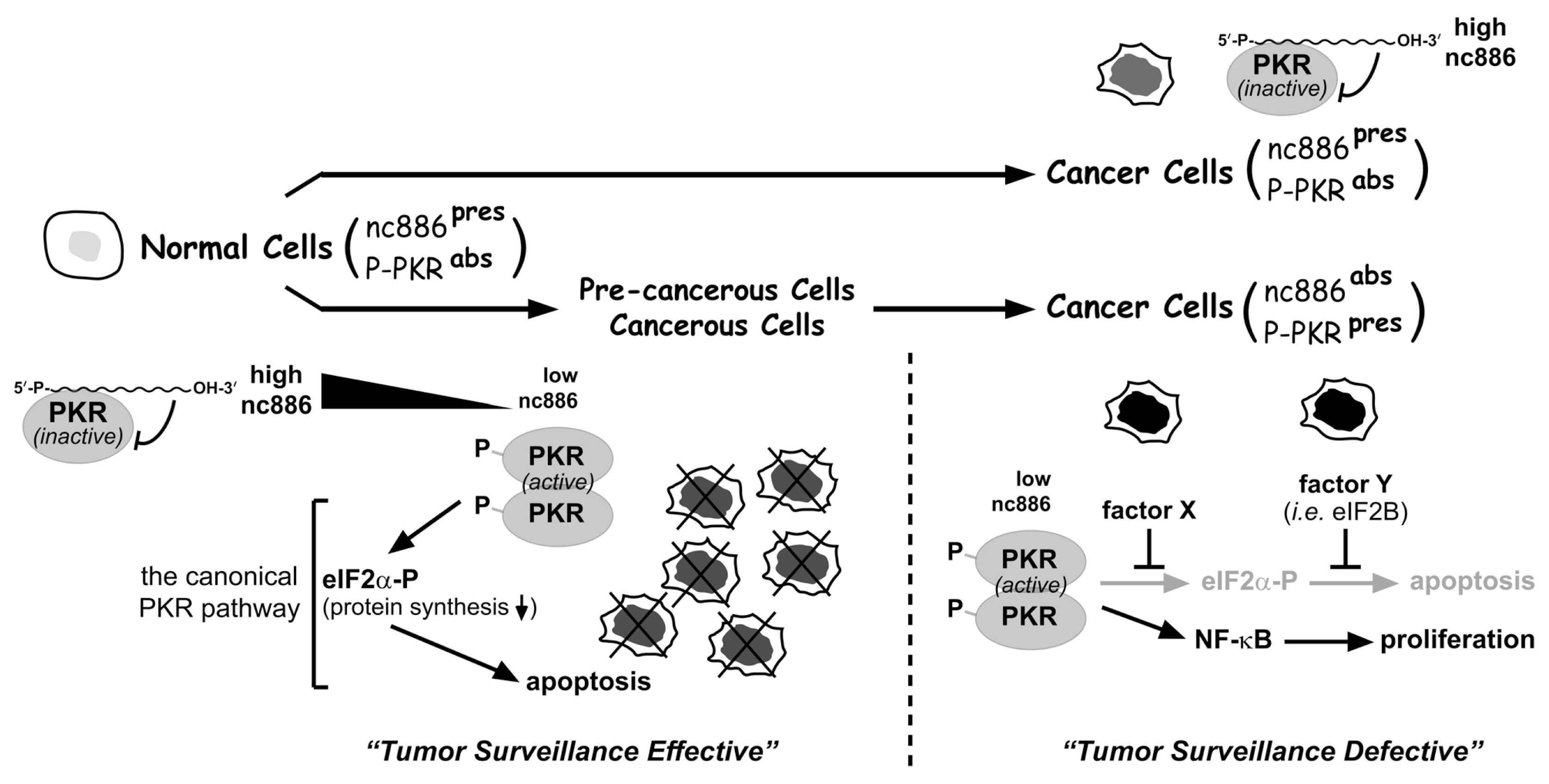
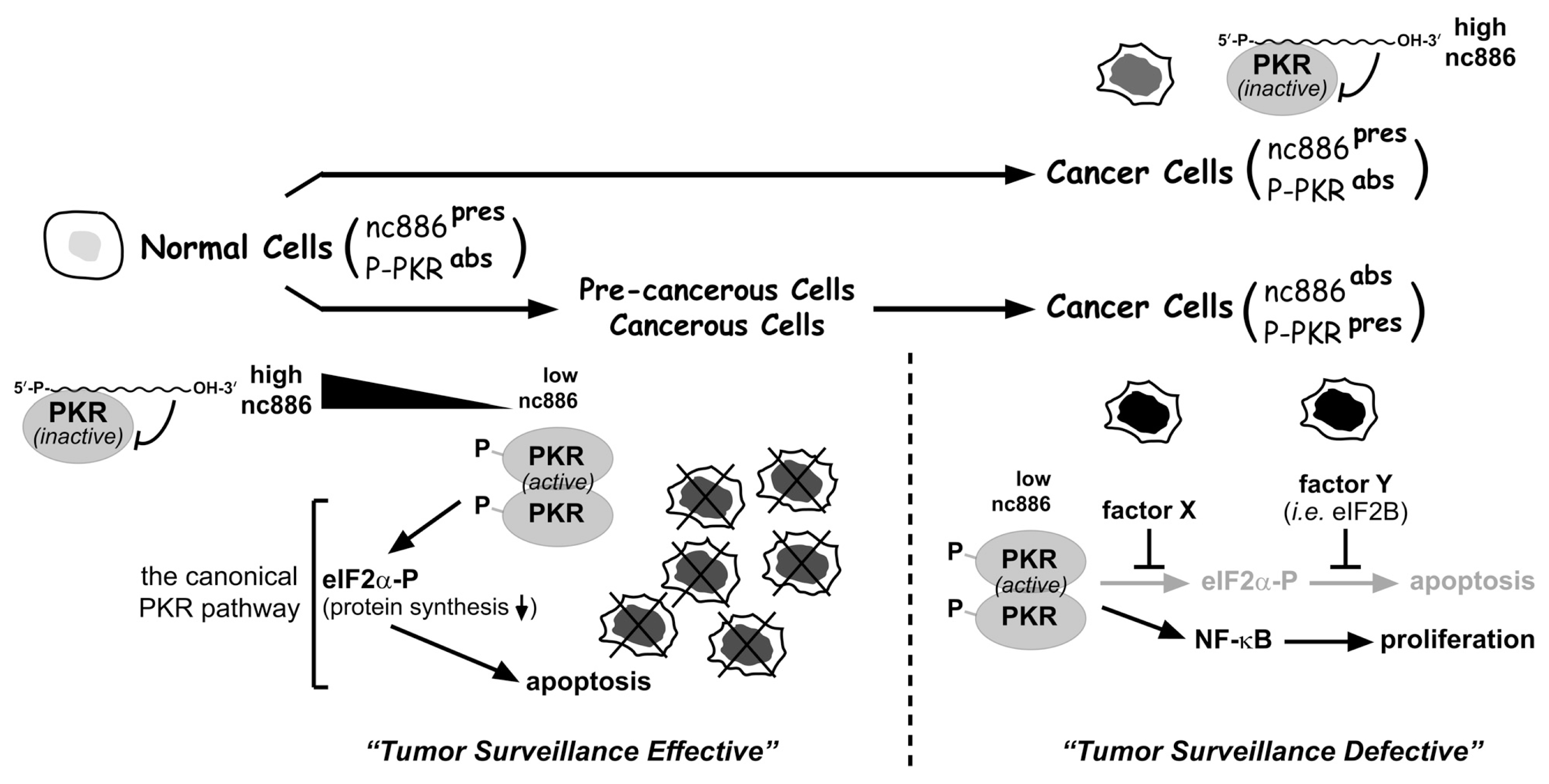
4. A Tumor Surveillance Model Involving nc886 and PKR
5. Concluding Remarks
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin 2011, 61, 212–236. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet 1993, 9, 138–141. [Google Scholar]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar]
- Malmberg, K.J.; Ljunggren, H.G. Escape from immune- and nonimmune-mediated tumor surveillance. Semin. Cancer Biol 2006, 16, 16–31. [Google Scholar]
- Klein, G.; Klein, E. Immune surveillance against virus-induced tumors and nonrejectability of spontaneous tumors: Contrasting consequences of host versus tumor evolution. Proc. Natl. Acad. Sci. USA 1977, 74, 2121–2125. [Google Scholar]
- Hoglund, P. DNA damage and tumor surveillance: One trigger for two pathways. Sci. STKE 2006, 2006. [Google Scholar] [CrossRef]
- Klein, G. Cancer, apoptosis, and nonimmune surveillance. Cell Death. Differ 2004, 11, 13–17. [Google Scholar]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar]
- Nandy, C.; Mrazek, J.; Stoiber, H.; Grasser, F.A.; Huttenhofer, A.; Polacek, N. Epstein-barr virus-induced expression of a novel human vault RNA. J. Mol. Biol 2009, 388, 776–784. [Google Scholar]
- Stadler, P.F.; Chen, J.J.; Hackermuller, J.; Hoffmann, S.; Horn, F.; Khaitovich, P.; Kretzschmar, A.K.; Mosig, A.; Prohaska, S.J.; Qi, X.; et al. Evolution of vault RNAs. Mol. Biol. Evol 2009, 26, 1975–1991. [Google Scholar]
- Kunkeaw, N.; Jeon, S.H.; Lee, K.; Johnson, B.H.; Tanasanvimon, S.; Javle, M.; Pairojkul, C.; Chamgramol, Y.; Wongfieng, W.; Gong, B.; et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene 2012. [Google Scholar] [CrossRef]
- Jeon, S.H.; Lee, K.; Lee, K.S.; Kunkeaw, N.; Johnson, B.H.; Holthauzen, L.M.; Gong, B.; Leelayuwat, C.; Lee, Y.S. Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett 2012, 586, 3477–3484. [Google Scholar]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar]
- Treppendahl, M.B.; Qiu, X.; Sogaard, A.; Yang, X.; Nandrup-Bus, C.; Hother, C.; Andersen, M.K.; Kjeldsen, L.; Mollgaard, L.; Hellstrom-Lindberg, E.; et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood 2012, 119, 206–216. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jeon, S.H.; Johnson, B.H.; Lee, Y.S. A Tumor Surveillance Model: A Non-Coding RNA Senses Neoplastic Cells and Its Protein Partner Signals Cell Death. Int. J. Mol. Sci. 2012, 13, 13134-13139. https://doi.org/10.3390/ijms131013134
Jeon SH, Johnson BH, Lee YS. A Tumor Surveillance Model: A Non-Coding RNA Senses Neoplastic Cells and Its Protein Partner Signals Cell Death. International Journal of Molecular Sciences. 2012; 13(10):13134-13139. https://doi.org/10.3390/ijms131013134
Chicago/Turabian StyleJeon, Sung Ho, Betty H. Johnson, and Yong Sun Lee. 2012. "A Tumor Surveillance Model: A Non-Coding RNA Senses Neoplastic Cells and Its Protein Partner Signals Cell Death" International Journal of Molecular Sciences 13, no. 10: 13134-13139. https://doi.org/10.3390/ijms131013134