Involvement of Oxidative Stress in Suppression of Insulin Biosynthesis under Diabetic Conditions

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Natural History of Pancreatic β-Cell Failure Which Is Often Observed in Type 2 Diabetes
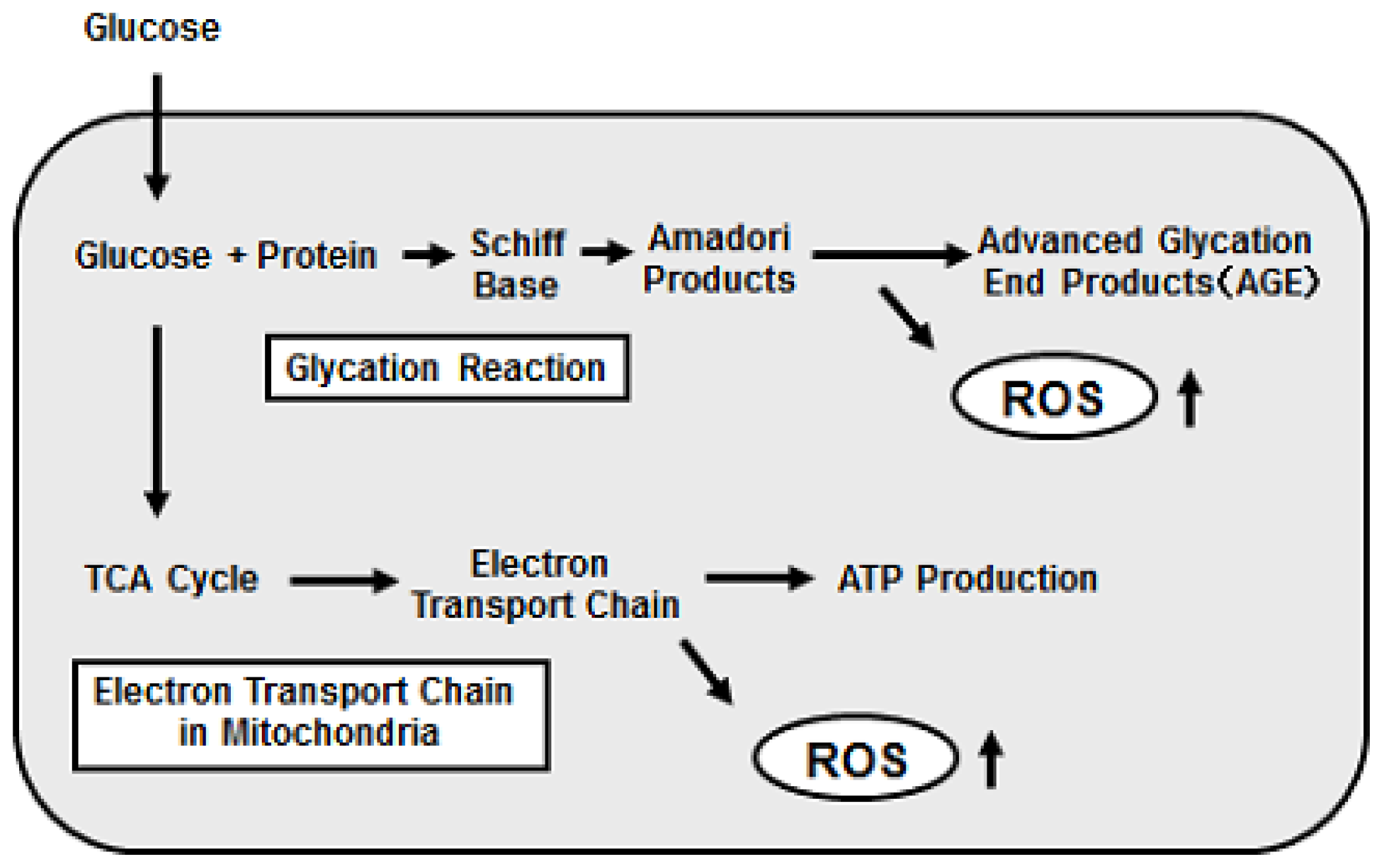
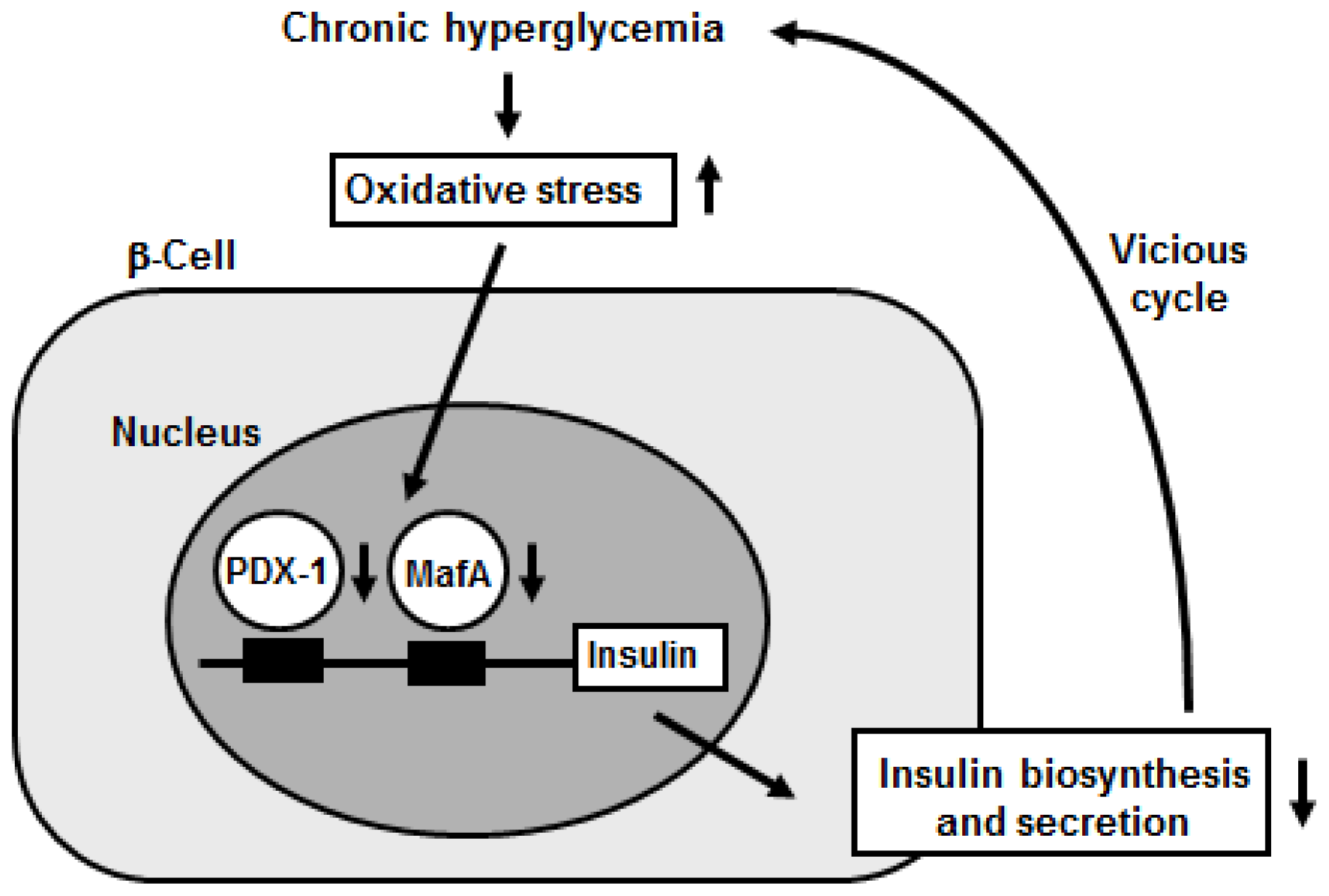
2. Oxidative Stress Is involved in Pancreatic β-Cell Glucose Toxicity, Which Is Often Observed in Type 2 Diabetes
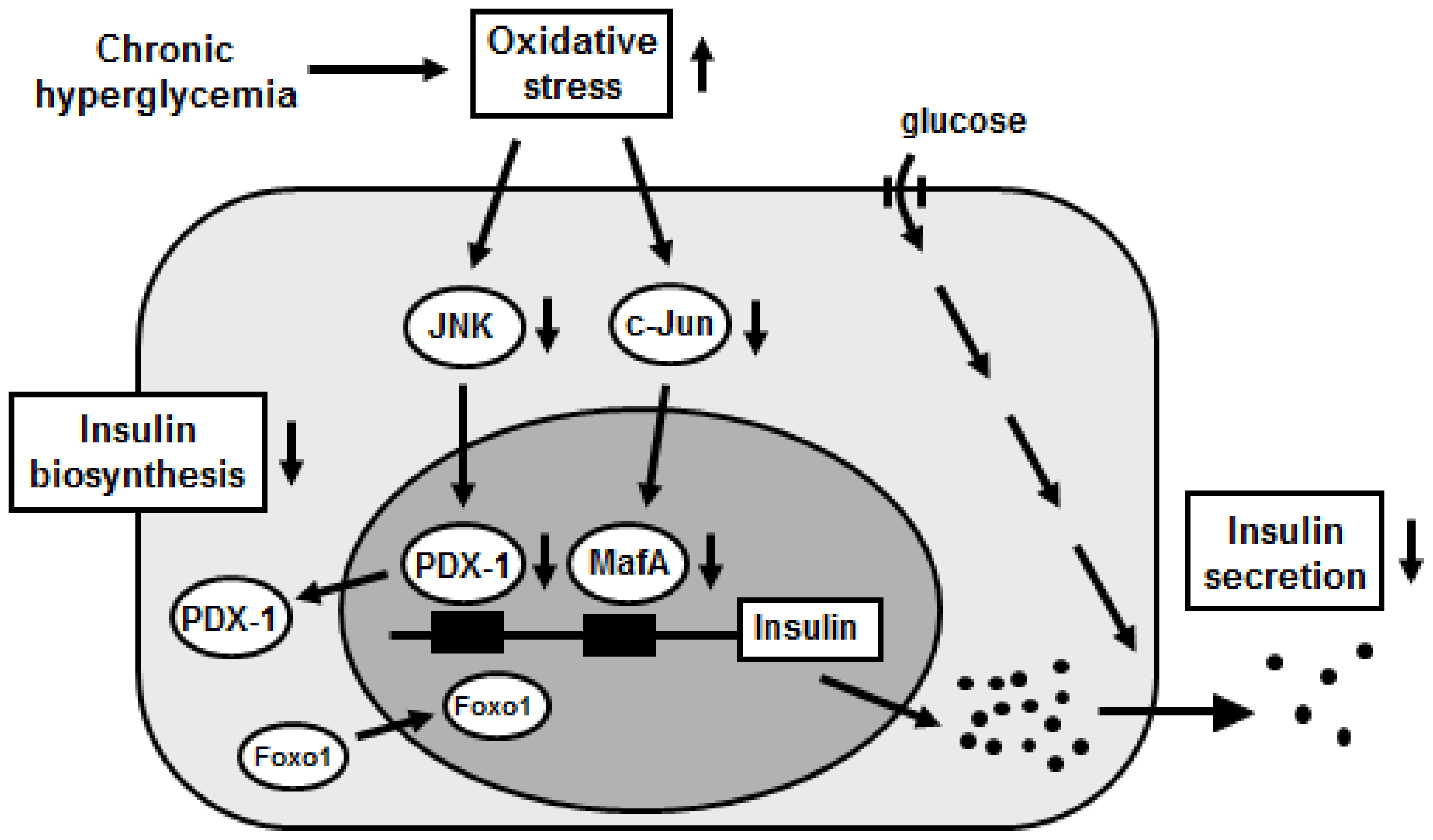
3. Activation of the JNK Pathway Is Involved in Pancreatic β-Cell Glucose Toxicity
4. Induction of c-Jun Expression Is Involved in Pancreatic β-Cell Glucose Toxicity
5. Conclusions
References
- Carlsson, C.; Borg, L.A.; Welsh, N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 1999, 140, 3422–3428. [Google Scholar]
- Joseph, J.W.; Koshkin, V.; Saleh, M.C.; Sivitz, W.I.; Zhang, C.Y.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Free fatty acid-induced β-cell defects are dependent on uncoupling protein 2 expression. J. Biol. Chem 2004, 279, 51049–51056. [Google Scholar]
- Wang, X.; Li, H.; de Leo, D.; Guo, W.; Koshkin, V.; Fantus, I.G.; Giacca, A.; Chan, C.B.; Der, S.; Wheeler, M.B. Gene and protein kinase expression profiling of reactive oxygen species-associated lipotoxicity in the pancreatic β-cell line MIN6. Diabetes 2004, 53, 129–140. [Google Scholar]
- Oprescu, A.I.; Bikopoulos, G.; Naassan, A.; Allister, E.M.; Tang, C.; Park, E.; Uchino, H.; Lewis, G.F.; Fantus, I.G.; Rozakis-Adcock, M.; et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes 2007, 56, 2927–2937. [Google Scholar]
- Bikopoulos, G.; da Silva Pimenta, A.; Lee, S.C.; Lakey, J.R.; Der, S.D.; Chan, C.B.; Ceddia, R.B.; Wheeler, M.B.; Rozakis-Adcock, M. Ex vivo transcriptional profiling of human pancreatic islets following chronic exposure to monounsaturated fatty acids. J. Endocrinol 2008, 196, 455–464. [Google Scholar]
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Invest 1997, 46, 1276–1280. [Google Scholar]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. β-Cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar]
- Poitout, V.; Robertson, R.P. Minireview: Secondary beta-cell failure in type 2 diabetes—A convergence of glucotoxicity and lipotoxicity. Endocrinology 2002, 143, 339–342. [Google Scholar]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in β-cells: Type 2 diabetes, good radicals gone bad and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Invest 2006, 116, 1802–1812. [Google Scholar]
- Kaneto, H.; Matsuoka, T.; Nakatani, Y.; Kawamori, D.; Miyatsuka, T.; Matsuhisa, M.; Yamasaki, Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med 2005, 83, 429–439. [Google Scholar]
- Robertson, R.P.; Zhang, H.J.; Pyzdrowski, K.L.; Walseth, T.F. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J. Clin. Invest 1992, 90, 320–325. [Google Scholar]
- Olson, L.K.; Redmon, J.B.; Towle, H.C.; Robertson, R.P. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J. Clin. Invest 1993, 92, 514–519. [Google Scholar]
- Olson, L.K.; Sharma, A.; Peshavaria, M.; Wright, C.V.; Towle, H.C.; Robertson, R.P.; Stein, R. Reduction of insulin gene transcription in HIT-T15 β-cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. Proc. Natl. Acad. Sci. USA 1995, 92, 514–519. [Google Scholar]
- Poitout, V.; Olson, L.K.; Robertson, R.P. Chronic exposure of βTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J. Clin. Invest 1996, 97, 1041–1046. [Google Scholar]
- Sharma, A.; Fusco-DeMane, D.; Henderson, E.; Efrat, S.; Stein, R. The role of the insulin control element and RIPE3b1 activators in glucose-stimulated transcription of the insulin gene. Mol. Endocrinol 1995, 9, 1468–1488. [Google Scholar]
- Moran, A.; Zhang, H.-J.; Olson, L.K.; Harmon, J.S.; Poitout, V.; Robertson, R.P. Differentiation of glucose toxicity from beta cell exhaustion during the evolution of defective insulin gene expression in the pancreatic islet cell line, HIT-T15. J. Clin. Invest 1997, 99, 534–539. [Google Scholar]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.T.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar]
- Tanaka, Y.; Tran, P.O.T.; Harmon, J.; Robertson, R.P. A role of glutathione peroxidase in protecting pancreatic β cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar]
- Kaneto, H.; Fujii, J.; Myint, T.; Islam, K.N.; Miyazawa, N.; Suzuki, K.; Kawasaki, Y.; Nakamura, M.; Tatsumi, H.; Yamasaki, Y.; et al. Reducing sugar triggers oxidative modification and apoptosis in pancreatic β-cells by provoking oxidative stress through the glycation reaction. Biochem. J 1996, 320, 855–863. [Google Scholar]
- Matsuoka, T.; Kajimoto, Y.; Watada, H.; Kaneto, H.; Kishimoto, M.; Umayahara, Y.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J. Clin. Invest 1997, 99, 144–150. [Google Scholar]
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic β-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932. [Google Scholar]
- Kajimoto, Y.; Matsuoka, T.; Kaneto, H.; Watada, H.; Fujitani, Y.; Kishimoto, M.; Sakamoto, K.; Matsuhisa, M.; Kawamori, R.; Yamasaki, Y.; et al. Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 1999, 42, 1417–1424. [Google Scholar]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem 1999, 274, 27905–27913. [Google Scholar]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants for diabetes: Possible protection of pancreatic β-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar]
- Kaneto, H.; Xu, G.; Song, K.H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the hexosamine pathway leads to deterioration of pancreatic β-cell function by provoking oxidative stress. J. Biol. Chem 2001, 276, 31099–31104. [Google Scholar]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem. Biophys. Res. Commun 2003, 300, 216–222. [Google Scholar]
- Gorogawa, S.; Kajimoto, Y.; Umayahara, U.; Kaneto, H.; Watada, H.; Kuroda, A.; Kawamori, D.; Yasuda, T.; Matsuhisa, M.; Yamasaki, Y.; et al. Probucol preserves pancreatic β-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res. Clin. Prac 2002, 57, 1–10. [Google Scholar]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem 2005, 280, 11107–11113. [Google Scholar]
- Harmon, J.S.; Bogdani, M.; Parazolli, S.D.; Mak, S.S.; Oseid, E.A.; Berghmans, M.; LeBoeuf, R.C.; Robertson, R.P. β-cell specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009, 150, 4855–4862. [Google Scholar]
- Yamamoto, M.; Yamato, E.; Toyoda, S.; Tashiro, F.; Ikegami, H.; Yodoi, J.; Miyazaki, J. Transgenic expression of antioxidant protein thioredoxin in pancreatic β cells prevents progression of type 2 diabetes mellitus. Antioxid. Redox. Signal 2008, 10, 43–49. [Google Scholar]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar]
- Sakurai, T.; Tsuchiya, S. Superoxide production from nonenzymatically glycated protein. FEBS Lett 1988, 236, 406–410. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar]
- Grankvist, K; Marklund, S.L.; Taljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J 1981, 199, 393–398. [Google Scholar]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar]
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing-transactivator of the insulin gene. EMBO J 1993, 12, 4251–4259. [Google Scholar]
- Leonard, J.; Peers, B.; Johnson, T.; Ferreri, K.; Lee, S.; Montminy, M.R. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol. Endocrinol 1993, 7, 1275–1283. [Google Scholar]
- Miller, C.P.; McGehee, R.E.; Habener, J.F. IDX-1: A new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J 1994, 13, 1145–1156. [Google Scholar]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 37, 606–609. [Google Scholar]
- Stoffers, D.A.; Zinkin, N.T.; Stanojevic, V.; Clarke, W.L.; Habener, J.F. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet 1997, 15, 106–110. [Google Scholar]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. β-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev 1998, 12, 1763–1768. [Google Scholar]
- Ferber, S.; Halkin, A.; Cohen, H.; Ber, I.; Einav, Y.; Goldberg, I.; Barshack, I.; Seijffers, R.; Kopolovic, J.; Kaiser, N.; et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat. Med 2000, 6, 568–572. [Google Scholar]
- Holland, A.M.; Hale, M.A.; Kagami, H.; Hammer, R.E.; MacDonald, R.J. Experimental control of pancreatic development and maintenance. Proc. Natl. Acad. Sci. USA 2002, 99, 12236–12241. [Google Scholar]
- Noguchi, H.; Kaneto, H.; Weir, G.C.; Bonner-Weir, S. PDX-1 protein containing its own Antennapedia-like protein transduction domain can transduce pancreatic duct and islet cells. Diabetes 2003, 52, 1732–1737. [Google Scholar]
- Miyazaki, S.; Yamato, E.; Miyazaki, J. Regulated expression of pdx-1 promotes in vitro differentiation of insulin-producing cells from embryonic stem cells. Diabetes 2004, 53, 1030–1037. [Google Scholar]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes 2005, 54, 1009–1022. [Google Scholar]
- Kaneto, H.; Miyatsuka, T.; Shiraiwa, T.; Yamamoto, K.; Kato, K.; Fujitani, Y.; Matsuoka, T. Crucial role of PDX-1 in pancreas development, β-cell differentiation, and induction of surrogate β-cells. Curr. Med. Chem 2007, 14, 103–112. [Google Scholar]
- Kaneto, H.; Miyatsuka, T.; Kawamori, D.; Yamamoto, K.; Kato, K.; Shiraiwa, T.; Katakami, N.; Yamasaki, Y.; Matsuhisa, M.; Matsuoka, T. PDX-1 and MafA play a crucial role in pancreatic β-cell differentiation and maintenance of mature β-cell function. Endocr. J 2008, 55, 235–252. [Google Scholar]
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742. [Google Scholar]
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem 2002, 277, 49903–49910. [Google Scholar]
- Matsuoka, T.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol. Cell. Biol 2003, 23, 6049–6062. [Google Scholar]
- Matsuoka, T.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar]
- Kaneto, H.; Matsuoka, T.; Nakatani, Y.; Miyatsuka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. A crucial role of MafA as a novel therapeutic target for diabetes. J. Biol. Chem 2005, 280, 15047–15052. [Google Scholar]
- Matsuoka, T.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet β cell function. Mol. Endocrinol 2007, 21, 2764–2774. [Google Scholar]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem 2002, 277, 30010–30018. [Google Scholar]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun N-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar]
- Nakae, J.; Biggs, W.H., III; Kitamura, T.; Cavenee, W.K.; Wright, C.V.; Arden, K.C.; Accili, D. Regulation of insulin action and pancreatic b-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet 2002, 32, 245–253. [Google Scholar]
- Biggs, W.H., III; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H., III; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J. Clin. Invest 2002, 110, 1839–1847. [Google Scholar]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem 2006, 281, 1091–1098. [Google Scholar]
- Matsuoka, T.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic β-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar]
- Butler, A.E.; Robertson, R.P.; Hernandez, R.; Matveyenko, A.V.; Gurlo, T.; Butler, P.C. Beta cell nuclear musculoaponeurotic fibrosarcoma oncogene family A (MafA) is deficient in type 2 diabetes. Diabetologia 2012. [Google Scholar] [CrossRef]
- Inagaki, N.; Maekawa, T; Sudo, T.; Ishii, S.; Seino, Y.; Imura, H. c-Jun represses the human insulin promoter activity that depends on multiple cAMP response elements. Proc. Natl. Acad. Sci. USA 1992, 89, 1045–1049. [Google Scholar]
- Henderson, E.; Stein, R. c-jun inhibits transcriptional activation by the insulin enhancer; the insulin control element is the target of control. Mol. Cell. Biol 1994, 14, 655–662. [Google Scholar]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol 2005, 25, 4969–4976. [Google Scholar]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaneto, H.; Matsuoka, T.-a. Involvement of Oxidative Stress in Suppression of Insulin Biosynthesis under Diabetic Conditions. Int. J. Mol. Sci. 2012, 13, 13680-13690. https://doi.org/10.3390/ijms131013680
Kaneto H, Matsuoka T-a. Involvement of Oxidative Stress in Suppression of Insulin Biosynthesis under Diabetic Conditions. International Journal of Molecular Sciences. 2012; 13(10):13680-13690. https://doi.org/10.3390/ijms131013680
Chicago/Turabian StyleKaneto, Hideaki, and Taka-aki Matsuoka. 2012. "Involvement of Oxidative Stress in Suppression of Insulin Biosynthesis under Diabetic Conditions" International Journal of Molecular Sciences 13, no. 10: 13680-13690. https://doi.org/10.3390/ijms131013680