Molecular Mechanisms of Pharmaceutical Drug Binding into Calsequestrin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
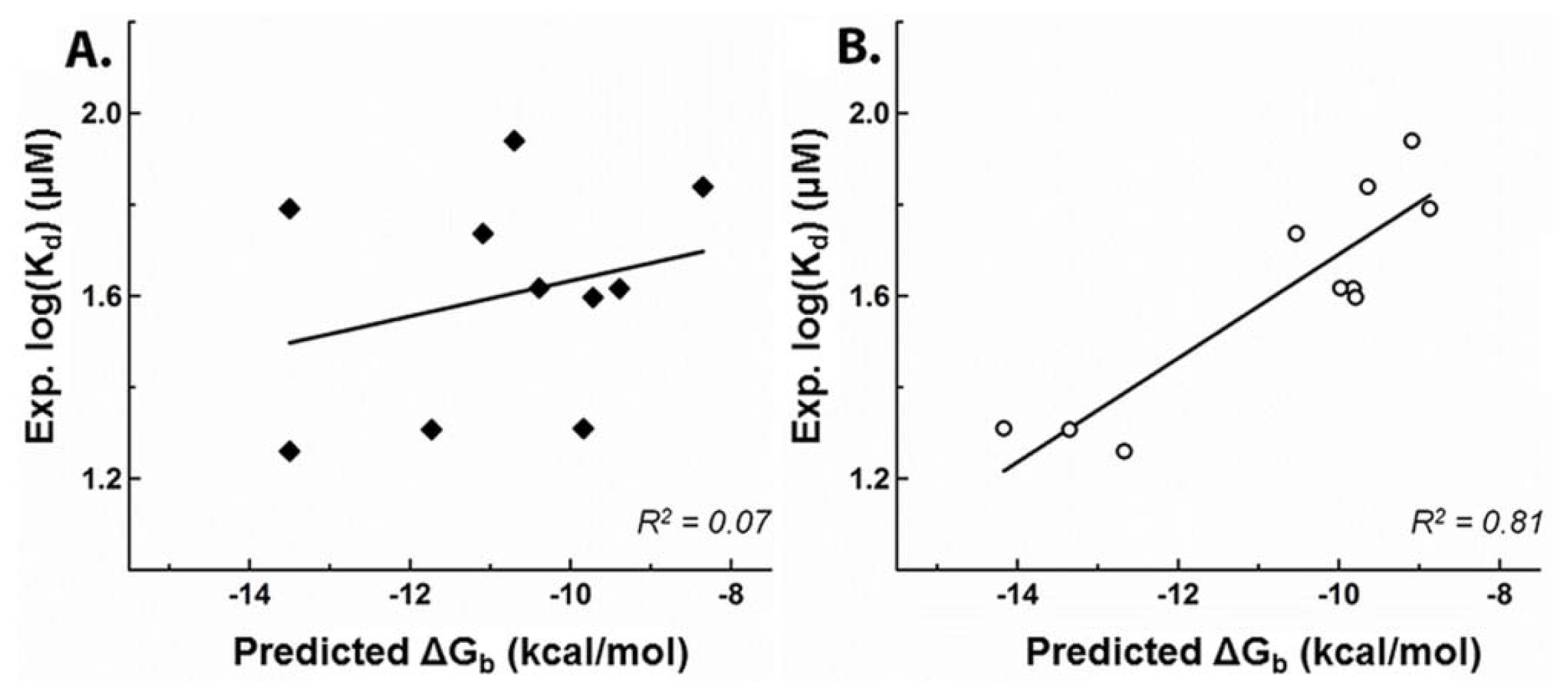
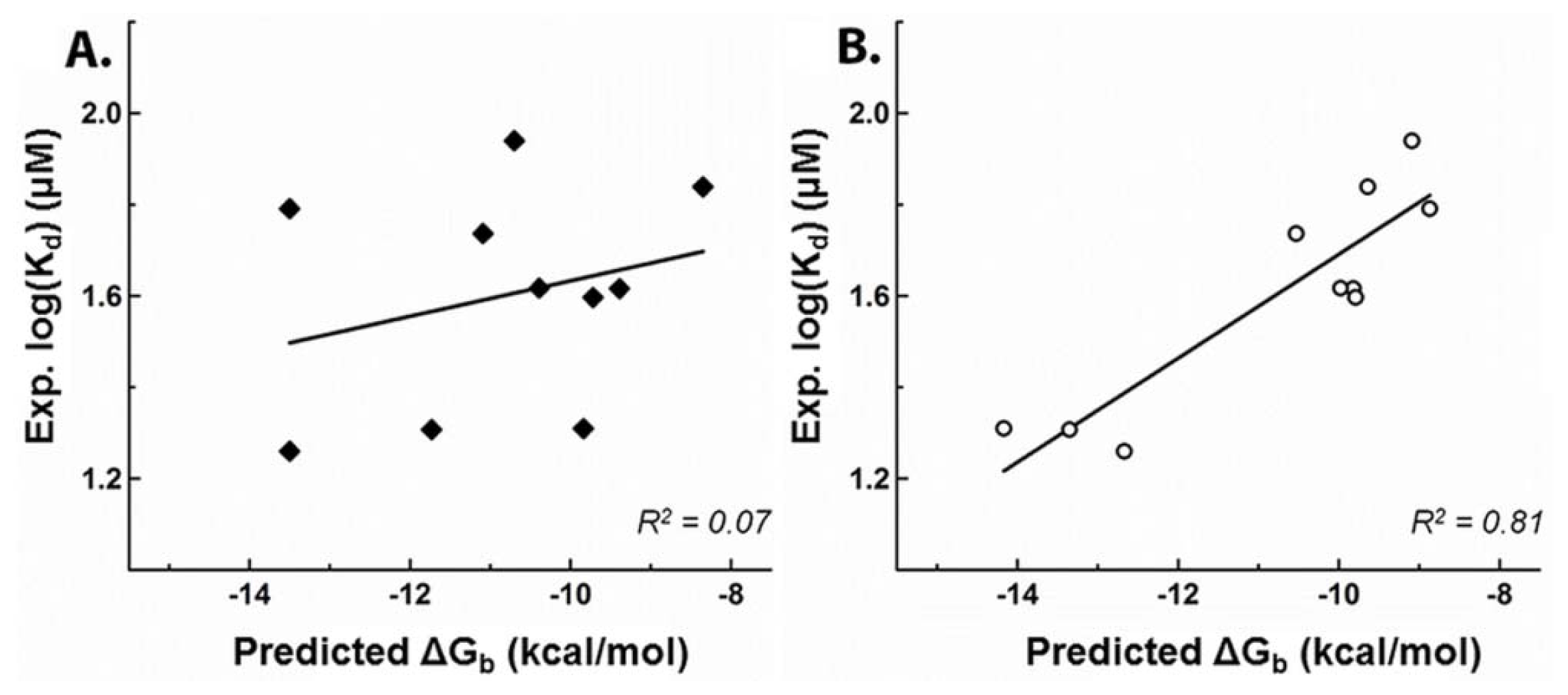
2.1. Development of a Highly-Predictive Molecular Docking Model for CASQ2-Ligand Binding
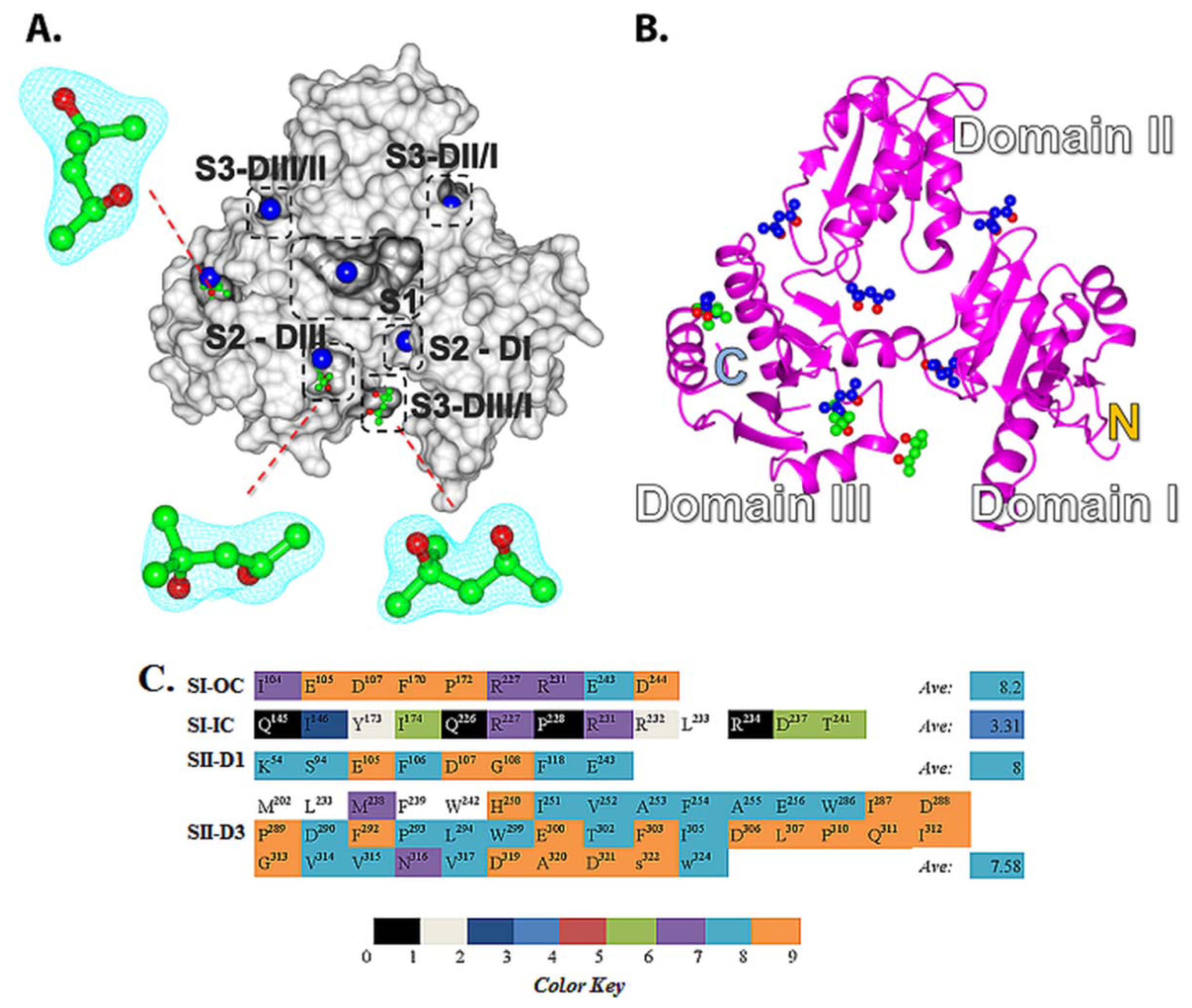
2.2. Prediction of Small Molecule Binding Pockets in CASQ2
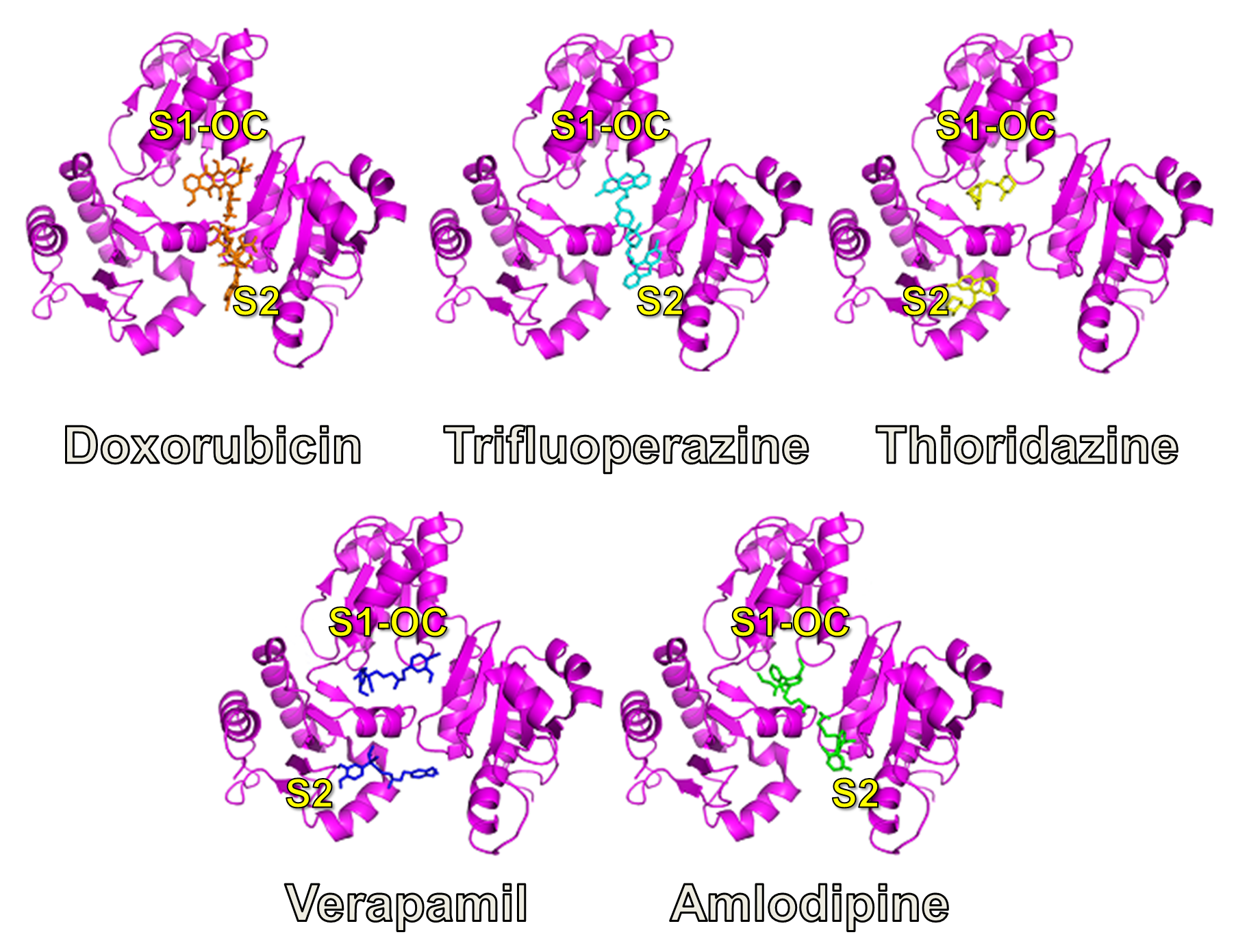
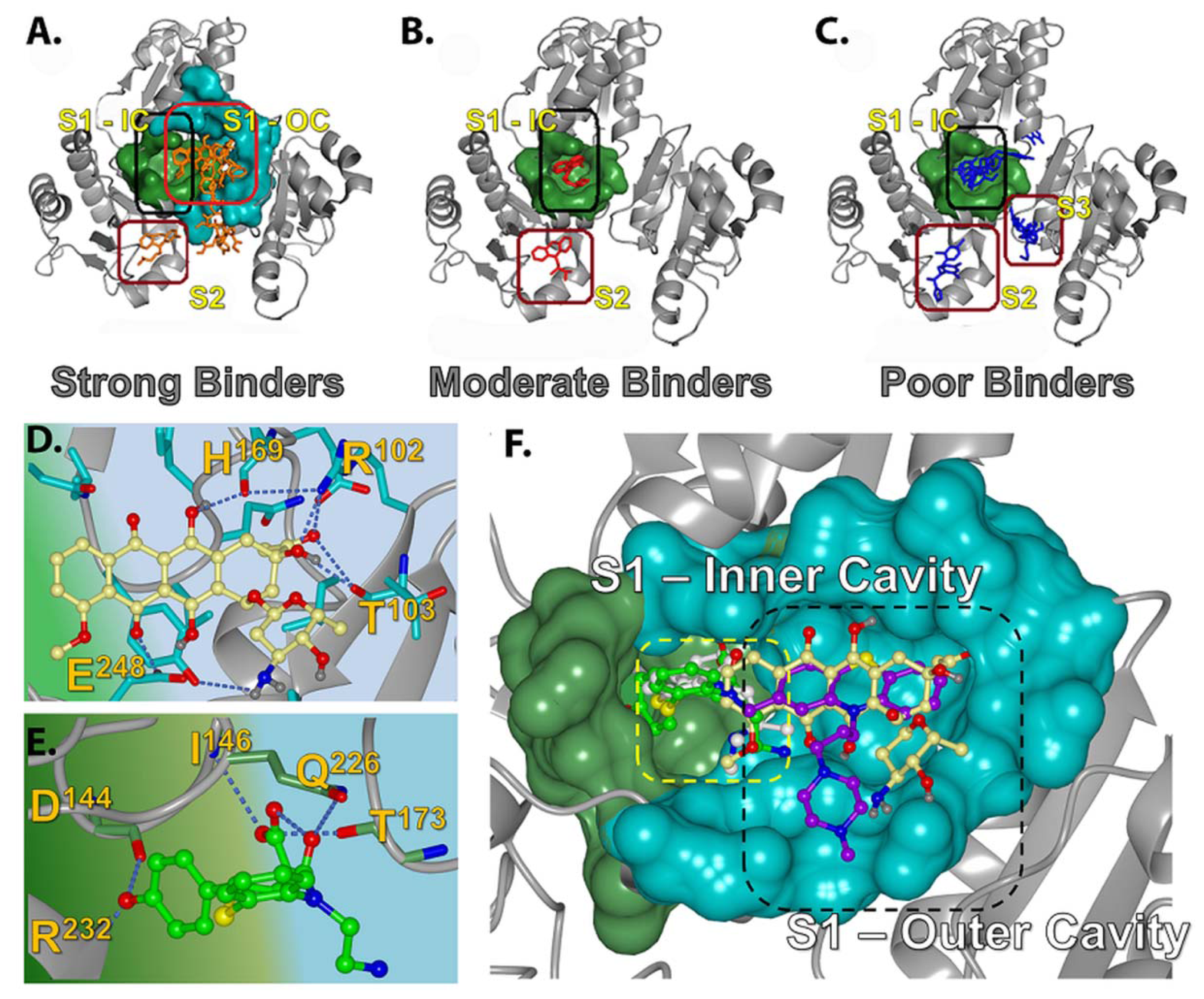
2.2.1. Prediction of Binding Pockets for Different Classes of Drugs—Molecular Docking
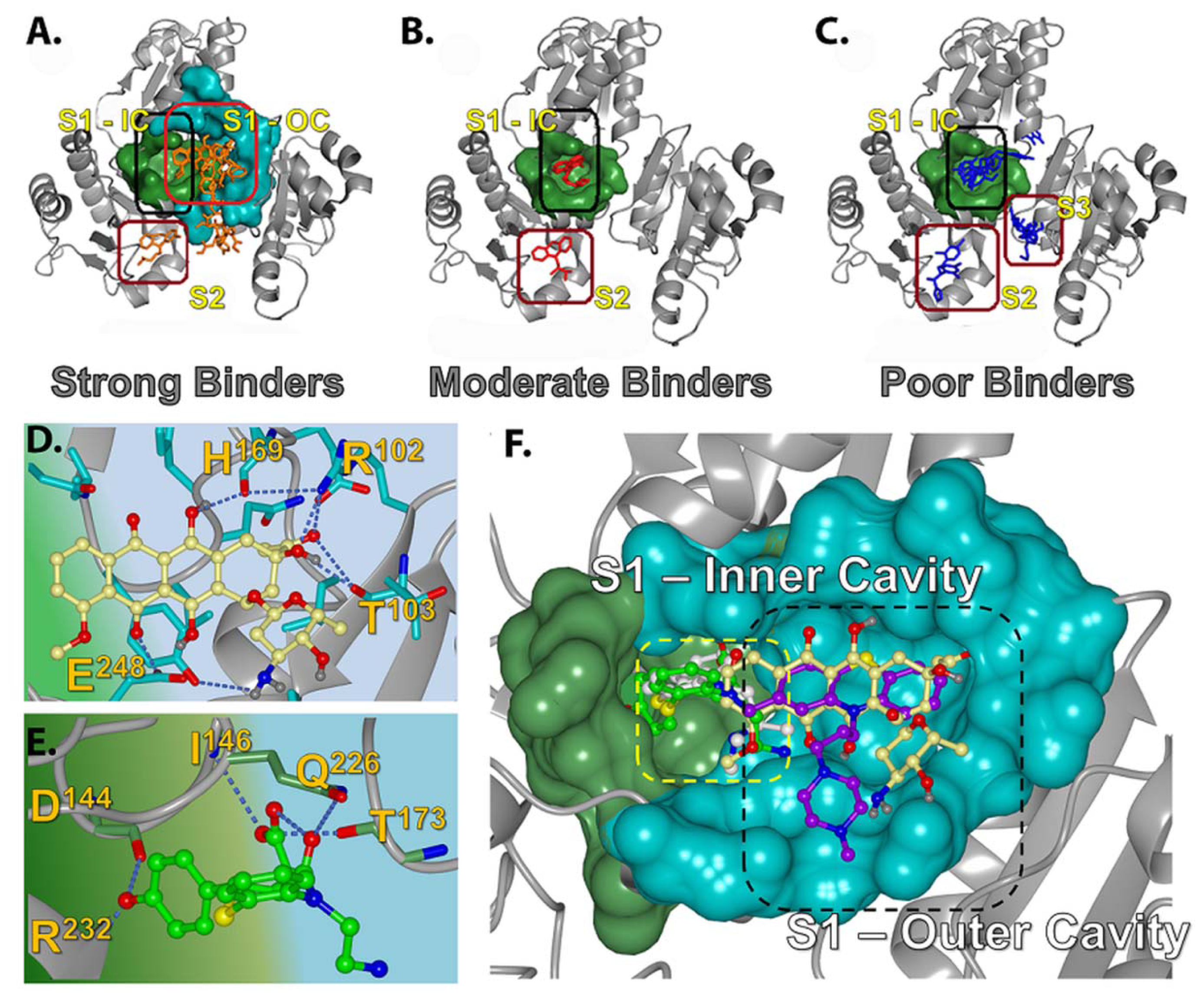
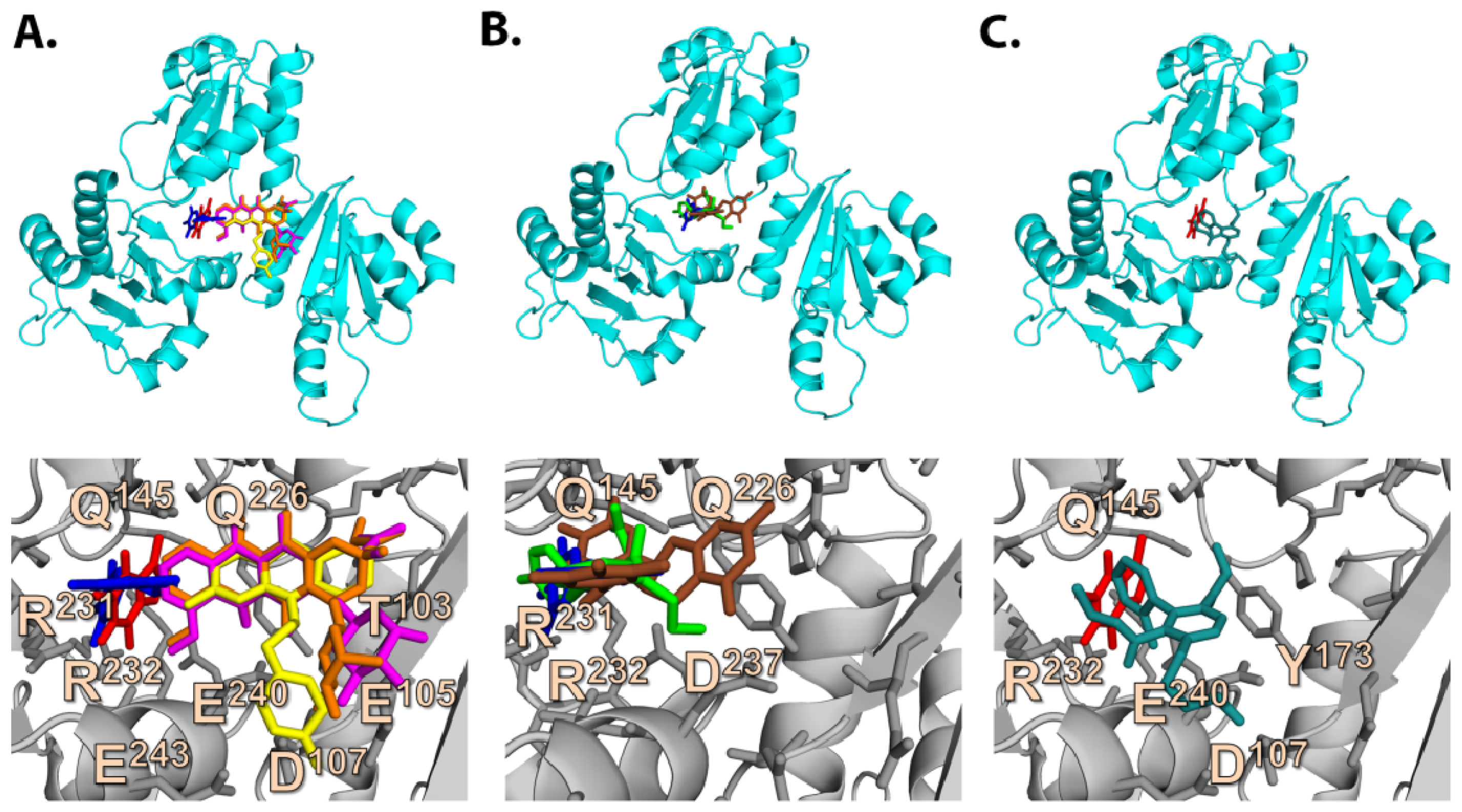
2.2.2. Strong Binders
2.2.3. Moderate Binders
2.2.4. Poor Binders
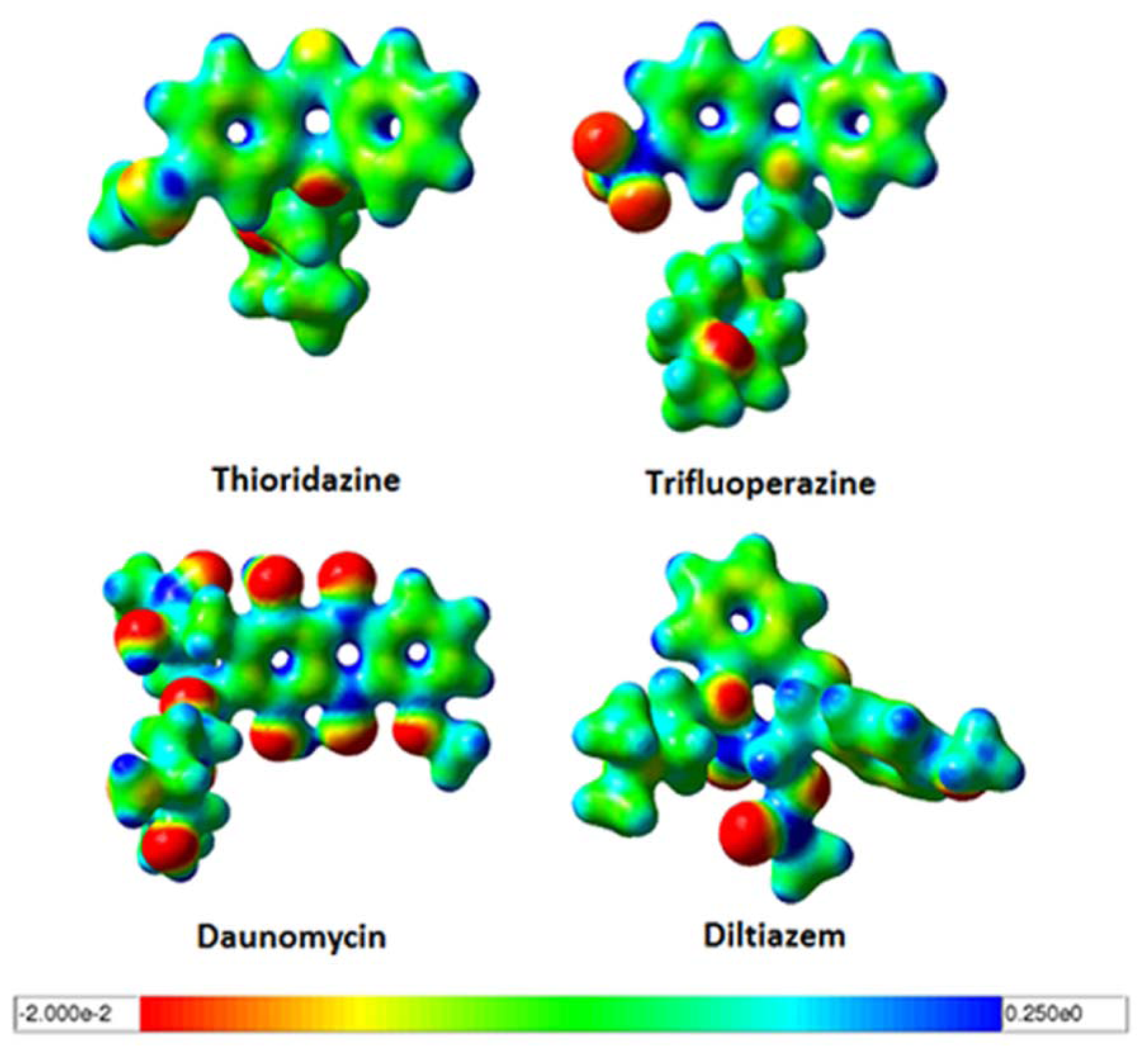
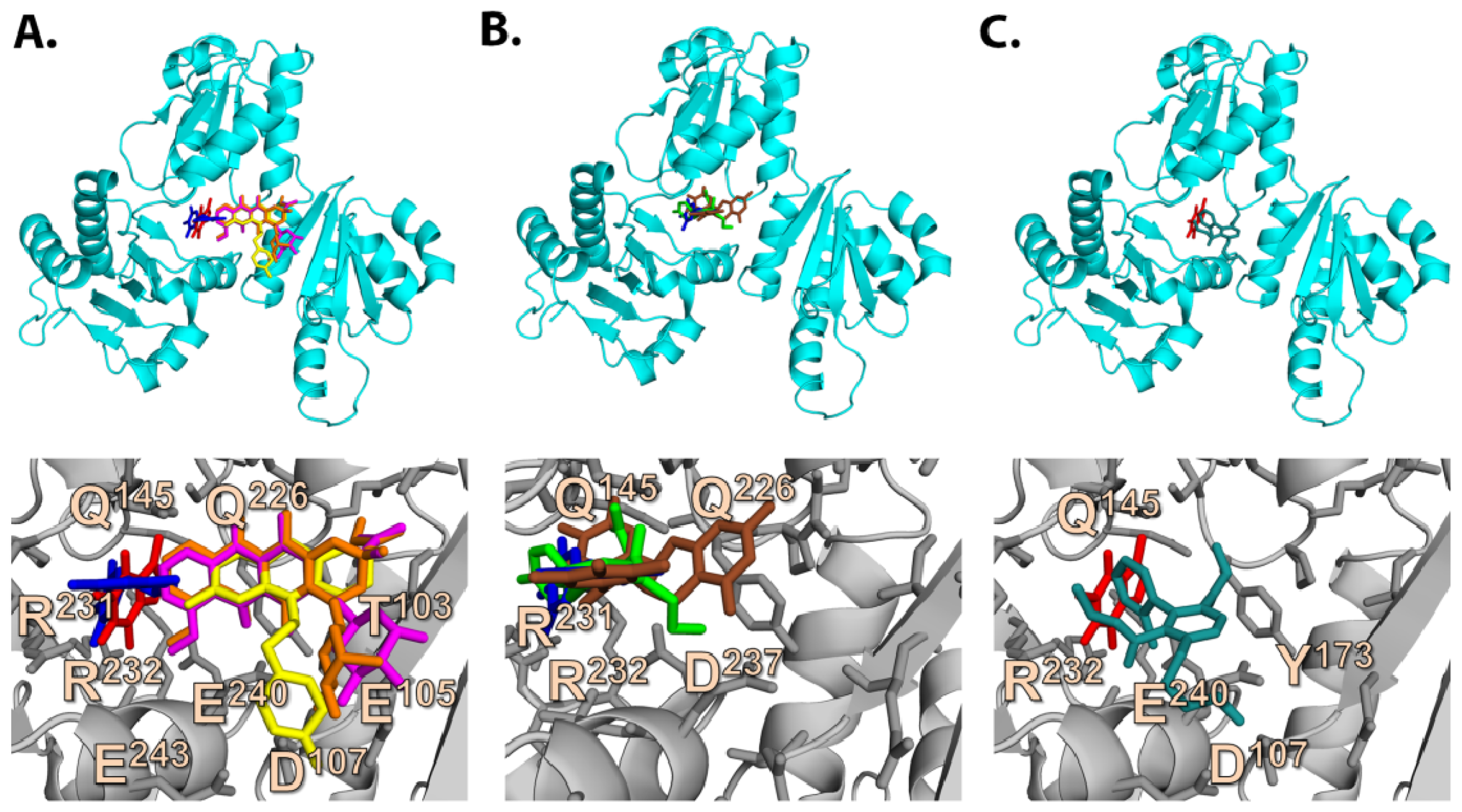
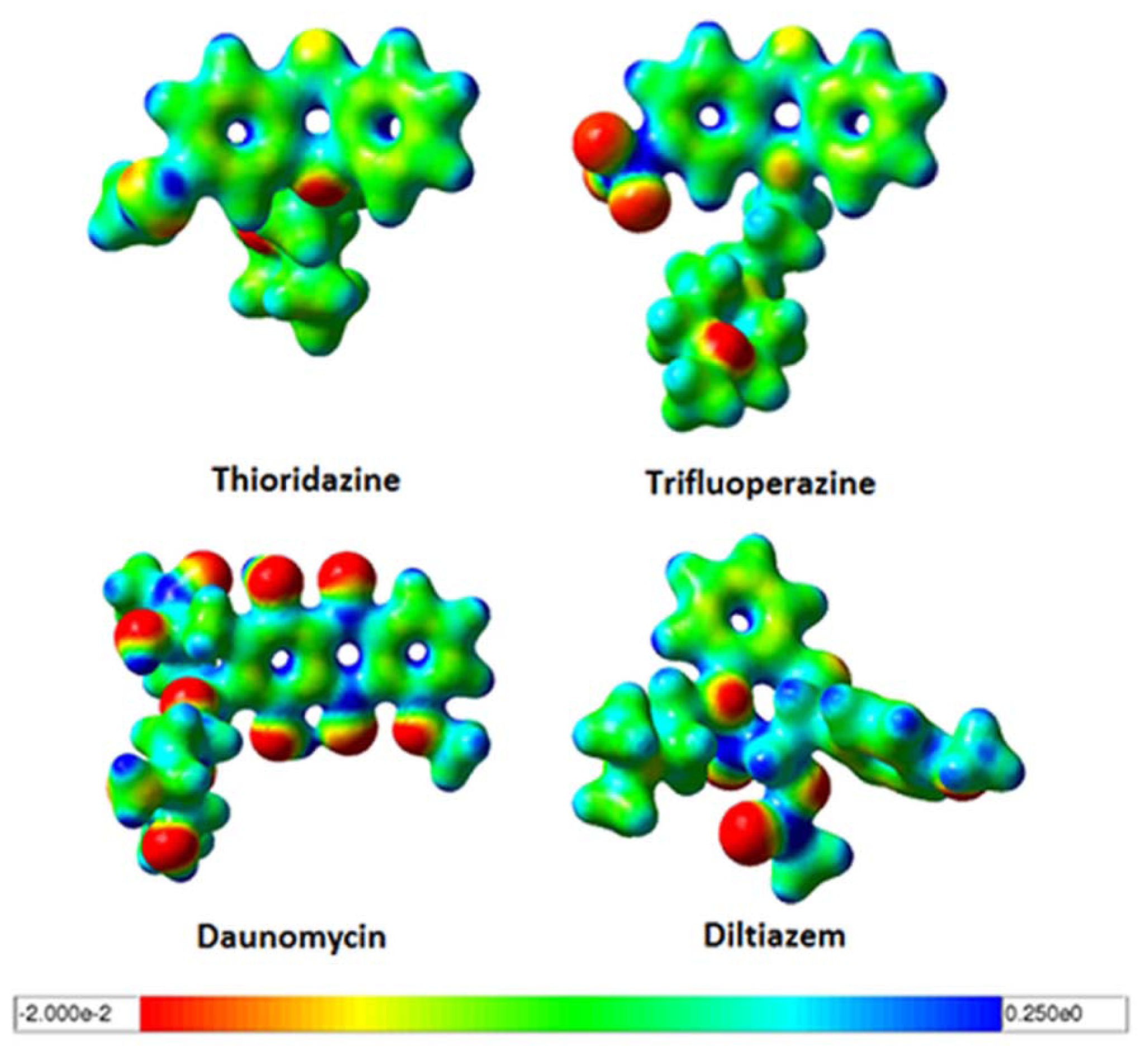
2.3. Quantum Mechanics Studies of Ligand Charge Distributions: Comparison between Strong Binders Predicted to Bind into the S1-OC of hCASQ2
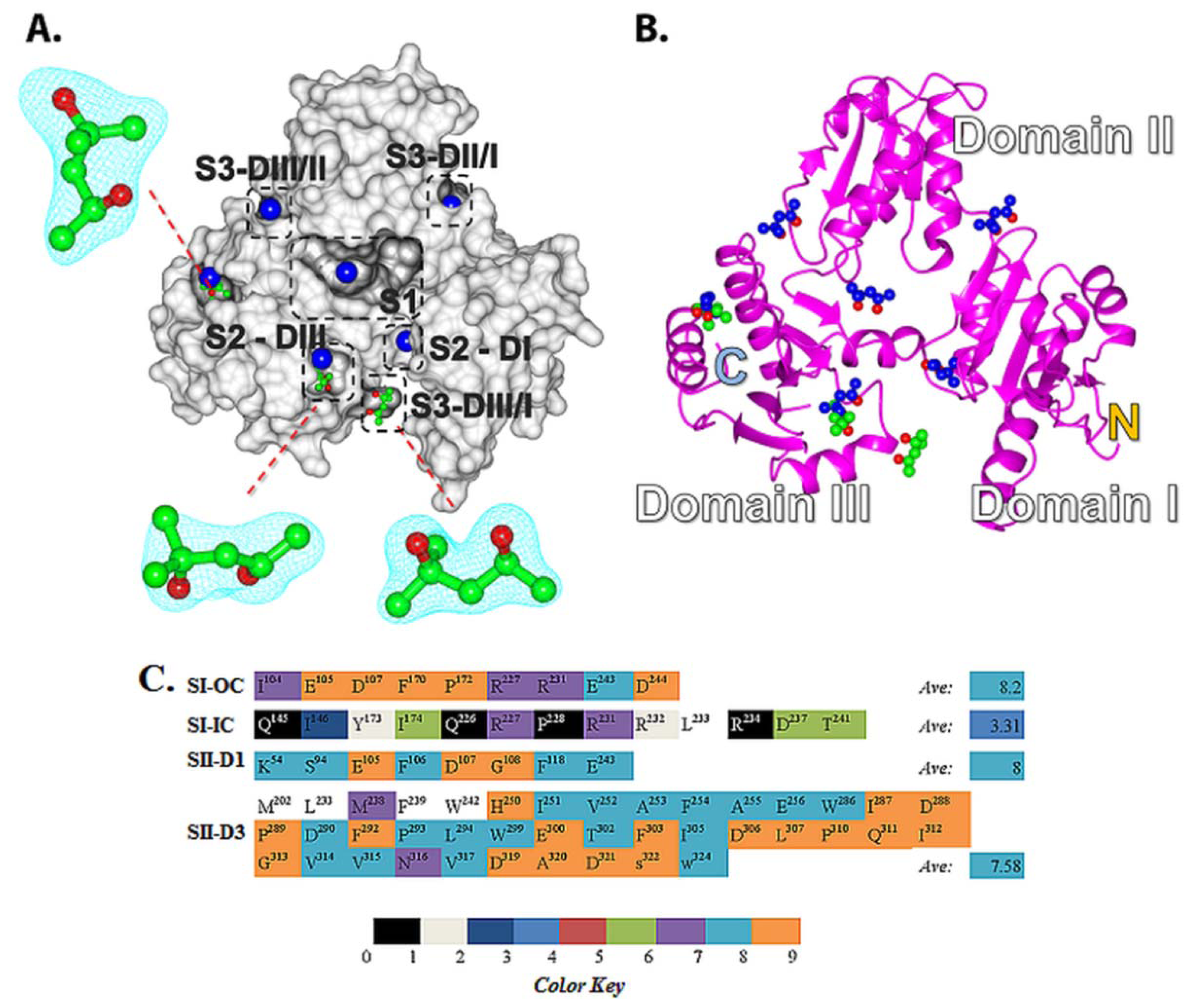
2.4. Amino Acid Conservation Analysis of Potential Small Molecule Binding Pockets in CASQ
2.5. Structural Basis of 2-Methyl-2,4-pentanediol (MPD) Binding into rCASQ1: Crystal Structure Reveals Simultaneous Multiple Binding into S2-DIII Pocket Predicted by Molecular Docking
2.6. Development of a Structure Model for Competitive Inhibition of Pharmaceutical Drugs by Caffeine (CAF) and Epigallocatechin (EGC)
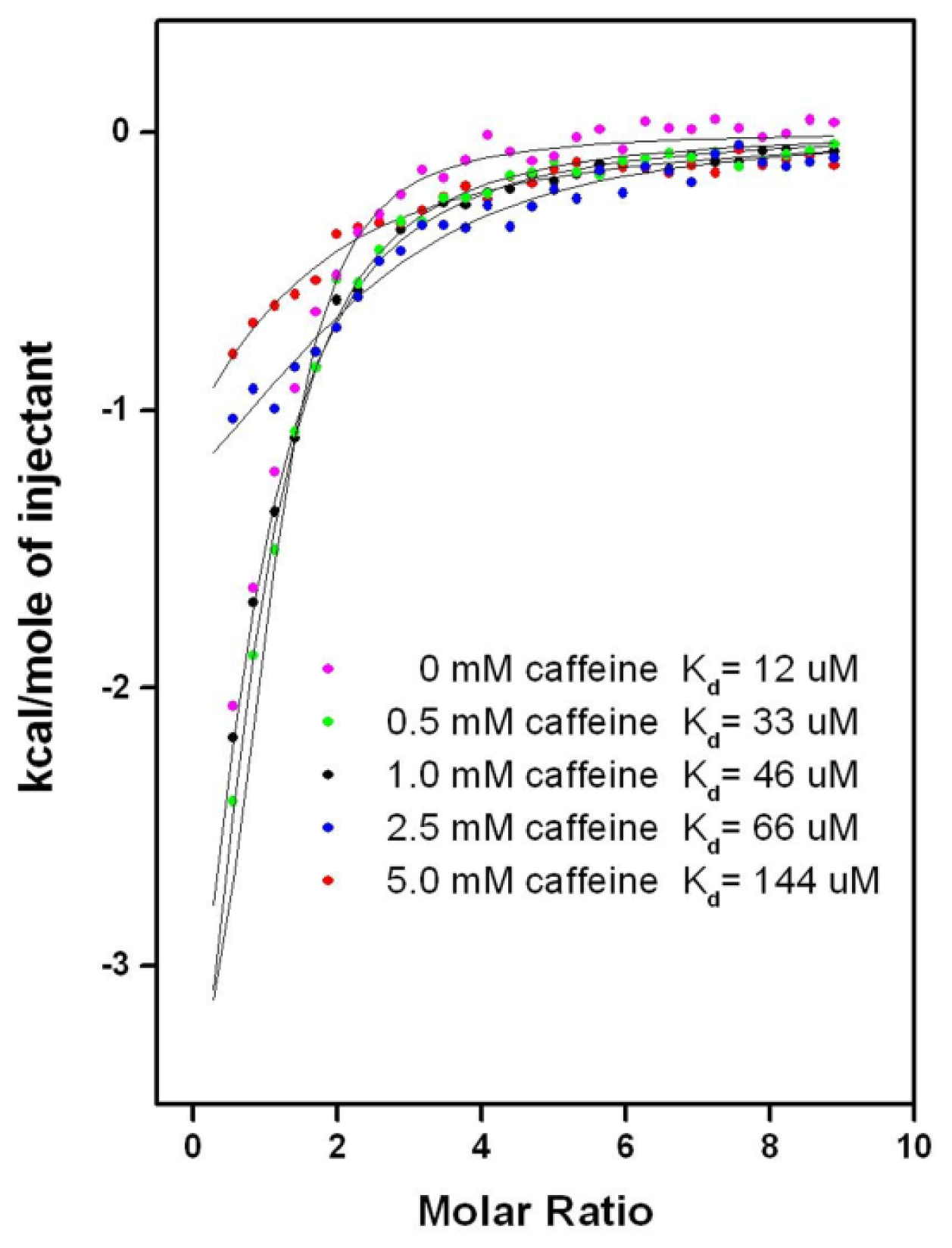
2.6.1. CAF/EGC Mediated Competitive Inhibition—Molecular Docking Studies
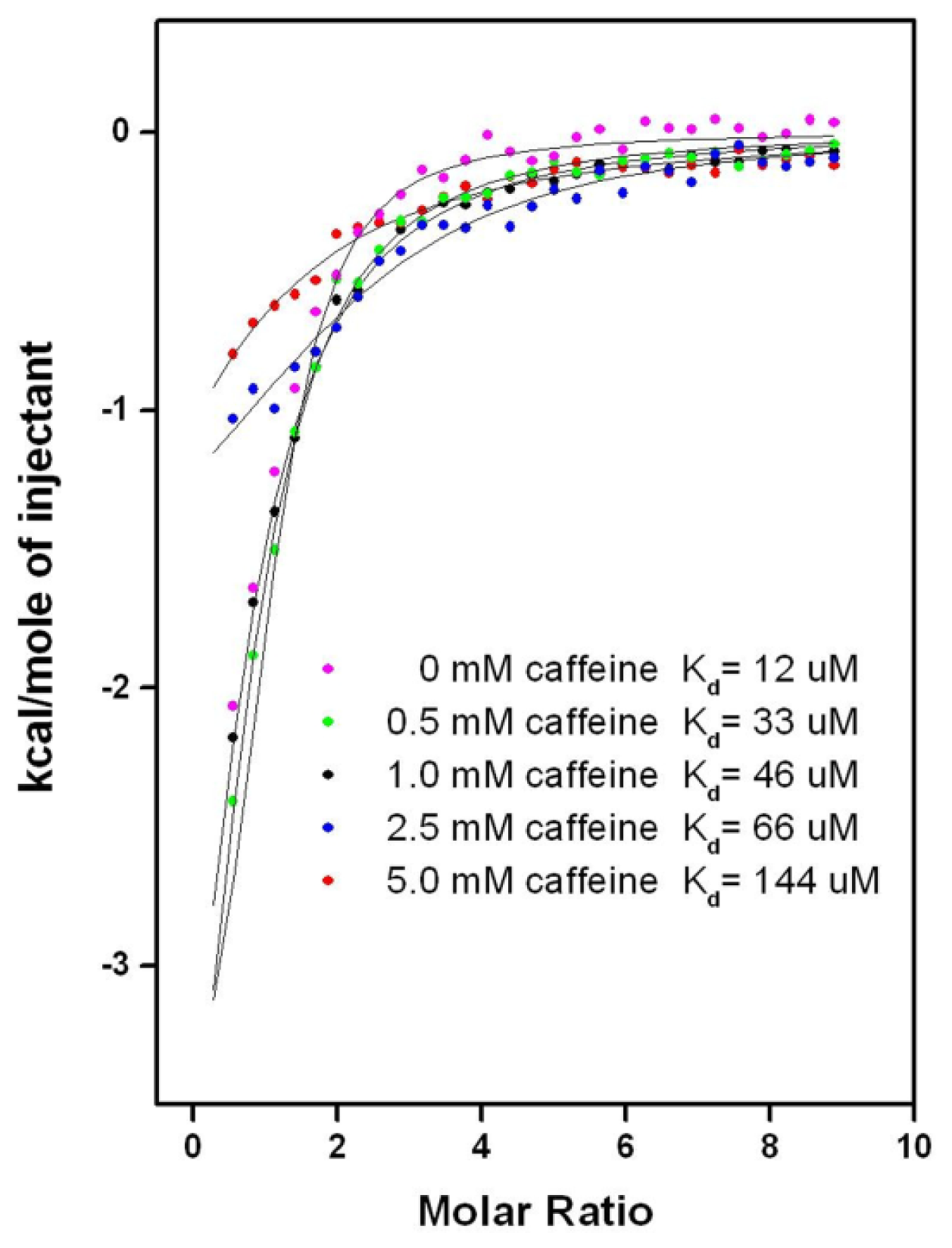
2.6.2. CAF/EGC Mediated Competitive Inhibition—Isothermal Calorimetry Assays
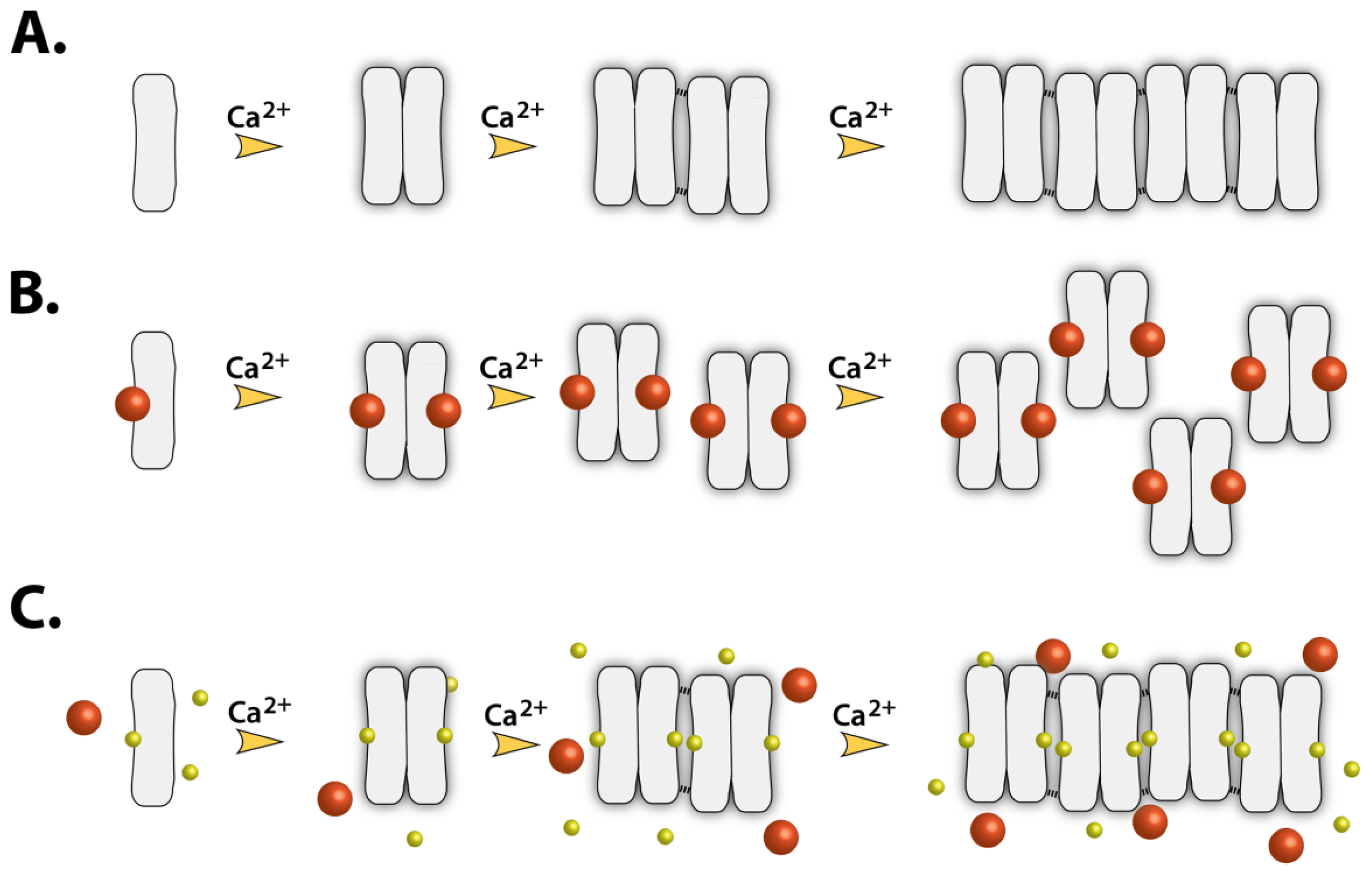
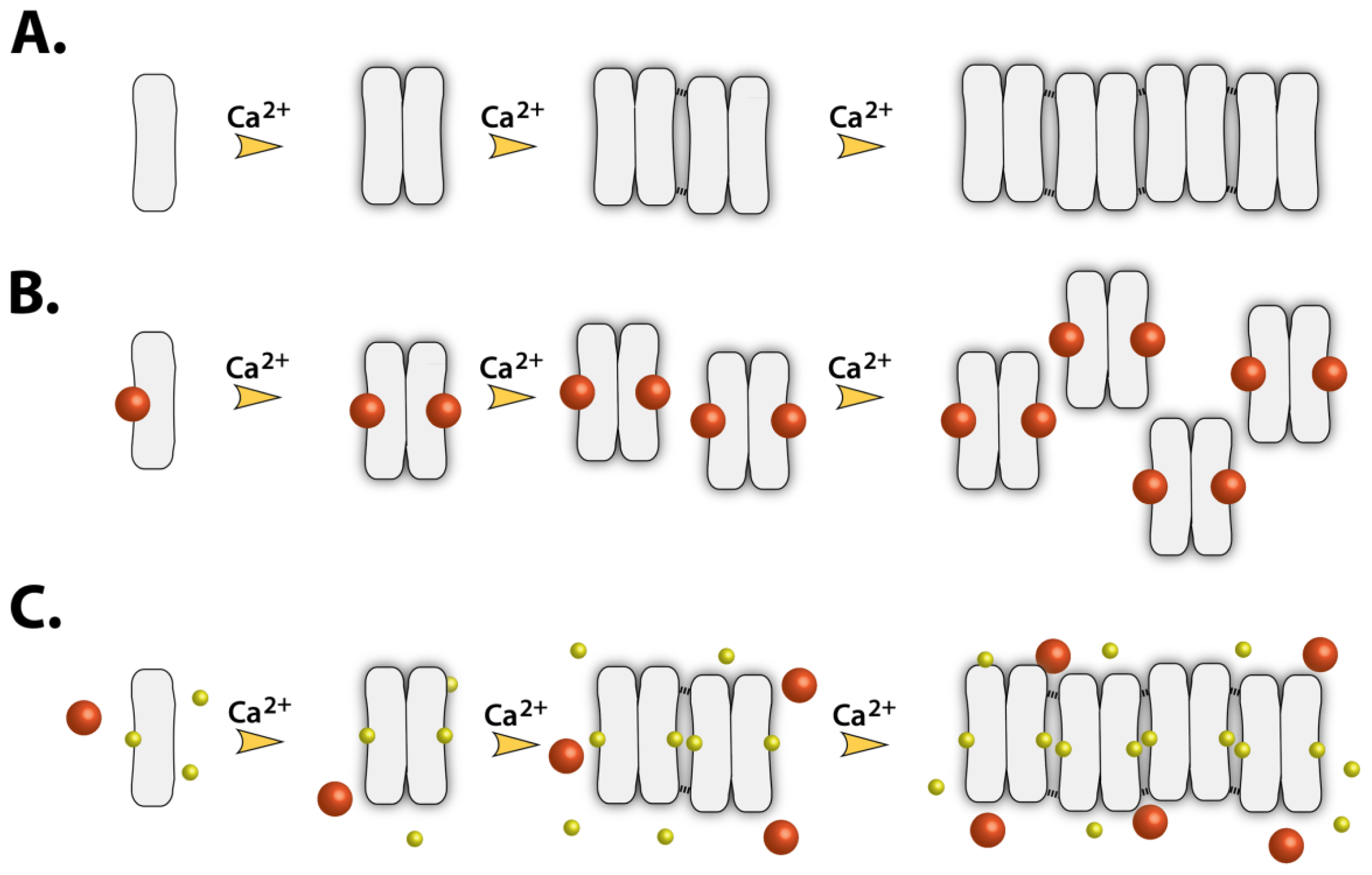
2.7. Implications of Drug Binding on the CASQ Function
3. Experimental Section
3.1. Preparation of Native Rabbit Calsequestrin (rCASQ1)
3.2. Crystallization of rCASQ1 and Structure Refinement
3.3. Iso-Density Surface Calculation
3.4. Isothermal Titration Calorimetry (ITC)
3.5. Computational Molecular Docking
4. Conclusions
Supplementary Materials
ijms-13-14326-s001.pdfSupplementary Materials
ijms-13-14326-s002.pdfAcknowledgments
- Conflicts of InterestThe authors declare no conflict of interest.
References
- Rivera, E. Liposomal anthracyclines in metastatic breast cancer: Clinical update. Oncologist 2003, 8, 3–9. [Google Scholar]
- Arora, R.; Liebo, M.; Maldonado, F. Statin-induced myopathy: The two faces of Janus. J. Cardiovasc. Pharmacol. Ther 2006, 11, 105–112. [Google Scholar]
- McClure, D.; Valuck, R.; Glanz, M.; Hokanson, J. Systematic review and meta-analysis of clinically relevant adverse events from HMG CoA reductase inhibitor trials worldwide from 1982 to present. Pharmacoepidemiol. Drug Saf 2007, 16, 132–143. [Google Scholar]
- MacLennan, D.; Abu-Abed, N.; Kang, C. Structure-Function Relationships in Ca2+ Cycling proteins. J. Mol. Cell. Cardiol 2002, 34, 897–918. [Google Scholar]
- Xu, L.; Tripathy, A.; Pasek, D.; Meissner, G. Potential for pharmacology of ryanodine receptor/calcium release channels. Ann. N. Y. Acad. Sci 1998, 16, 130–148. [Google Scholar]
- Qin, J.; Zima, A.; Porta, M.; Blatter, L.; Fill, M. Trifluoperazine: A ryanodine receptor agonist. Pflugers Arch 2009, 458, 643–651. [Google Scholar]
- Dulhunty, A.; Casarotto, M.; Beard, N. The ryanodine receptor: A pivotal Ca2+ regulatory protein and potential therapeutic drug target. Curr. Drug Targets 2011, 12, 709–723. [Google Scholar]
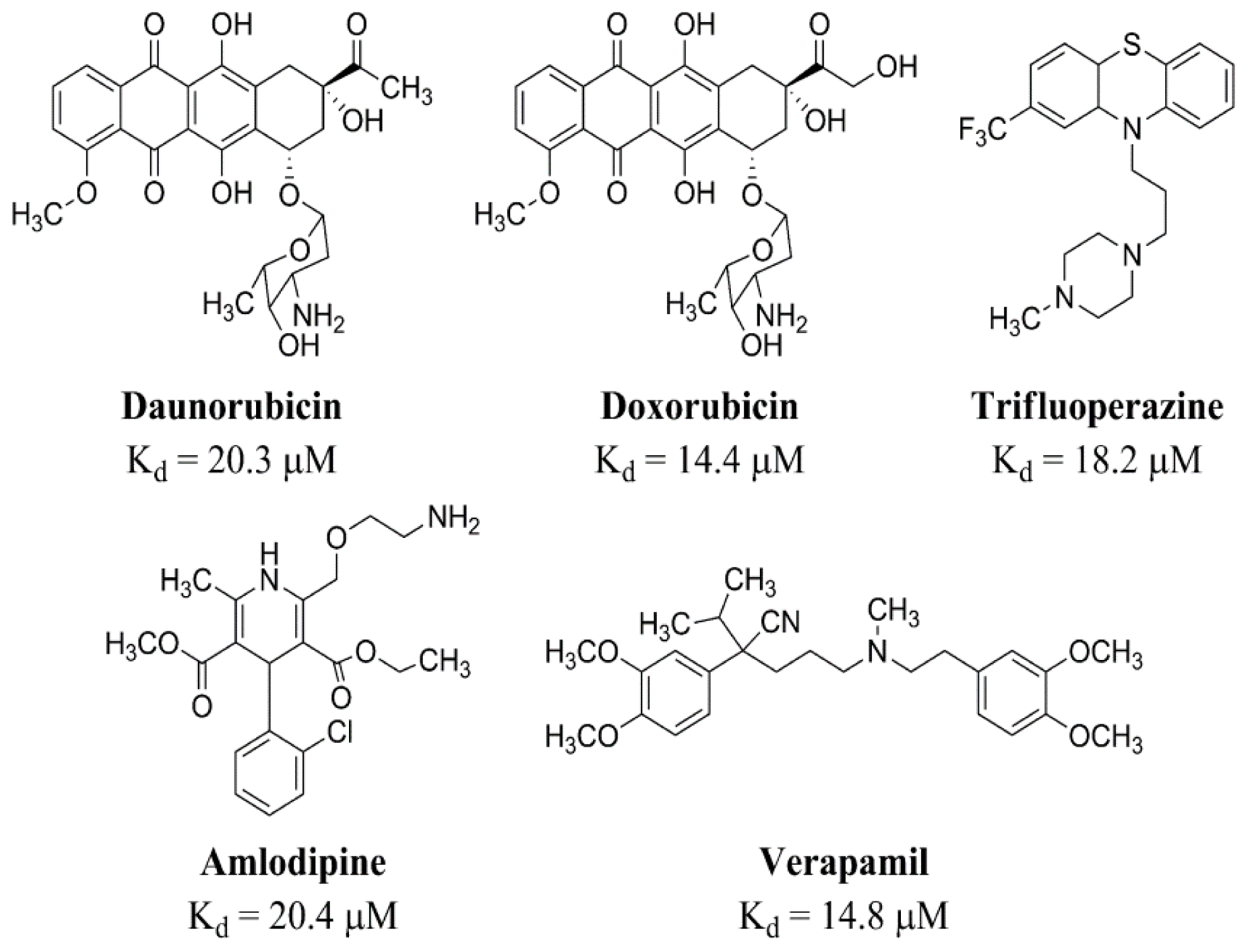
- Charlier, H., Jr; Olson, R.; Thornock, C.; Mercer, W.; Olson, D.; Broyles, T.; Muhlestein, D.; Larson, C.; Cusack, B.; Shadle, S. Investigations of calsequestrin as a target for anthracyclines: Comparison of functional effects of daunorubicin and daunorubicinol and trifluoperazine. Mol. Pharmacol. 2005, 67, 1505–1512. [Google Scholar]
- Kang, C.; Nissen, M.; Sanchez, E.; Milting, H. Potential adverse interactions of human cardiac calsequestrin. Eur. J. Pharmacol 2010, 646, 12–21. [Google Scholar]
- Kim, E.; Tam, M.; Siems, W.F.; Kang, C. Effects of drugs with muscle-related side effects and affinity for calsequestrin on the calcium regulatory function of sarcoplasmic reticulum microsomes. Mol. Pharmacol 2005, 68, 1708–1715. [Google Scholar]
- Park, I.Y.; Kim, E.J.; Park, H.; Fields, K.; Dunker, A.K.; Kang, C. Interaction between cardiac Calsequestrin and drugs with known cardiotoxicity. Mol. Pharmacol 2005, 67, 97–104. [Google Scholar]
- Park, H.; Park, I.-Y.; Kim, E.; Youn, B.; Fields, K.; Dunker, A. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: A proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem 2004, 279, 18026–18033. [Google Scholar]
- Wang, S.; Trumble, W.; Liao, H.; Wesson, C.; Dunker, A.; Kang, C. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol 1998, 5, 476–483. [Google Scholar]
- Sanchez, E.; Lewis, K.; Munske, G.; Nissen, M.; Kang, C. Glycosylation of skeletal calsequestrin, implication for its function. J. Biol. Chem 2012, 287, 3042–3050. [Google Scholar]
- Sanchez, E.; Lewis, K.; Danna, B.; Kang, C. High-capacity Ca2+-binding of human skeletal calsequestrin. J. Biol. Chem 2012, 287, 11592–11601. [Google Scholar]
- Branden, C. Relation between structure and function of proteins. Q. Rev. Biophys 1980, 13, 317–338. [Google Scholar]
- Katti, S.; LeMaster, D.; Eklund, H. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J. Mol. Biol 1990, 212, 167–184. [Google Scholar]
- MacLennan, D.; Chen, S. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J. Physiol 2009, 587, 3113–3115. [Google Scholar]
- Bast, A.; Kaiserová, H.; den Hartog, G.; Haenen, G.; van der Vijgh, W. Protectors against doxorubicin-induced cardiotoxicity: Flavonoids. Cell Biol. Toxicol 2007, 23, 39–47. [Google Scholar]
- Das, A.; Durrant, D.; Mitchell, C.; Mayton, E.; Hoke, N.; Salloum, F.; Park, M.; Quershi, I.; Lee, R.; Dent, P.; et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 18202–18207. [Google Scholar]
- Sadzuka, Y.; Mochizuki, E.; Takino, Y. Caffeine modulates the antitumor activity and toxic side effects of adriamycin. Jpn. J. Cancer Res 1993, 84, 348–353. [Google Scholar]
- Zheng, J.; Lee, H.; Bin Sattar, M.; Huang, Y.; Bian, J. Cardioprotective effects of epigallocatechin-3- gallate against doxorubicin-induced cardiomyocyte injury. Eur. J. Pharmacol 2011, 652, 82–88. [Google Scholar]
- Perola, E.; Walters, P.; Charifson, P. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. [Google Scholar]
- Altschul, S.; Madden, T.; Schäffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 2010, 38, W529–W533. [Google Scholar]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 2005, 33, W299–W302. [Google Scholar]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.; Bechor, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of Functional Regions in Proteins by Surface-Mapping of Phylogenetic Information. Bioinformatics 2003, 19, 163–164. [Google Scholar]
- Györke, S.; Stevens, S.; Terentyev, D. Cardiac calsequestrin: Quest inside the SR. J. Physiol 2009, 587, 3091–3094. [Google Scholar]
- Lee, K.; Maeng, J.; Choi, J.; Lee, Y.; Hwang, C.; Park, S.; Park, K.; Chung, B.; Lee, S.; Kim, Y.; et al. Role of Junctin Protein Interactions in Cellular Dynamics of Calsequestrin Polymer upon Calcium Perturbation. J. Biol. Chem 2012, 287, 1679–1687. [Google Scholar]
- McFarland, T.; Milstein, M.; Cala, S. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J. Mol. Cell. Cardiol 2010, 49, 556–564. [Google Scholar]
- Adams, P.; Afonine, P.; Bunkóczi, G.; Chen, V.; Davis, I.; Echols, N.; Headd, J.; Hung, L.-W.; Kapral, G.; Grosse-Kunstleve, R.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys. Rev 1988, B37, 785–789. [Google Scholar]
- Becke, A. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford, CT, UK, 2004. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5; J. Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Morris, G.; Goodsell, D.; Halliday, R.; Huey, R.; Hart, W.; Belew, R.; Olson, A. Automated molecular docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 18, 1639–1662. [Google Scholar]
- Bolton, E.; Wang, Y.; Thiessen, P.; Bryant, S. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem 2008, 4, 217–241. [Google Scholar]
- Discovery Studio Modeling Environment, Release 3.0; Accelrys Software Inc.: San Diego, CA, USA, 2007.
- Sanner, M. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model 1999, 17, 57–61. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Subra, A.K.; Nissen, M.S.; Lewis, K.M.; Muralidharan, A.K.; Sanchez, E.J.; Milting, H.; Kang, C. Molecular Mechanisms of Pharmaceutical Drug Binding into Calsequestrin. Int. J. Mol. Sci. 2012, 13, 14326-14343. https://doi.org/10.3390/ijms131114326
Subra AK, Nissen MS, Lewis KM, Muralidharan AK, Sanchez EJ, Milting H, Kang C. Molecular Mechanisms of Pharmaceutical Drug Binding into Calsequestrin. International Journal of Molecular Sciences. 2012; 13(11):14326-14343. https://doi.org/10.3390/ijms131114326
Chicago/Turabian StyleSubra, Arun K., Mark S. Nissen, Kevin M. Lewis, Ashwin K. Muralidharan, Emiliano J. Sanchez, Hendrik Milting, and ChulHee Kang. 2012. "Molecular Mechanisms of Pharmaceutical Drug Binding into Calsequestrin" International Journal of Molecular Sciences 13, no. 11: 14326-14343. https://doi.org/10.3390/ijms131114326