Roles of Nitric Oxide and Asymmetric Dimethylarginine in Pregnancy and Fetal Programming



{kind=link}
Abstract
:1. Introduction
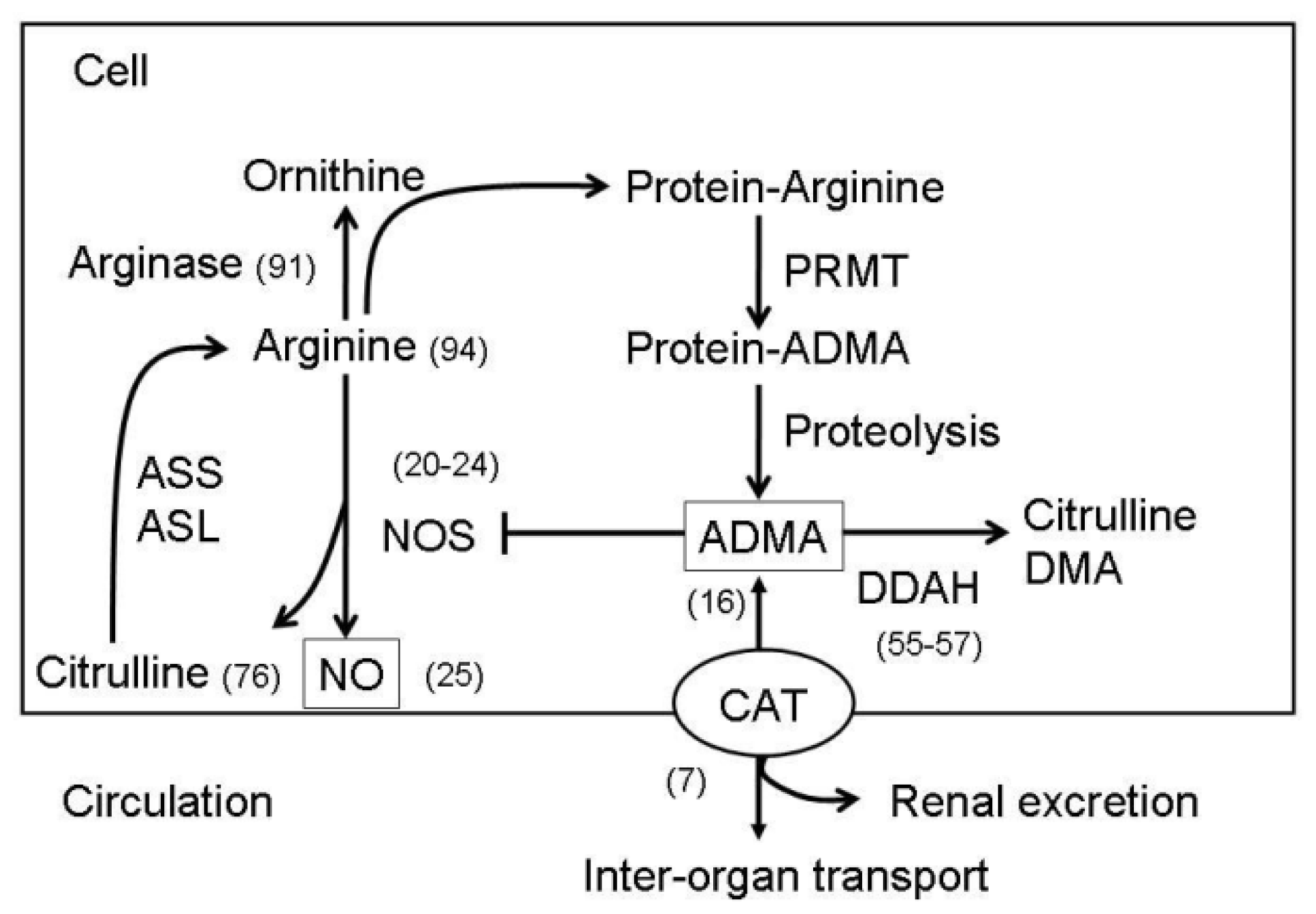
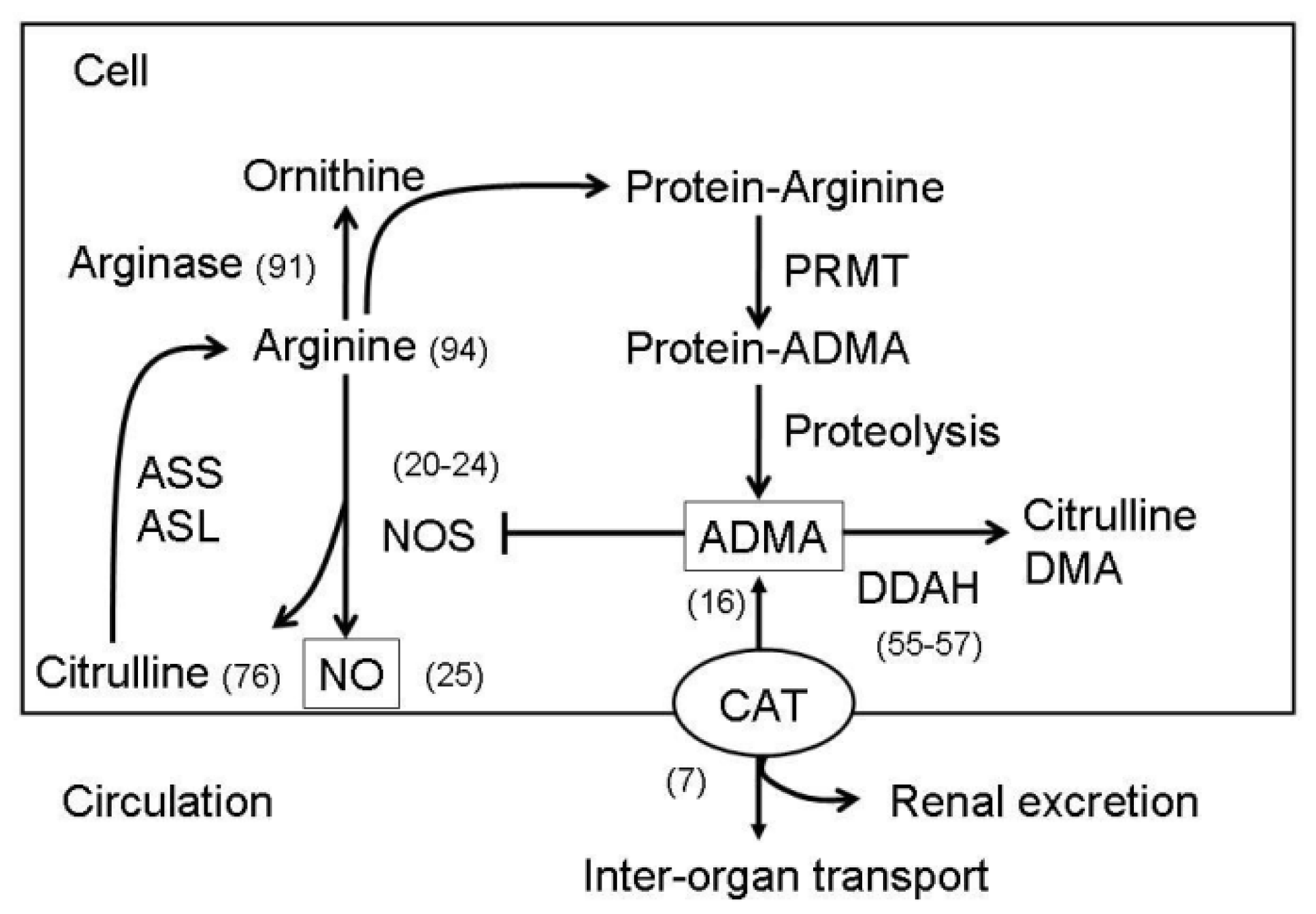
2. Metabolisms of NO and ADMA
3. Roles of NO and ADMA in Normal Pregnancies
4. Roles of NO and ADMA in Compromised Pregnancies
4.1. Preeclampsia
4.2. Gestational Diabetes Mellitus
4.3. Prenatal Malnutrition
4.4. Prenatal Glucocorticoid and Stress Exposure
5. Placental Insufficiency and Developmental Programming
5.1. Role of the Placental Nitrergic System in Epigenetic Fetal Programming
5.2. Manipulations of the ADMA-NO Pathway to Prevent Compromised Pregnancies and Fetal Programming
6. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| CAT | Cationic amino acid transporters |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| FGF | Fibroblast growth factor |
| GDM | Gestational diabetes mellitus |
| HDAC | Histone deacetylase |
| HUVEC | Human umbilical cord endothelial cells |
| IUGR | Intrauterine growth restriction |
| MMP | Matrix metalloproteinases |
| NO | Nitric oxide |
| NOS1 or nNOS | Nitric oxide synthase 1 |
| NOS2 or iNOS | Nitric oxide synthase 2 |
| NOS3 or eNOS | Nitric oxide synthase 3 |
| PRMT | Protein arginine methyltransferases |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
References
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol 2006, 572, 25–30. [Google Scholar]
- Reynolds, L.P.; Borowicz, P.P.; Caton, J.S.; Vonnahme, K.A.; Luther, J.S.; Buchanan, D.S.; Hafez, S.A.; Grazul-Bilska, A.T.; Redmer, D.A. Uteroplacental vascular development and placental function: an update. Int. J. Dev. Biol 2010, 54, 355–366. [Google Scholar]
- Krause, B.J.; Hanson, M.A.; Casanello, P. Role of nitric oxide in placental vascular development and function. Placenta 2011, 32, 797–805. [Google Scholar]
- Godfrey, M. The role of the placenta in fetal programming—A review. Placenta 2002, 23, S20–S27. [Google Scholar]
- Reynolds, L.P.; Caton, J.S.; Redmer, D.A.; Grazul-Bilska, A.T.; Vonnahme, K.A.; Borowicz, P.P.; Luther, J.S.; Wallace, J.M.; Wu, G.; Spencer, T.E. Evidence for altered placental blood flow and vascularity in compromised pregnancies. J. Physiol 2006, 572, 51–58. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1023–1030. [Google Scholar]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res 2009, 60, 448–460. [Google Scholar]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev 2010, 6, 82–90. [Google Scholar]
- Fickling, S.A.; Williams, D.; Vallance, P.; Nussey, S.S.; Whitley, G.S. Plasma of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and pre-eclampsia. Lancet 1993, 342, 242–243. [Google Scholar]
- Holden, D.P.; Fickling, S.A.; Whitley, G.S.; Nussey, S.S. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am. J. Obstet. Gynecol 1998, 178, 551–556. [Google Scholar]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. (Lond.) 2007, 113, 1–13. [Google Scholar]
- Reynolds, L.P.; Caton, J.S. Role of the pre- and post-natal environment in developmental programming of health and productivity. Mol. Cell Endocrinol 2012, 354, 54–59. [Google Scholar]
- Rugg-Gunn, P.J. Epigenetic features of the mouse trophoblast. Reprod. Biomed. Online 2012, 25, 21–30. [Google Scholar]
- Illi, B.; Colussi, C.; Grasselli, A.; Farsetti, A.; Capogrossi, M.C.; Gaetano, C. NO sparks off chromatin: Tales of a multifaceted epigenetic regulator. Pharmacol. Ther 2009, 123, 344–352. [Google Scholar]
- Nott, A.; Riccio, A. Nitric oxide-mediated epigenetic mechanisms in developing neurons. Cell Cycle 2009, 8, 725–730. [Google Scholar]
- Tain, Y.L.; Huang, L.T. Asymmetric dimethylarginine: Clinical applications in pediatric medicine. J. Formos. Med. Assoc 2011, 110, 70–77. [Google Scholar]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Boger, S.M. Plasma asymmetric dimethylarginine concentration during the perinatal period. Neonatology 2007, 92, 8–13. [Google Scholar]
- Lucke, T.; Kanzelmeyer, N.; Kemper, M.J.; Tsikas, D.; Das, A.M. Developmental changes in the l-arginine/nitric oxide pathway from infancy to adulthood: Plasma asymmetric dimethylarginine levels decrease with age. Clin. Chem. Lab. Med 2007, 45, 1525–1530. [Google Scholar]
- Horowitz, J.D.; Heresztyn, T. An overview of plasma concentrations of asymmetric dimethylarginine (ADMA) in health and disease and in clinical studies: Methodological considerations. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2007, 851, 42–50. [Google Scholar]
- Rossmanith, W.G.; Hoffmeister, U.; Wolfahrt, S.; Kleine, B.; McLean, M.; Jacobs, R.A.; Grossman, A.B. Expression and functional analysis of endothelial nitric oxide synthase (eNOS) in human placenta. Mol. Hum. Reprod 1999, 5, 487–494. [Google Scholar]
- Ariel, I.; Hochberg, A.; Shochina, M. Endothelial nitric oxide synthase immunoreactivity in early gestation and in trophoblastic disease. J. Clin. Pathol 1998, 51, 427–431. [Google Scholar]
- Suzuki, T.; Ikeda, Y.; Yoshikawa, H.; Tanaka, K.; Morita, H.; Yamamoto, M.; Takizawa, T. Gestational changes in production of NO and expression of NOS mRNA isoforms in the rat placenta. J. Vet. Med. Sci 2009, 71, 495–498. [Google Scholar]
- Suzuki, T.; Mori, C.; Yoshikawa, H.; Miyazaki, Y.; Kansaku, N.; Tanaka, K.; Morita, H.; Takizawa, T. Changes in nitric oxide production levels and expression of nitric oxide synthase isoforms in the rat uterus during pregnancy. Biosci. Biotechnol. Biochem 2009, 73, 2163–2166. [Google Scholar]
- Purcell, T.L.; Given, R.; Chwalisz, K.; Garfield, R.E. Nitric oxide synthase distribution during implantation in the mouse. Mol. Hum. Reprod 1999, 5, 467–475. [Google Scholar]
- Seligman, S.P.; Buyon, J.P.; Clancy, R.M.; Young, B.K.; Abramson, S.B. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol 1994, 171, 944–948. [Google Scholar]
- Xiao, D.; Pearce, W.; Zhang, L. Pregnancy enhances endothelium-dependent relaxation of ovine uterine artery: Role of NO and intracellular Ca2+. Am. J. Physiol. Heart Circ. Physiol 2001, 281, H183–H190. [Google Scholar]
- Kaufmann, P.; Mayhew, T.M.; Charnock-Jones, D.S. Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 2004, 25, 114–126. [Google Scholar]
- Demir, R.; Kayisli, U.A.; Cayli, S.; Huppertz, B. Sequential steps during vasculogenesis and angiogenesis in the very early human placenta. Placenta 2006, 27, 535–539. [Google Scholar]
- Shizukuda, Y.; Tang, S.; Yokota, R.; Ware, J.A. Vascular endothelial growth factor-induced endothelial cell migration and proliferation depend on a nitric oxide-mediated decrease in protein kinase C activity. Circ. Res 1999, 85, 247–256. [Google Scholar]
- Nath, A.K.; Enciso, J.; Kuniyasu, M.; Hao, X.Y.; Madri, J.A.; Pinter, E. Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development 2004, 131, 2485–2496. [Google Scholar]
- Frank, S.; Stallmeyer, B.; Kampfer, H.; Schaffner, C.; Pfeilschifter, J. Differential regulation of vascular endothelial growth factor and its receptor fms-like-tyrosine kinase is mediated by nitric oxide in rat renal mesangial cells. Biochem. J 1999, 338, 367–374. [Google Scholar]
- Han, R.N.; Stewart, D.J. Defective lung vascular development in endothelial nitric oxide synthasedeficient mice. Trends Cardiovasc. Med 2006, 16, 29–34. [Google Scholar]
- Zhao, X.; Lu, X.; Feng, Q. Deficiency in endothelial nitric oxide synthase impairs myocardial angiogenesis. Am. J. Physiol. Heart Circ. Physiol 2002, 283, H2371–H2378. [Google Scholar]
- Kon, K.; Fujii, S.; Kosaka, H.; Fujiwara, T. Nitric oxide synthase inhibition by N(G)-nitro-l-arginine methyl ester retards vascular sprouting in angiogenesis. Microvasc. Res 2003, 65, 2–8. [Google Scholar]
- Dulak, A.; Jozkowicz, J. Regulation of vascular endothelial growth factor synthesis by nitric oxide: Facts and controversies. Antioxid. Redox. Signal 2003, 5, 123–132. [Google Scholar]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. Birth by cesarean section is associated with elevated neonatal plasma levels of dimethylarginines. Pediatr. Int 2012, 54, 476–479. [Google Scholar]
- Reynolds, L.P.; Borowicz, P.P.; Vonnahme, K.A.; Johnson, M.L.; Grazul-Bilska, A.T.; Wallace, J.M.; Caton, J.S.; Redmer, D.A. Animal models of placental angiogenesis. Placenta 2005, 26, 689–708. [Google Scholar]
- Myatt, L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta 2010, 24, S66–S69. [Google Scholar]
- Vonnahme, K.A.; Wilson, M.E.; Li, Y.; Rupnow, H.L.; Phernetton, T.M.; Ford, S.P.; Magness, R.R. Circulating levels of nitric oxide and vascular endothelial growth factor throughout ovine pregnancy. J. Physiol 2005, 565, 101–109. [Google Scholar]
- Reynolds, L.P.; Redmer, D.A. Angiogenesis in the placenta. Biol. Reprod 2001, 64, 1033–1040. [Google Scholar]
- Bird, I.M.; Zhang, L.; Magness, R.R. Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 284, R245–R258. [Google Scholar]
- Khullar, S.; Greenwood, S.L.; McCord, N.; Glazier, J.D.; Ayuk, P.T. Nitric oxide and superoxide impair human placental amino acid uptake and increase Na+ permeability: Implications for fetal growth. Free Radic. Biol. Med 2004, 36, 271–277. [Google Scholar]
- Kossenjans, W.; Eis, A.; Sahay, R.; Brockman, D.; Myatt, L. Role of peroxynitrite in altered fetal-placental vascular reactivity in diabetes or preeclampsia. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1311–H1139. [Google Scholar]
- Roberts, V.H.; Webster, R.P.; Brockman, D.E.; Pitzer, B.A.; Myatt, L. Post-translational modifications of the P2X(4) purinergic receptor subtype in the human placenta are altered in preeclampsia. Placenta 2007, 28, 270–277. [Google Scholar]
- Webster, R.P.; Macha, S.; Brockman, D.; Myatt, L. Peroxynitrite treatment in vitro disables catalytic activity of recombinant p38 MAPK. Proteomics 2006, 6, 4838–4844. [Google Scholar]
- Savvidou, M.D.; Hingorani, A.D.; Tsikas, D.; Frolich, J.C.; Vallance, P.; Nicolaides, K.H. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet 2003, 361, 1511–1517. [Google Scholar]
- McCarthy, A.L.; Woolfson, R.G.; Evans, B.J.; Davies, D.R.; Raju, S.K.; Poston, L. Functional characteristics of small placental arteries. Am. J. Obstet. Gynecol 1994, 170, 945–951. [Google Scholar]
- Chang, J.K.; Roman, C.; Heymann, M.A. Effect of endothelium-derived relaxing factor inhibition on the umbilical-placental circulation in fetal lambs in utero. Am. J. Obstet. Gynecol 1992, 166, 727–734. [Google Scholar]
- Suzuki, T.; Nagamatsu, C.; Kushima, T.; Miyakoshi, R.; Tanaka, K.; Morita, H.; Sakaue, M.; Takizawa, T. Apoptosis caused by an inhibitor of NO production in the decidua of rat from mid-gestation. Exp. Biol. Med. (Maywood) 2010, 235, 455–462. [Google Scholar]
- Lowe, D.T. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide 2000, 4, 441–458. [Google Scholar]
- Baylis, C.; Beinder, E.; Suto, T.; August, P. Recent insights into the roles of nitric oxide and renin-angiotensin in the pathophysiology of preeclamptic pregnancy. Semin. Nephrol 1998, 18, 208–230. [Google Scholar]
- Noris, M.; Todeschini, M.; Cassis, P.; Pasta, F.; Cappellini, A.; Bonazzola, S.; Macconi, D.; Maucci, R.; Porrati, F.; Benigni, A.; et al. l-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension 2004, 43, 614–622. [Google Scholar]
- Myatt, L.; Eis, A.L.; Brockman, D.E.; Kossenjans, W.; Greer, I.A.; Lyall, F. Endothelial nitric oxide in placental villous tissue from normal, pre-eclamptic and intrauterine restricted pregnancies. Hum. Reprod 1997, 12, 714–718. [Google Scholar]
- Leiper, J.; MacAllister, R.; Whitley, G.; Santa Maria, J.; Chubb, A.; Charles, I.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology to microbial arginine deiminases. Biochem. J 1999, 343, 209–214. [Google Scholar]
- Anderssohn, M.; Maass, L.M.; Diemert, A.; Lüneburg, N.; Atzler, D.; Hecher, K.; Böger, R.H. Severely decreased activity of placental dimethylarginine dimethylaminohydrolase in pre-eclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol 2012, 161, 152–156. [Google Scholar]
- Akbar, F.; Heinonen, S.; Pirskanen, M.; Uimari, P.; Tuomainen, T.P.; Salonen, J.T. Haplotypic association of DDAH1 with susceptibility to pre-eclampsia. Mol. Hum. Reprod 2005, 11, 73–77. [Google Scholar]
- Kim, Y.J.; Park, B.H.; Park, H.; Jung, S.C.; Pang, M.G.; Ryu, H.M.; Lee, K.S.; Eom, S.M.; Park, H.Y. No association of the genetic polymorphisms of endothelial nitric oxide synthase, dimethylarginine dimethylaminohydrolase, and vascular endothelial growth factor with preeclampsia in Korean populations. Twin Res. Hum. Genet 2008, 11, 77–83. [Google Scholar]
- Sobrevia, L.; Mann, G.E. Dysfunction of the endothelial nitric oxide signalling pathway in diabetes and hyperglycaemia. Exp. Physiol 1997, 82, 423–452. [Google Scholar]
- Kucuk, M.; Doymaz, F. Placental weight and placental weight-to-birth weight ratio are increased in diet- and exercise-treated gestational diabetes mellitus subjects but not in subjects with one abnormal value on 100-g oral glucose tolerance test. J. Diabetes Complicat 2009, 23, 25–31. [Google Scholar]
- Daskalakis, G.; Marinopoulos, S.; Krielesi, V.; Papapanagiotou, A.; Papantoniou, N.; Mesogitis, S.; Antsaklis, A. Placental pathology in women with gestational diabetes. Acta. Obstet. Gynecol. Scand 2008, 87, 403–407. [Google Scholar]
- Sobrevia, L.; Abarzúa, F.; Nien, J.K.; Salomón, C.; Westermeier, F.; Puebla, C.; Cifuentes, F.; Guzmán-Gutiérrez, E.; Leiva, A.; Casanello, P. Review: Differential placental macrovascular and microvascular endothelial dysfunction in gestational diabetes. Placenta 2011, 25, S159–S164. [Google Scholar]
- Figueroa, R.; Martinez, E.; Fayngersh, R.P.; Tejani, N.; Mohazzab, H.K.M.; Wolin, M.S. Alterations in relaxation to lactate and H2O2 in human placental vessels from gestational diabetic pregnancies. Am. J. Physiol 2000, 278, H706–H713. [Google Scholar]
- Farías, M.; Puebla, C.; Westermeier, F.; Jo, M.J.; Pastor-Anglada, M.; Casanello, P.; Sobrevia, L. Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBPα in human umbilical vein endothelial cells from gestational diabetes. Cardiovas. Res 2010, 86, 45–54. [Google Scholar]
- Vásquez, G.; Sanhueza, F.; Vásquez, R.; González, M.; San Martín, R.; Casanello, P.; Sobrevia, L. Role of adenosine transport in gestational diabetes-induced l-arginine transport and nitric oxide synthesis in human umbilical vein endothelium. J. Physiol 2004, 560, 111–122. [Google Scholar]
- Stojanovic, N.; Lewandowski, K.; Salata, I.; Bienkiewicz, M.; Tuck, S.; Prelevic, G.; Press, M. Serum levels of matrix metalloproteinases MMP-2 and MMP-9 and their inhibitors in women with glucose intolerance in pregnancy and normal controls. Gynecol. Endocrinol 2010, 26, 201–207. [Google Scholar]
- Novaro, V.; Colman-Lerner, A.; Ortega, F.V.; Jawerbaum, A.; Paz, D.; Lo Nostro, F.; Pustovrh, C.; Gimeno, M.F.; González, E. Regulation of metalloproteinases by nitric oxide in human trophoblast cells in culture. Reprod. Fertil. Dev 2001, 13, 411–420. [Google Scholar]
- Akturk, M.; Altinova, A.; Mert, I.; Dincel, A.; Sargin, A.; Buyukkagnici, U.; Arslan, M.; Danisman, N. Asymmetric dimethylarginine concentrations are elevated in women with gestational diabetes. Endocrine 2010, 38, 134–141. [Google Scholar]
- Wu, G.; Pond, W.G.; Flynn, S.P.; Ott, T.L.; Bazer, F.W. Maternal dietary protein deficiency decreases nitric oxide synthase and ornithine decarboxylase activities in placenta and endometrium of pigs during early gestation. J. Nutr 1998, 128, 2395–2402. [Google Scholar]
- Kwon, H.; Ford, S.P.; Bazer, F.W.; Spencer, T.E.; Nthanielsz, P.W.; Nijland, M.J.; Hess, B.W.; Wu, G. Maternal undernutrition reduces concentrations of amino acids and polyamines in ovine fetal plasma and fluids. Biol. Reprod 2004, 71, 901–908. [Google Scholar]
- Ozaki, T.; Hawkins, P.; Nishina, H.; Steyn, C.; Poston, L.; Hanson, M.A. Effects of undernutrition in early pregnancy on systemic small artery function in late-gestation fetal sheep. Am. J. Obstet. Gynecol 2000, 183, 1301–1307. [Google Scholar]
- Redmer, D.A.; Wallace, J.M.; Reynolds, L.P. Effect of nutrient intake during pregnancy on fetal and placental growth and vascular development. Domest. Anim. Endocrinol 2004, 27, 199–217. [Google Scholar]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol 1998, 274, H1054–H1058. [Google Scholar]
- Babaei, S.; Teichert-Kuliszewska, K.; Monge, J.C.; Mohamed, F.; Bendeck, M.P.; Stewart, D.J. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ. Res 1998, 18, 1007–1015. [Google Scholar]
- Rutland, C.S.; Latunde-Dada, A.O.; Thorpe, A.; Plant, R.; Langley-Evans, S.; Leach, L. Effect of gestational nutrition on vascular integrity in the murine placenta. Placenta 2007, 28, 734–742. [Google Scholar]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar]
- Polyakov, A.; Cohen, S.; Baum, M.; Trickey, D.; Jolley, D.; Wallace, E.M. Patterns of antenatal corticosteroid prescribing 1998–2004. Aust. N. Z. J. Obstet. Gynaecol 2007, 47, 42–45. [Google Scholar]
- Huang, L.T. The link between perinatal glucocorticoids exposure and psychiatric disorders. Pediatr. Res 2011, 69, 19R–25R. [Google Scholar]
- Lui, C.C.; Wang, J.Y.; Tain, Y.L.; Chen, Y.C.; Chang, K.A.; Lai, M.C.; Huang, L.T. Prenatal stress in rat causes long-term spatial memory deficit and hippocampus MRI abnormality: Differential effects of postweaning enriched environment. Neurochem. Int 2011, 58, 434–441. [Google Scholar]
- Hewitt, D.P.; Mark, P.J.; Waddell, B.J. Glucocorticoids prevent the normal increase in placental vascular endothelial growth factor expression and placental vascularity during late pregnancy in the rat. Endocrinology 2006, 147, 5566–5574. [Google Scholar]
- Parenti, A.; Morbidelli, L.; Cui, X.L.; Douglas, J.G.; Hood, J.D.; Granger, H.J.; Ledda, F.; Ziche, M. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J. Biol. Chem 1998, 273, 4220–4226. [Google Scholar]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar]
- Bussolati, B.; Dunk, C.; Grohman, M.; Kontos, C.D.; Mason, J.; Ahmed, A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am. J. Pathol 2001, 159, 993–1008. [Google Scholar]
- Kroll, J.; Waltenberger, J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem. Biophys. Res. Commun 1998, 252, 743–746. [Google Scholar]
- Ribatti, D.; Nico, B.; Crivellato, E. Morphological and molecular aspects of physiological vascular morphogenesis. Angiogenesis 2009, 12, 101–111. [Google Scholar]
- Rössig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Förstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res 2002, 91, 837–844. [Google Scholar]
- Zeng, L.; Xiao, Q.; Margariti, A.; Zhang, Z.; Zampetaki, A.; Patel, S.; Capogrossi, M.C.; Hu, Y.; Xu, Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell Biol 2006, 174, 1059–1069. [Google Scholar]
- Kuhn, P.; Xu, W. Protein arginine methyltransferases: Nuclear receptor coregulators and beyond. Prog. Mol. Biol. Transl. Sci 2009, 87, 299–342. [Google Scholar]
- Chen, W.; Cao, M.; Yang, Y.; Nagahama, Y.; Zhao, H. Expression pattern of prmt5 in adult fish and embryos of medaka, Oryzias latipes. Fish Physiol. Biochem 2009, 35, 325–332. [Google Scholar]
- Tain, Y.L.; Huang, L.T.; Lin, I.C.; Lau, Y.T.; Lin, C.Y. Melatonin prevents hypertension and increased asymmetric dimethylarginine in young spontaneous hypertensive rats. J. Pineal Res 2010, 49, 390–398. [Google Scholar]
- Piecha, G.; Koleganova, N.; Ritz, E.; Müller, A.; Fedorova, O.V.; Bagrov, A.Y.; Lutz, D.; Schirmacher, P.; Gross-Weissmann, M.L. High salt intake causes adverse fetal programming-vascular effects beyond blood pressure. Nephrol. Dial. Transplant 2012, 27, 3464–3476. [Google Scholar]
- Tain, Y.L.; Freshour, G.; Dikalova, A.; Griendling, K.; Baylis, C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am. J. Physiol. Renal Physiol 2007, 292, F1404–F1410. [Google Scholar]
- Tain, Y.L.; Kao, Y.H.; Hsieh, C.S.; Chen, C.C.; Sheen, J.M.; Lin, I.C.; Huang, L.T. Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Radic. Biol. Med 2010, 49, 1088–1098. [Google Scholar]
- Germain, A.M.; Valdés, G.; Romanik, M.C.; Reyes, M.S. Evidence supporting a beneficial role for long-term l-arginine supplementation in high-risk pregnancies. Hypertension 2004, 44, e1. [Google Scholar]
- Cynober, L.; Moinard, C.; De Bandt, J.P. The 2009 ESPEN Sir David Cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr 2010, 29, 545–551. [Google Scholar]
- Prieto, C.P.; Krause, B.J.; Quezada, C.; San Martin, R.; Sobrevia, L.; Casanello, P. Hypoxia-reduced nitric oxide synthase activity is partially explained by higher arginase-2 activity and cellular redistribution in human umbilical vein endothelium. Placenta 2011, 32, 932–940. [Google Scholar]
- Miller, S.L.; Wallace, E.M.; Walker, D.W. Antioxidant Therapies: A potential role in perinatal medicine. Neuroendocrinology 2012, 96, 13–23. [Google Scholar]
- Dennery, P.A. Oxidative stress in development: nature or nurture? Free Radic. Biol. Med 2010, 49, 1147–1151. [Google Scholar]
- Rumbold, A.; Duley, L.; Crowther, C.A.; Haslam, R.R. Antioxidants for preventing pre-eclampsia. Cochrane Database Syst. Rev. 2008, 1. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, L.-T.; Hsieh, C.-S.; Chang, K.-A.; Tain, Y.-L. Roles of Nitric Oxide and Asymmetric Dimethylarginine in Pregnancy and Fetal Programming. Int. J. Mol. Sci. 2012, 13, 14606-14622. https://doi.org/10.3390/ijms131114606
Huang L-T, Hsieh C-S, Chang K-A, Tain Y-L. Roles of Nitric Oxide and Asymmetric Dimethylarginine in Pregnancy and Fetal Programming. International Journal of Molecular Sciences. 2012; 13(11):14606-14622. https://doi.org/10.3390/ijms131114606
Chicago/Turabian StyleHuang, Li-Tung, Chih-Sung Hsieh, Kow-Aung Chang, and You-Lin Tain. 2012. "Roles of Nitric Oxide and Asymmetric Dimethylarginine in Pregnancy and Fetal Programming" International Journal of Molecular Sciences 13, no. 11: 14606-14622. https://doi.org/10.3390/ijms131114606