Nanosecond T-Jump Experiment in Poly(glutamic acid): A Circular Dichroism Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
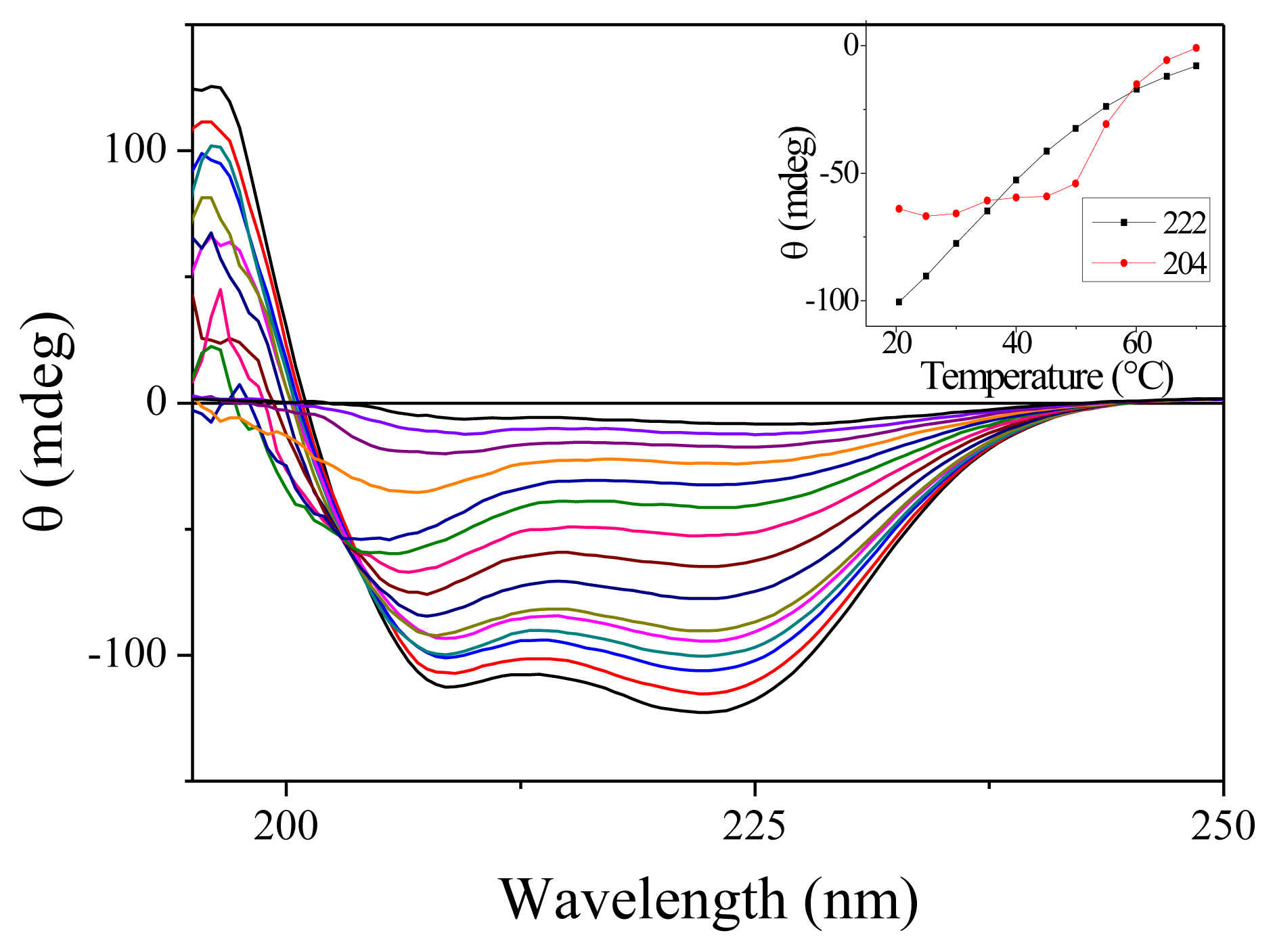
2.1. Steady-State Spectra
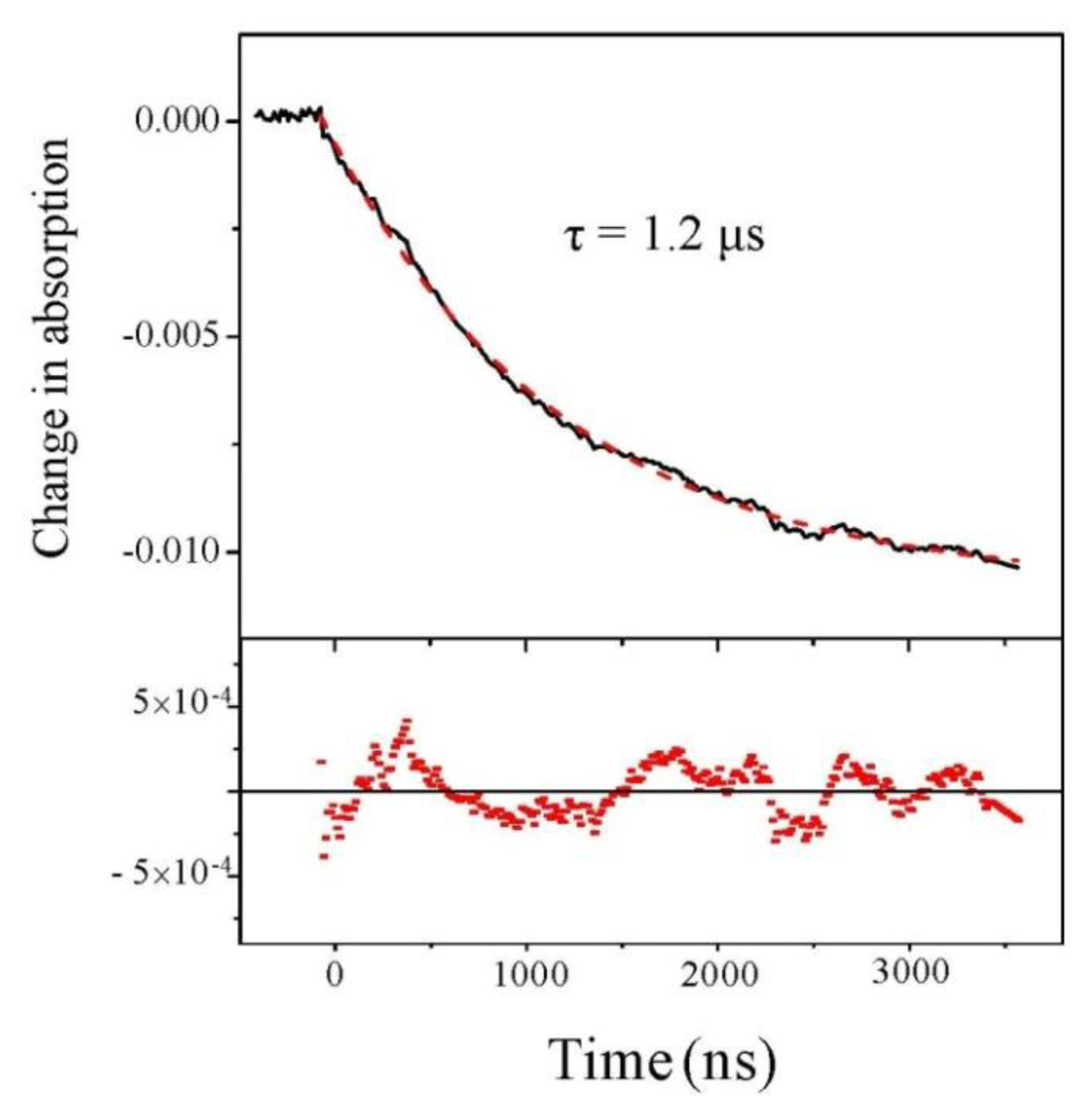
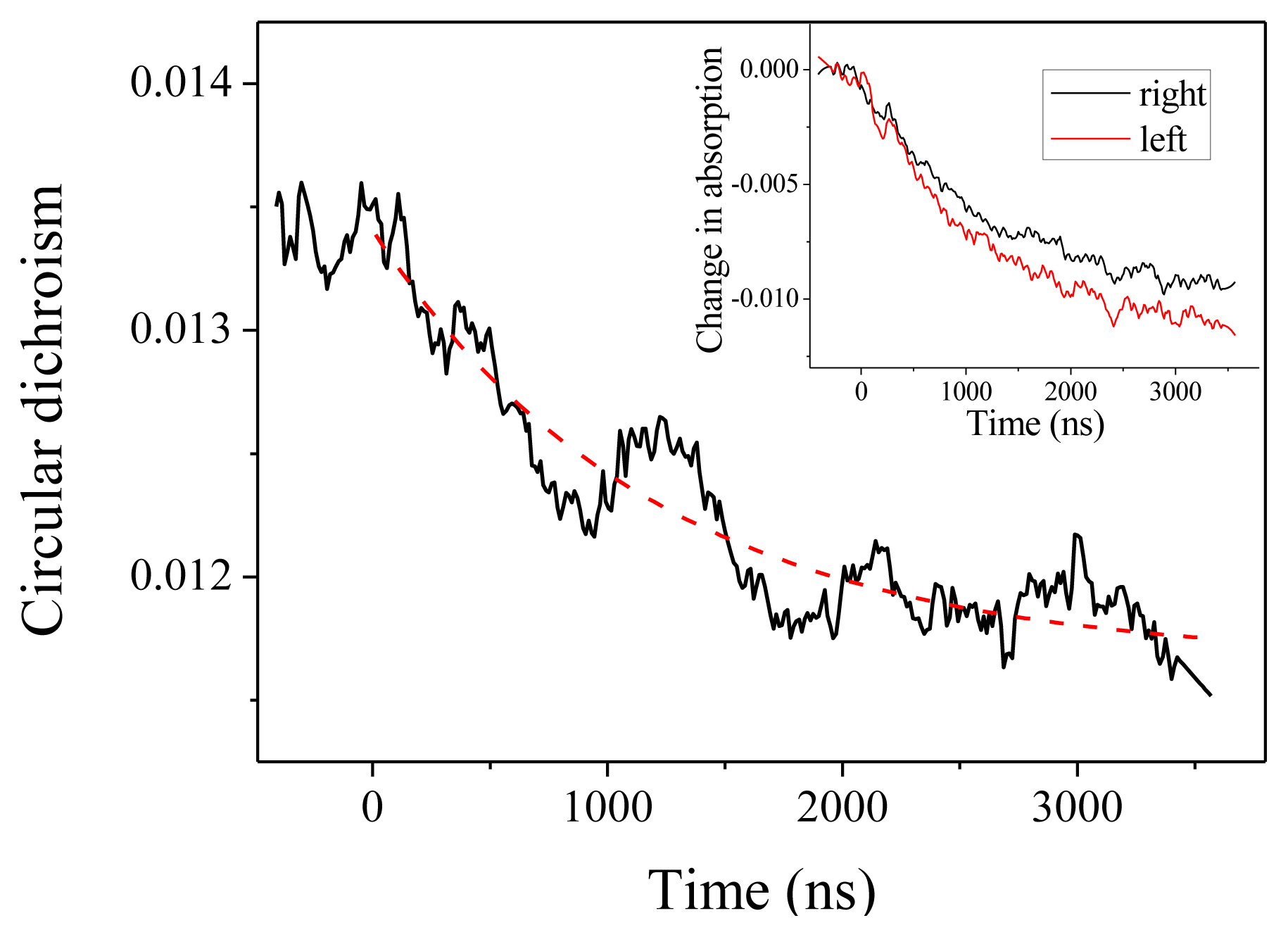
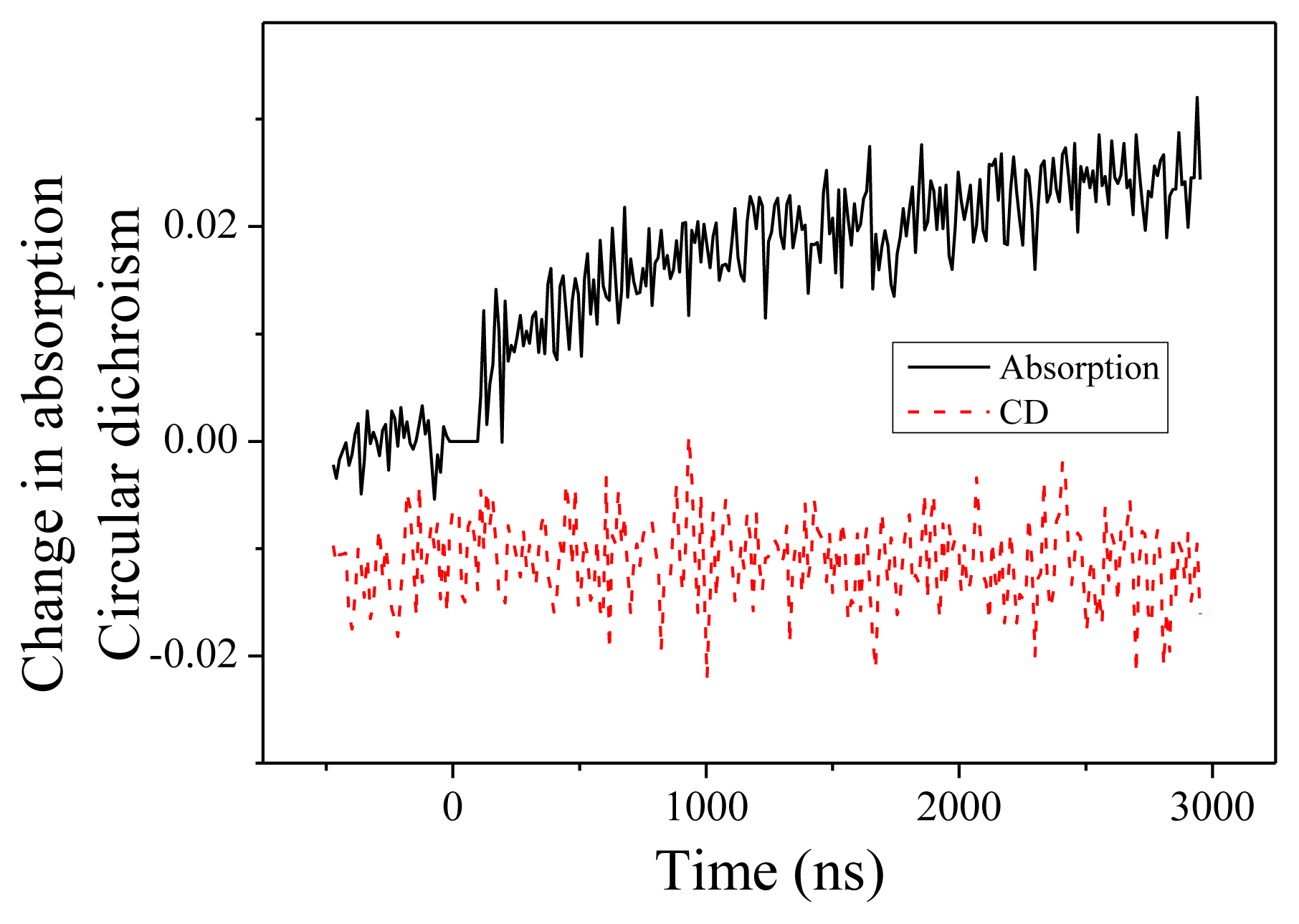
2.2. Experiments at 220 and 204 nm
2.4. Discussion
3. Experimental Section
3.1. Sample
3.2. Steady-State CD Measurements
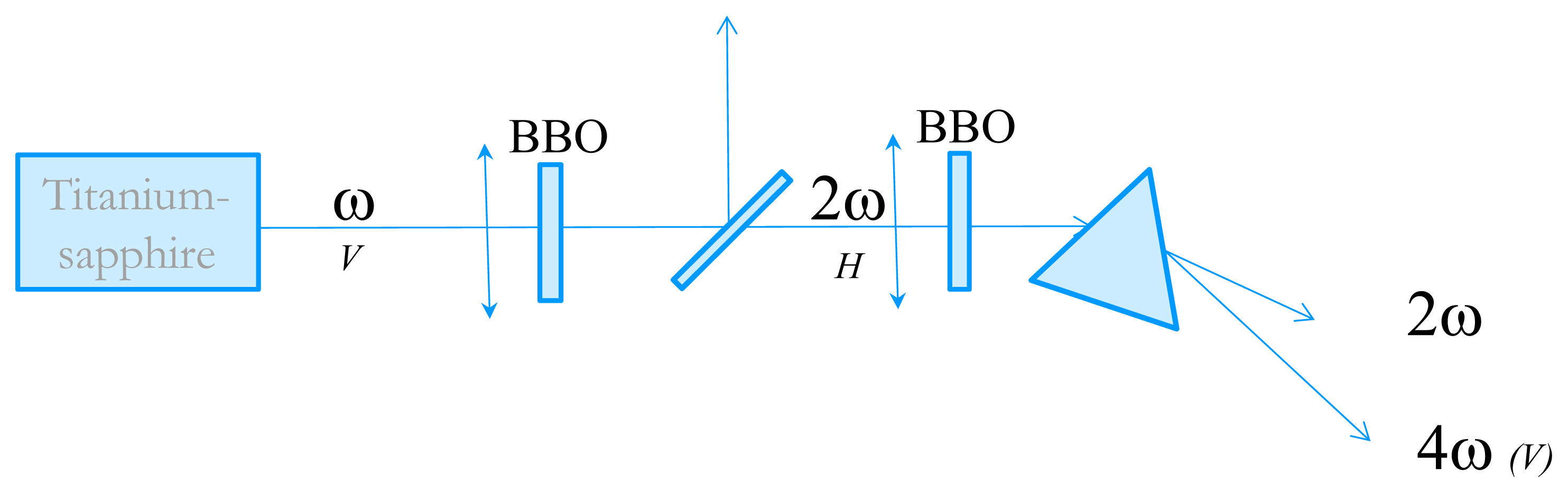
3.3. T-Jump Measurements
4. Conclusions
Acknowledgments
References
- Fersht, A.R. From the first protein structures to our current knowledge of protein folding: Delights and scepticisms. Nat. Rev. Mol. Cell Biol 2008, 9, 650–654. [Google Scholar]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The protein folding problem. Annu. Rev. Biophys 2008, 37, 289–316. [Google Scholar]
- Krieger, F.; Fierz, B.; Bieri, O.; Drewello, M.; Kiefhaber, T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J. Mol. Biol 2003, 332, 265–274. [Google Scholar]
- Eaton, W.A.; Muñoz, V.; Thompson, P.A.; Henry, E.R.; Hofrichter, J. Kinetics and dynamics of loops, a-helices, b-hairpins and fast-folding proteins. Acc. Chem. Res 1998, 31, 745–753. [Google Scholar]
- Marquese, S.; Robbins, V.H.; Baldwin, R.L. Unusually stable helix formation in short alanine-based peptides. Biochemistry 1989, 86, 5286–5290. [Google Scholar]
- Kimura, T.; Takahashi, S.; Akiyama, S.; Uzawa, T.; Ishimori, K.; Morishima, I. Direct observation of the multistep helix formation of poly-L-glutamic acid. J. Am. Chem. Soc 2002, 124, 11596–11597. [Google Scholar]
- Callender, R.H.; Dyer, R.B. Probing protein dynamics using temperature jump relaxation spectroscopy. Curr. Opin. Struct. Biol 2002, 12, 628–633. [Google Scholar]
- Pozo Ramajo, A.; Petty, S.A.; Starzyk, A.; Decatur, A.M.; Volk, M. The α-helix folds more rapidly at the C-terminus than at the N-terminus. J. Am. Chem. Soc 2005, 127, 13784–13785. [Google Scholar]
- Wang, T.; Du, D.; Gai, F. Helix-coil kinetics of two 14-residue peptides. Chem. Phys. Lett 2003, 370, 842–848. [Google Scholar]
- Shashilov, V.A.; Sikirzhytski, V.; Popova, L.A.; Lednev, I.K. Quantitative methods for structural characterization of proteins based on deep UV resonance Raman spectroscopy. Methods 2010, 52, 23–37. [Google Scholar]
- Fasman, G.D. Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA, 1996. [Google Scholar]
- Wallace, B.A.; Janes, R.W. Synchrotron radiation circular dichroism spectroscopy of proteins: Secondary structure, fold recognition and structural genomics. Curr. Opin. Chem. Biol 2001, 5, 567–571. [Google Scholar]
- Rohl, C.A.; Baldwin, R.L. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of the helix—Coil transition in peptides. Biochemistry 1997, 36, 8435–8442. [Google Scholar]
- Jennings, B.R.; Spach, G.; Schuster, T.M. Specific aggregation of poly(α-l-Glutamic acid) and hysteresis effects in aqueous solutions. I. Influence of temperature-dependent aggregation on the optical rotation of PGA. Biopolymers 1968, 6, 635–652. [Google Scholar]
- Krejtschi, C.; Hauser, K. Stability and folding dynamics of polyglutamic acid. Eur. Biophys. J 2011, 40, 673–685. [Google Scholar]
- Rosenheck, K.; Doty, P. The far ultraviolet absorption spectra of polypeptide and protein solutions and their dependence on conformation. Proc. Natl. Acad. Sci. USA 1961, 47, 1775–1785. [Google Scholar]
- Lednev, I.K.; Karnoup, A.S.; Sparrow, M.C.; Asher, S.A. α-helix peptide folding and unfolding activation barriers: A nanosecond UV resonance Raman study. J. Am. Chem. Soc 1999, 121, 8074–8086. [Google Scholar]
- Sharma, S. Helix-Coil Transition Dynamics of α-Helical Peptides. In PhD thesis; University of Liverpool: Liverpool, UK, 12 October 2006. [Google Scholar]
- Khuc, M.T.; Mendonça, L.; Sharma, S.; Solinas, X.; Volk, M.; Hache, F. Measurement of circular dichroism dynamics in a nanosecond temperature-jump experiment. Rev. Sci. Instrum 2011, 82, 054302:1–054302:8. [Google Scholar]
- Wen, Y.X.; Chen, E.; Lewis, J.W.; Kliger, D.S. Nanosecond time-resolved circular dichroism measurements using an upconverted Ti:sapphire laser. Rev. Sci. Instrum 1996, 67, 3010–3016. [Google Scholar]





© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mendonça, L.; Hache, F. Nanosecond T-Jump Experiment in Poly(glutamic acid): A Circular Dichroism Study. Int. J. Mol. Sci. 2012, 13, 2239-2248. https://doi.org/10.3390/ijms13022239
Mendonça L, Hache F. Nanosecond T-Jump Experiment in Poly(glutamic acid): A Circular Dichroism Study. International Journal of Molecular Sciences. 2012; 13(2):2239-2248. https://doi.org/10.3390/ijms13022239
Chicago/Turabian StyleMendonça, Lucille, and François Hache. 2012. "Nanosecond T-Jump Experiment in Poly(glutamic acid): A Circular Dichroism Study" International Journal of Molecular Sciences 13, no. 2: 2239-2248. https://doi.org/10.3390/ijms13022239
APA StyleMendonça, L., & Hache, F. (2012). Nanosecond T-Jump Experiment in Poly(glutamic acid): A Circular Dichroism Study. International Journal of Molecular Sciences, 13(2), 2239-2248. https://doi.org/10.3390/ijms13022239