Development of On-Line High Performance Liquid Chromatography (HPLC)-Biochemical Detection Methods as Tools in the Identification of Bioactives


Abstract
:1. Introduction
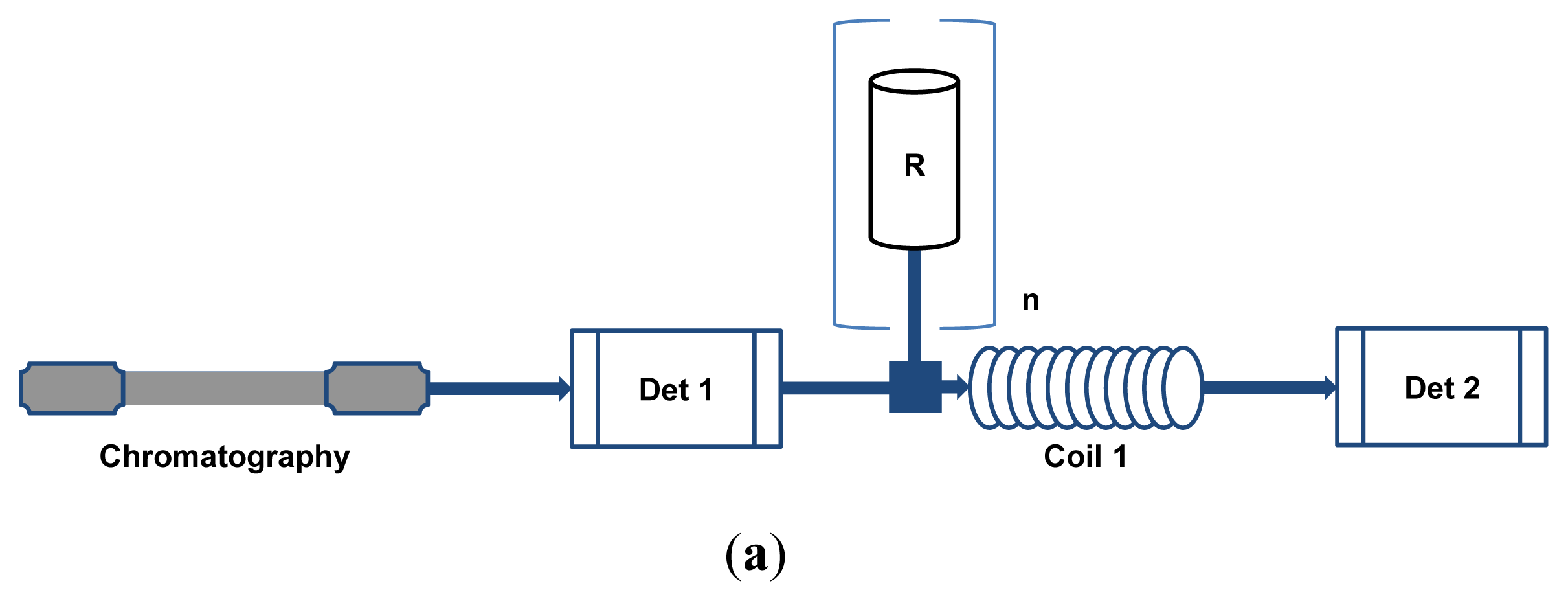
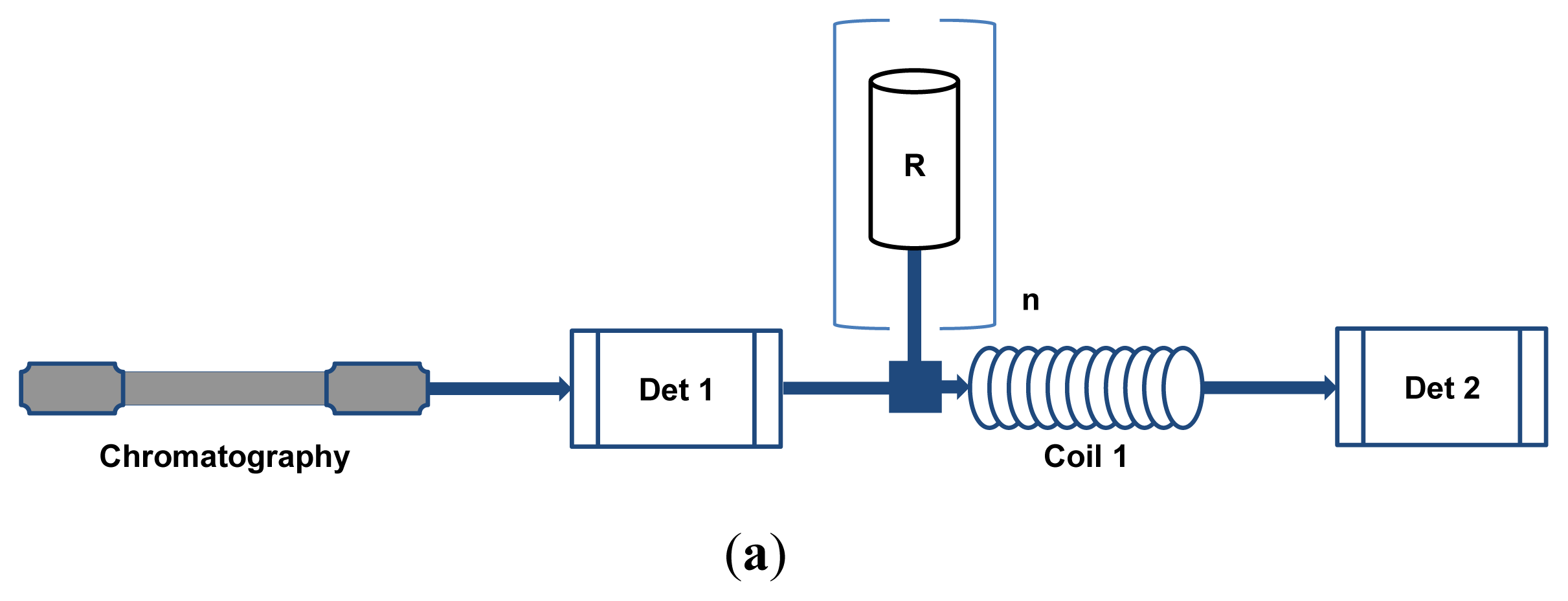
2. HPLC-BCD Configurations and General Requirements
3. HPLC Coupled On-Line to EAD and RAD Assays and Their Application in Metabolic Profiling
3.1. EAD Assays
3.2. RAD Assays
3.3. Application of EAD and RAD Assays in Metabolic Profiling Assays
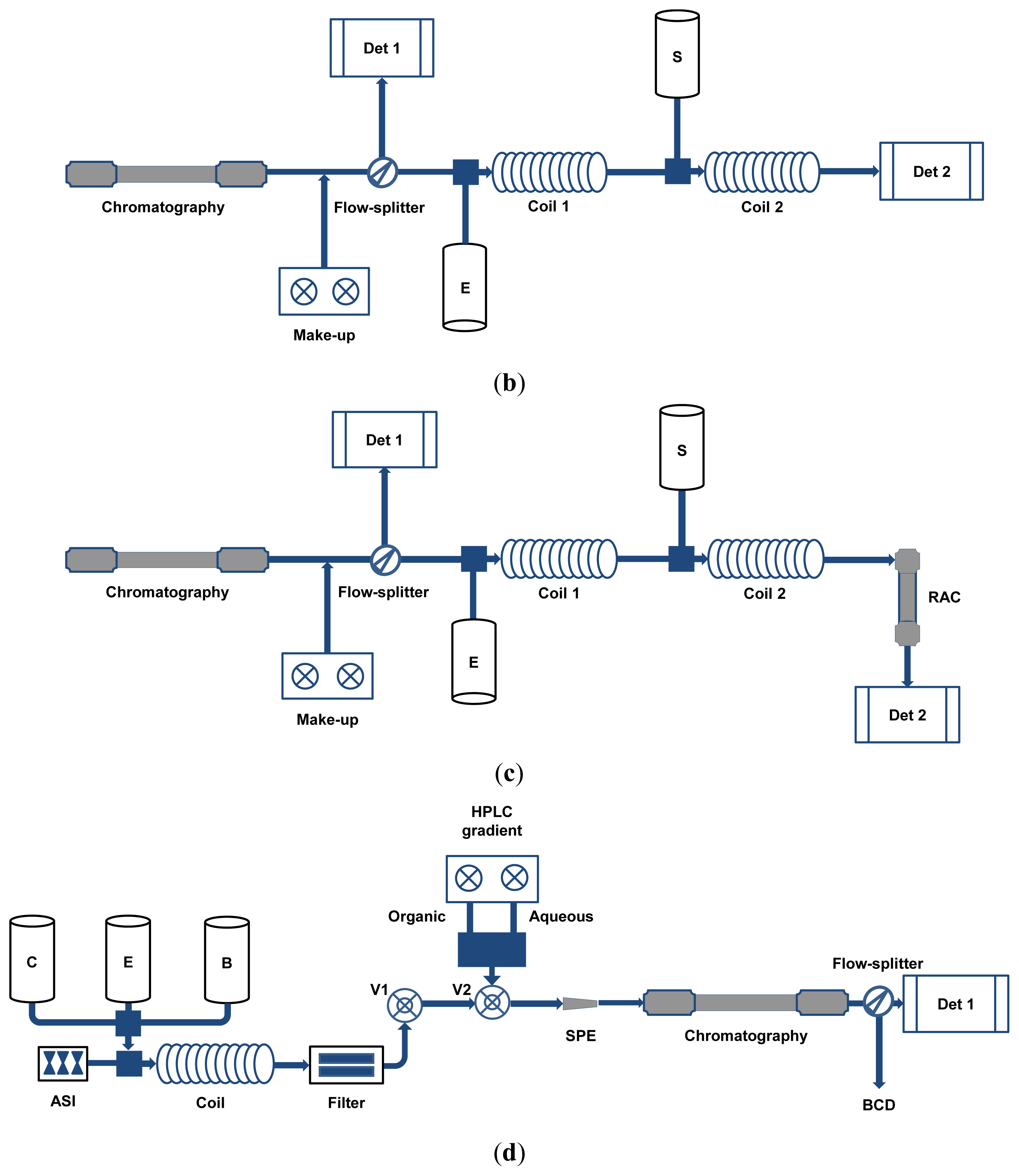
4. HPLC Coupled to On-Line Antioxidant Detection
4.1. On-Line Antioxidant Assays Based on Stable Oxidizing Reagents
4.2. On-Line Antioxidant Assays Based on Physiologically-Relevant ROS
5. Conclusions
Acknowledgments
References
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod 2004, 67, 2141–2153. [Google Scholar]
- Potterat, O.; Hamburger, M. Natural products in drug discovery—Concepts and approaches for tracking bioactivity. Curr. Org. Chem 2006, 10, 899–920. [Google Scholar]
- Balunas, M.J.; Kinghorn, A.D. Drug discovery from medicinal plants. Life Sci 2005, 278, 431–441. [Google Scholar]
- de Beer, D.; Joubert, E.; Malherbe, C.J.; Brand, D.J. Use of countercurrent chromatography during isolation of 6-hydroxyluteolin-7-O-β-glucoside, a major antioxidant of Athrixia phylicoides. J. Chromatogr. A 2011, 1218, 6179–6186. [Google Scholar]
- Koleva, I.I.; Niederländer, H.A.G.; van Beek, T.A. An on-line HPLC method for detection of radical scavenging compounds in complex mixtures. Anal. Chem 2000, 72, 2323–2328. [Google Scholar]
- Ingkaninan, K.; de Best, C.M.; van der Heijden, R.; Hofte, A.J.P.; Karabatak, B.; Irth, H.; Tjaden, U.R.; van der Greef, J.; Verpoorte, R. High-performance liquid chromatography with on-line coupled UV, mass spectrometric and biochemical detection for identification of acetylcholinesterase inhibitors from natural products. J. Chromatogr. A 2000, 872, 61–73. [Google Scholar]
- Ingkaninan, K.; Hazekamp, A.; de Best, C.M.; Irth, H.; Tjaden, U.R.; van der Heijden, R.; van der Greef, J.; Verpoorte, R. The application of HPLC with on-line coupled UV/MS-biochemical detection for isolation of an acetylcholinesterase inhibitor from Narcissus “Sir Winston Churchill”. J. Nat. Prod 2000, 63, 803–806. [Google Scholar]
- de Vlieger, J.S.B.; Kolkman, A.J.; Ampt, K.A.M.; Commandeur, J.N.M.; Vermeulen, N.P.E.; Kool, J.; Wijmenga, S.S.; Niessen, W.M.A.; Irth, H.; Honing, M. Determination and identification of estrogenic compounds generated with biosynthetic enzymes using hyphenated screening assays, high resolution mass spectrometry and off-line NMR. J. Chromatogr. B 2010, 878, 667–674. [Google Scholar]
- Kool, J.; van Liempd, S.M.; Harmsen, S.; Schenk, T.; Irth, H.; Commandeur, J.N.M.; Vermeulen, N.P.E. An on-line post-column detection system for the detection of reactive-oxygen-species-producing compounds and antioxidants in mixtures. Anal. Bioanal. Chem 2007, 388, 871–879. [Google Scholar]
- Van Elswijk, D.A.; Diefenbach, O.; van der Berg, S.; Irth, H.; Tjaden, U.R.; van der Greef, J. Rapid detection and identification of angiotensin-converting enzyme inhibitors by on-line liquid chromatography-biochemical detection, coupled to electrospray mass spectrometry. J. Chromatogr. A 2003, 1020, 45–58. [Google Scholar]
- Schobel, U.; Frenay, M.; van Elswijk, D.A.; McAndrews, J.M.; Long, K.R.; Olson, L.M.; Bobzin, S.C.; Irth, H. High resolution screening of plant natural product extracts for estrogen receptor α and β binding activity using an online HPLC-MS biochemical detection system. J. Biomol. Screen 2001, 6, 291–303. [Google Scholar]
- Oosterkamp, A.J.; van der Hoeven, R.; Glässgen, W.; König, B.; Tjaden, U.R.; van der Greef, J.; Irth, H. Gradient reversed-phase liquid chromatography coupled on-line to receptor-affinity detection based on the urokinase receptor. J. Chromatogr. B 1998, 715, 331–338. [Google Scholar]
- van Liempd, S.M.; Kool, J.; Niessen, W.M.A.; van Elswijk, D.E.; Irth, H.; Vermeulen, N.P.E. On-line formation, separation, and estrogen receptor affinity screening of cytochrome P450-derived metabolites of selective estrogen receptor modulators. Drug Metab. Dispos 2006, 34, 1640–1649. [Google Scholar]
- Li, D.-Q.; Qian, Z.-M.; Li, S.-P. Inhibition of three selected beverage extracts on α-glucosidase and rapid identification of their active compounds using HPLC-DAD-MS/MS and biochemical detection. J. Agric. Food Chem 2010, 58, 6608–6613. [Google Scholar]
- Kool, J.; van Liempd, S.M.; Ramautar, R.; Schenk, T.; Meerman, J.H.N.; Irth, H.; Commandeur, J.N.M.; Vermeulen, N.P.E. Development of a novel cytochrome P450 bioaffinity detection system coupled online to gradient reversed-phase high-performance liquid chromatography. J. Biomol. Screen 2005, 10, 427–436. [Google Scholar]
- Kool, J.; Eggink, M.; van Rossum, H.; van Liempd, S.M.; van Elswijk, D.A.; Irth, H.; Commandeur, J.N.M.; Meerman, J.H.N.; Vermeulen, N.P.E. Online biochemical detection of glutathione-S-transferase P1-specific inhibitors in complex mixtures. J. Biomol. Screen 2007, 12, 396–405. [Google Scholar]
- de Jong, L.A.A.; Uges, D.R.A.; Franke, J.P.; Bischoff, R. Receptor-ligand binding assays: Technologies and applications. J. Chromatogr. B 2005, 829, 1–25. [Google Scholar]
- Emnéus, J.; Marko-Varga, G. Biospecific detection in liquid chromatography. J. Chromatogr. A 1995, 703, 191–243. [Google Scholar]
- Entzeroth, M. Emerging trends in high-throughput screening. Curr. Opin. Pharmacol 2003, 3, 522–529. [Google Scholar]
- Shi, S.-Y.; Zhang, Y.-P.; Jiang, X.-Y.; Chen, X.-Q.; Huang, K.-L.; Zhou, H.-H.; Jiang, X.-Y. Coupling HPLC to on-line, post-column (bio)chemical assays for high-resolution screening of bioactive compounds from complex mixtures. TrAC Trends Anal. Chem 2009, 28, 865–877. [Google Scholar]
- van Elswijk, D.A.; Irth, H. Analytical tools for the detection and characterization of biologically active compounds from nature. Phytochem. Rev 2002, 1, 427–439. [Google Scholar]
- Oosterkamp, A.J.; Irth, H.; Tjaden, U.R.; van der Greef, J. Online coupling of liquid chromatography to biochemical assays based on fluorescent-labeled ligands. Anal. Chem 1994, 66, 4295–4301. [Google Scholar]
- Oosterkamp, A.J.; Irth, H.; Tjaden, U.R.; van der Greef, J. Theoretical concepts of on-line liquid chromatographic-biochemical detection systems II. Detection systems based on labelled affinity proteins. J. Chromatogr. A 1997, 787, 37–46. [Google Scholar]
- Oosterkamp, A.J.; Irth, H.; Herraiz, M.T.V.; Tjaden, U.R.; van der Greef, J. Theoretical concepts of on-line liquid chromatographic-biochemical detection systems I. Detection systems based on labelled ligands. J. Chromatogr. A 1997, 787, 27–35. [Google Scholar]
- van Bommel, M.R.; de Jong, A.P.J.M.; Tjaden, U.R.; Irth, H.; van der Greef, J. Enzyme amplification as detection tool in continuous-flow systems: I. Development of an enzyme-amplified biochemical detection system coupled on-line to flow-injection analysis. J. Chromatogr. A 1999, 855, 383–396. [Google Scholar]
- van Bommel, M.R.; de Jong, A.P.J.M.; Tjaden, U.R.; Irth, H.; van der Greef, J. Enzyme amplification as detection tool in continuous-flow systems: II. On-line coupling of liquid chromatography to enzyme-amplified biochemical detection after pre-column derivatization with biotin. J. Chromatogr. A 1999, 855, 397–409. [Google Scholar]
- de Boer, A.R.; Lingeman, H.; Niessen, W.M.A.; Irth, H. Mass spectrometry-based biochemical assays for enzyme-inhibitor screening. TrAC Trends Anal. Chem 2007, 26, 867–883. [Google Scholar]
- Kool, J.; Giera, M.; Irth, H.; Niessen, W.M.A. Advances in mass spectrometry-based post-column bioaffinity profiling of mixtures. Anal. Bioanal. Chem 2011, 399, 2655–2668. [Google Scholar]
- van Liempd, S.M.; Kool, J.; Reinen, J.; Schenk, T.; Meerman, J.H.N.; Irth, H.; Vermeulen, N.P.E. Development and validation of a microsomal online cytochrome P450 bioreactor coupled to solid-phase extraction and reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1075, 205–212. [Google Scholar]
- van Liempd, S.M.; Kool, J.; Meerman, J.H.; Irth, H.; Vermeulen, N.P. Metabolic profiling of endocrine-disrupting compounds by on-line cytochrome P450 bioreaction coupled to on-line receptor affinity screening. Chem. Res. Toxicol 2007, 20, 1825–1832. [Google Scholar]
- van Midwoud, P.M.; Janssen, J.; Merema, M.T.; de Graaf, I.A.M.; Groothuis, G.M.M.; Verpoorte, E. On-line HPLC analysis system for metabolism and inhibition studies in precision-cut liver slices. Anal. Chem 2011, 83, 84–91. [Google Scholar]
- Derks, R.J.E.; Hogenboom, A.C.; van der Zwan, G.; Irth, H. On-line continuous-flow, multi-protein biochemical assays for the characterization of bioaffinity compounds using electrospray quadrupole time-of-flight mass spectrometry. Anal. Chem 2003, 75, 3376–3384. [Google Scholar]
- German, I.; Kennedy, R.T. Reversed-phase capillary liquid chromatography coupled on-line to capillary electrophoresis immunoassays. Anal. Chem 2000, 72, 5365–5372. [Google Scholar]
- Hogenboom, A.C.; de Boer, A.R.; Derks, R.J.E.; Irth, H. Continuous-flow, on-line monitoring of biospecific interactions using electrospray mass spectrometry. Anal. Chem 2001, 73, 3816–3823. [Google Scholar]
- Irth, H.; Oosterkamp, A.J.; Tjaden, U.R.; van der Greef, J. Strategies for on-line coupling of immunoassays to high-performance liquid chromatography. TrAC Trends Anal. Chem 1995, 14, 355–361. [Google Scholar]
- Önnerfjord, P.; Eremin, S.A.; Emnéus, J.; Marko-Varga, G. High sample throughput flow immunoassay utilising restricted access columns for the separation of bound and free label. J. Chromatogr. A 1998, 800, 219–230. [Google Scholar]
- Shahdeo, K.; Karnes, H.T. Combining immunoassays with chromatographic and electrophoretic separation techniques—A review. Microchim. Acta 1998, 129, 19–27. [Google Scholar]
- Fabel, S.; Niessner, R.; Weller, M.G. Effect-directed analysis by high-performance liquid chromatography with gas-segmented enzyme inhibition. J. Chromatogr. A 2005, 1099, 103–110. [Google Scholar]
- Rhee, I.K.; Appels, N.; Luijendijk, T.; Irth, H.; Verpoorte, R. Determining acetylcholinesterase inhibitory activity in plant extracts using a fluorimetric flow assay. Phytochem. Anal 2003, 14, 145–149. [Google Scholar]
- Marques, L.A.; Kool, J.; de Kanter, F.; Lingeman, H.; Niessen, W.; Irth, H. Production and on-line acetylcholinesterase bioactivity profiling of chemical and biological degradation products of tacrine. J. Pharm. Biomed. Anal 2010, 53, 609–616. [Google Scholar]
- de Jong, C.F.; Derks, R.J.E.; Bruyneel, B.; Niessen, W.; Irth, H. High-performance liquid chromatography-mass spectrometry-based acetylcholinesterase assay for the screening of inhibitors in natural extracts. J. Chromatogr. A 2006, 1112, 303–310. [Google Scholar]
- Li, S.P.; Zhao, J.; Yang, B. Strategies for quality control of Chinese medicines. J. Pharm. Biomed. Anal 2011, 55, 802–809. [Google Scholar]
- Kool, J.; van Liempd, S.M.; van Rossum, H.; van Elswijk, D.A.; Irth, H.; Commandeur, J.N.M.; Vermeulen, N.P.E. Development of three parallel cytochrome P450 enzyme affinity detection systems coupled on-line to gradient high-performance liquid chromatography. Drug Metab. Dispos 2007, 35, 640–648. [Google Scholar]
- Jeurissen, S.M.F.; Claassen, F.W.; Havlik, J.; Bouwmans, E.E.; Cnubben, N.H.P.; Sudhölter, E.J.R.; Rietjens, I.M.C.M.; van Beek, T.A. Development of an on-line high performance liquid chromatography detection system for human cytochrome P450 1A2 inhibitors in extracts of natural products. J. Chromatogr. A 2007, 1141, 81–89. [Google Scholar]
- Reinen, J.; Ferman, S.; Vottero, E.; Vermeulen, N.P.E.; Commandeur, J.N.M. Application of a fluorescence-based continuous-flow bioassay to screen for diversity of cytochrome P450 BM3 mutant libraries. J. Biomol. Screen 2011, 16, 239–250. [Google Scholar]
- Schebb, N.; Faber, H.; Maul, R.; Heus, F.; Kool, J.; Irth, H.; Karst, U. Analysis of glutathione adducts of patulin by means of liquid chromatography (HPLC) with biochemical detection (BCD) and electrospray ionization tandem mass spectrometry (ESI-MS/MS). Anal. Bioanal. Chem 2009, 394, 1361–1373. [Google Scholar]
- Schebb, N.H.; Heus, F.; Saenger, T.; Karst, U.; Irth, H.; Kool, J. Development of a countergradient parking system for gradient liquid chromatography with online biochemical detection of serine protease inhibitors. Anal. Chem 2008, 80, 6764–6772. [Google Scholar]
- Hirata, J.; Ariese, F.; Gooijer, C.; Irth, H. Continuous-flow protease assay based on fluorescence resonance energy transfer. Anal. Chim. Acta 2003, 478, 1–10. [Google Scholar]
- Hirata, J.; Chung, L.P.; Ariese, F.; Irth, H.; Gooijer, C. Coupling of size-exclusion chromatography to a continuous assay for subtilisin using a fluorescence resonance energy transfer peptide substrate: Testing of two standard inhibitors. J. Chromatogr. A 2005, 1081, 140–144. [Google Scholar]
- Schenk, T.; Breel, G.J.; Koevoets, P.; van den Berg, S.; Hogenboom, A.C.; Irth, H.; Tjaden, U.R.; van der Greef, J. Screening of natural products extracts for the presence of phosphodiesterase inhibitors using liquid chromatography coupled online to parallel biochemical detection and chemical characterization. J. Biomol. Screen 2003, 8, 421–429. [Google Scholar]
- Schenk, T.; Appels, N.M.G.M.; van Elswijk, D.A.; Irth, H.; Tjaden, U.R.; van der Greef, J. A generic assay for phosphate-consuming or -releasing enzymes coupled on-line to liquid chromatography for lead finding in natural products. Anal. Biochem 2003, 316, 118–126. [Google Scholar]
- Falck, D.; de Vlieger, J.; Niessen, W.; Kool, J.; Honing, M.; Giera, M.; Irth, H. Development of an online p38α mitogen-activated protein kinase binding assay and integration of LC-HR-MS. Anal. Bioanal. Chem 2010, 398, 1771–1780. [Google Scholar]
- Oosterkamp, A.J.; Herraiz, M.T.V.; Irth, H.; Tjaden, U.R.; van der Greef, J. Reversed-phase liquid chromatography coupled on-line to receptor affinity detection based on the human estrogen receptor. Anal. Chem 1996, 68, 1201–1206. [Google Scholar]
- Kool, J.; Ramautar, R.; van Liempd, S.M.; Beckman, J.; de Kanter, F.J.J.; Meerman, J.H.N.; Schenk, T.; Irth, H.; Commandeur, J.N.M.; Vermeulen, N.P.E. Rapid on-line profiling of estrogen receptor binding metabolites of tamoxifen. J. Med. Chem 2006, 49, 3287–3292. [Google Scholar]
- Reinen, J.; Kool, J.; Vermeulen, N.P.E. Reversed-phase liquid chromatography coupled on-line to estrogen receptor bioaffinity detection based on fluorescence polarization. Anal. Bioanal. Chem 2008, 390, 1987–1998. [Google Scholar]
- Mukherjee, P.K.; Kumar, V.; Mal, M.; Houghton, P.J. Acetylcholinesterase inhibitors from plants. Phytomedicine 2007, 14, 289–300. [Google Scholar]
- de Boer, A.R.; Alcaide-Hidalgo, J.M.; Krabbe, J.G.; Kolkman, J.; van Emde Boas, C.N.; Niessen, W.M.A.; Lingeman, H.; Irth, H. High-temperature liquid chromatography coupled on-line to a continuous-flow biochemical screening assay with electrospray ionization mass spectrometric detection. Anal. Chem 2005, 77, 7894–7900. [Google Scholar]
- de Boer, A.R.; Letzel, T.; van Elswijk, D.A.; Lingeman, H.; Niessen, W.M.A.; Irth, H. On-line coupling of high-performance liquid chromatography to a continuous-flow enzyme assay based on electrospray ionization mass spectrometry. Anal. Chem 2004, 76, 3155–3161. [Google Scholar]
- Kool, J.; Heus, F.; de Kloe, G.; Lingeman, H.; Smit, A.B.; Leurs, R.; Edink, E.; de Esch, I.J.P.; Irth, H.; Niessen, W.M.A. High-resolution bioactivity profiling of mixtures toward the acetylcholine binding protein using a nanofractionation spotter technology. J. Biomol. Screen 2011, 16, 917–924. [Google Scholar]
- Casirola, D.M.; Ferraris, R.P. α-Glucosidase inhibitors prevent diet-induced increases in intestinal sugar transport in diabetic mice. Metab. Clin. Exp 2006, 55, 832–841. [Google Scholar]
- Kim, J.-S.; Kwon, Y.-S.; Chun, W.-J.; Kim, T.-Y.; Sun, J.; Yu, C.-Y.; Kim, M.-J. Rhus verniciflua Stokes flavonoid extracts have anti-oxidant, anti-microbial and α-glucosidase inhibitory effect. Food Chem 2010, 120, 539–543. [Google Scholar]
- Carey, R.M.; Siragy, H.M. Newly recognized components of the renin-angiotensin system: Potential roles in cardiovascular and renal regulation. Endocr. Rev 2003, 24, 261–271. [Google Scholar]
- Araujo, M.C.; Melo, R.L.; Cesari, M.H.; Juliano, M.A.; Juliano, L.; Carmona, A.K. Peptidase specificity characterization of C- and N-terminal catalytic cites of angiotensin I-converting enzyme. Biochemistry 2000, 39, 8519–8525. [Google Scholar]
- Rudzki, P.J.; Bus, K.; Ksycinska, H.; Kobylinska, K. An overview of chromatographic methods coupled with mass spectrometric detection for determination of angiotensin-converting enzyme inhibitors in biological material. J. Pharm. Biomed. Anal 2007, 44, 356–367. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol 2008, 21, 70–83. [Google Scholar]
- van Beek, T.; Tetala, K.; Koleva, I.; Dapkevicius, A.; Exarchou, V.; Jeurissen, S.; Claassen, F.; van der Klift, E. Recent developments in the rapid analysis of plants and tracking their bioactive constituents. Phytochem. Rev 2009, 8, 387–399. [Google Scholar]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol 2005, 45, 51–88. [Google Scholar]
- Huntington, J.A.; Baglin, T.P. Targeting thrombin—Rational drug design from natural mechanisms. Trends Pharmacol. Sci 2003, 24, 589–595. [Google Scholar]
- Schebb, N.H.; Vielhaber, T.; Jousset, A.; Karst, U. Development of a liquid chromatography-based screening methodology for proteolytic enzyme activity. J. Chromatogr. A 2009, 1216, 4407–4415. [Google Scholar]
- Dunford, H.B. Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology and Physiology, 2nd ed; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010. [Google Scholar]
- Haselberg, R.; Hempen, C.; van Leeuwen, S.M.; Vogel, M.; Karst, U. Analysis of microperoxidases using liquid chromatography, post-column substrate conversion and fluorescence detection. J. Chromatogr. B 2006, 830, 47–53. [Google Scholar]
- Houslay, M.D. Adaptation in cyclic AMP signalling processes: A central role for cyclic AMP phosphodiesterases. Semin. Cell Dev. Biol 1998, 9, 161–167. [Google Scholar]
- Kool, J.; van Marle, A.; Hulscher, S.; Selman, M.; van Iperen, D.J.; van Altena, K.; Gillard, M.; Bakker, R.A.; Irth, H.; Leurs, R.; et al. A flow-through fluorescence polarization detection system for measuring GPCR-mediated modulation of cAMP production. J. Biomol. Screen 2007, 12, 1074–1083. [Google Scholar]
- Ichijo, H. From receptors to stress-activated MAP kinases. Oncogene 1999, 18, 6087–6093. [Google Scholar]
- Oosterkamp, A.J.; Hock, B.; Seifert, M.; Irth, H. Novel monitoring strategies for xenoestrogens. TrAC Trends Anal. Chem 1997, 16, 544–553. [Google Scholar]
- van Elswijk, D.A.; Schobel, U.P.; Lansky, E.P.; Irth, H.; van der Greef, J. Rapid dereplication of estrogenic compounds in pomegranate (Punica granatum) using on-line biochemical detection coupled to mass spectrometry. Phytochemistry 2004, 65, 233–241. [Google Scholar]
- Preissner, K.T.; Kanse, S.M.; May, A.E. Urokinase receptor: A molecular organizer in cellular communication. Curr. Opin. Cell Biol 2000, 12, 621–628. [Google Scholar]
- Niederländer, H.A.G.; van Beek, T.A.; Bartasiute, A.; Koleva, I.I. Antioxidant activity assays on-line with liquid chromatography. J. Chromatogr. A 2008, 1210, 121–134. [Google Scholar]
- Zhang, Q.; van der Klift, E.J.C.; Janssen, H.G.; van Beek, T.A. An on-line normal-phase high performance liquid chromatography method for the rapid detection of radical scavengers in non-polar food matrixes. J. Chromatogr. A 2009, 1216, 7268–7274. [Google Scholar]
- Cardeñosa, R.; Mohamed, R.; Pineda, M.; Aguilar, M. On-line HPLC detection of tocopherols and other antioxidants through the formation of a phosphomolybdenum complex. J. Agric. Food Chem 2002, 50, 3390–3395. [Google Scholar]
- Kusznierewicz, B.; Piasek, A.; Bartoszek, A.; Namiesnik, J. Application of a commercially available derivatization instrument and commonly used reagents to HPLC on-line determination of antioxidants. J. Food Compos. Anal 2011, 24, 1073–1080. [Google Scholar]
- Kusznierewicz, B.; Piasek, A.; Bartoszek, A.; Namiesnik, J. The optimisation of analytical parameters for routine profiling of antioxidants in complex mixtures by HPLC coupled post-column derivatisation. Phytochem. Anal 2011, 22, 392–402. [Google Scholar]
- Çelik, S.E.; Özyürek, M.; Güçlü, K.; Apak, R. Determination of antioxidants by a novel on-line HPLC-cupric reducing antioxidant capacity (CUPRAC) assay with post-column detection. Anal. Chim. Acta 2010, 674, 79–88. [Google Scholar]
- Mnatsakanyan, M.; Goodie, T.A.; Conlan, X.A.; Francis, P.S.; McDermott, G.P.; Barnett, N.W.; Shock, D.; Gritti, F.; Guiochon, G.; Shalliker, R.A. High performance liquid chromatography with two simultaneous on-line antioxidant assays: Evaluation and comparison of espresso coffees. Talanta 2010, 81, 837–842. [Google Scholar]
- Mnatsakanyan, M.; Stevenson, P.G.; Conlan, X.A.; Francis, P.S.; Goodie, T.A.; McDermott, G.P.; Barnett, N.W.; Shalliker, R.A. The analysis of café espresso using two-dimensional reversed phase-reversed phase high performance liquid chromatography with UV-absorbance and chemiluminescence detection. Talanta 2010, 82, 1358–1363. [Google Scholar]
- McDermott, G.P.; Conlan, X.A.; Noonan, L.K.; Costin, J.W.; Mnatsakanyan, M.; Shalliker, R.A.; Barnett, N.W.; Francis, P.S. Screening for antioxidants in complex matrices using high performance liquid chromatography with acidic potassium permanganate chemiluminescence detection. Anal. Chim. Acta 2011, 684, 134–141. [Google Scholar]
- Dapkevicius, A.; van Beek, T.A.; Niederländer, H.A.G. Evaluation and comparison of two improved techniques for the on-line detection of antioxidants in liquid chromatography eluates. J. Chromatogr. A 2001, 912, 73–82. [Google Scholar]
- He, W.; Gao, Y.; Yuan, F.; Bao, Y.; Liu, F.; Dong, J. Optimization of supercritical carbon dioxide extraction of Gardenia fruit oil and the analysis of functional components. J. Am. Oil Chem. Soc 2010, 87, 1071–1079. [Google Scholar]
- Koleva, I.I.; Niederländer, H.A.G.; van Beek, T.A. Application of ABTS radical cation for selective on-line detection of radical scavengers in HPLC eluates. Anal. Chem 2001, 73, 3373–3381. [Google Scholar]
- Francis, P.S.; Costin, J.W.; Conlan, X.A.; Bellomarino, S.A.; Barnett, J.A.; Barnett, N.W. A rapid antioxidant assay based on acidic potassium permanganate chemiluminescence. Food Chem 2010, 122, 926–929. [Google Scholar]
- Bartasiute, A.; Westerink, B.H.C.; Verpoorte, E.; Niederländer, H.A.G. Improving the in vivo predictability of an on-line HPLC stable free radical decoloration assay for antioxidant activity in methanol-buffer medium. Free Radic. Biol. Med 2007, 42, 413–423. [Google Scholar]
- McDermott, G.P.; Noonan, L.K.; Mnatsakanyan, M.; Shalliker, R.A.; Conlan, X.A.; Barnett, N.W.; Francis, P.S. High-performance liquid chromatography with post-column 2,2'-diphenyl-1-picrylhydrazyl radical scavenging assay: Methodological considerations and application to complex samples. Anal. Chim. Acta 2010, 675, 76–82. [Google Scholar]
- Arthur, H.; Joubert, E.; de Beer, D.; Malherbe, C.J.; Witthuhn, R.C. Phenylethanoid glycosides as major antioxidants in Lippia multiflora herbal infusion and their stability during steam pasteurisation of plant material. Food Chem 2011, 127, 581–588. [Google Scholar]
- Koşar, M.; Dorman, H.J.D.; Bachmayer, O. An improved on-line HPLC-DPPH method for the screening of free radical scavenging compounds in water extracts of Lamiaceae plants. Chem. Nat. Compd 2003, 39, 118–122. [Google Scholar]
- Oki, T.; Kobayashi, M.; Nakamura, T.; Okuyama, A.; Masuda, M.; Shiratsuchi, H.; Suda, I. Changes in radical-scavenging activity and components of mulberry fruit during maturation. J. Food Sci 2006, 71, C18–C22. [Google Scholar]
- Nuengchamnong, N.; de Jong, C.F.; Bruyneel, B.; Niessen, W.M.A.; Irth, H.; Ingkaninan, K. HPLC coupled on-line to ESI-MS and a DPPH-based assay for the rapid identification of anti-oxidants in Butea superba. Phytochem. Anal 2005, 16, 422–428. [Google Scholar]
- Nuengchamnong, N.; Ingkaninan, K. On-line characterization of phenolic antioxidants in fruit wines from family Myrtaceae by liquid chromatography combined with electrospray ionization tandem mass spectrometry and radical scavenging detection. LWT Food Sci. Technol 2009, 42, 297–302. [Google Scholar]
- Nuengchamnong, N.; Krittasilp, K.; Ingkaninan, K. Rapid screening and identification of antioxidants in aqueous extracts of Houttuynia cordata using LC-ESI-MS coupled with DPPH assay. Food Chem 2009, 117, 750–756. [Google Scholar]
- Nuengchamnong, N.; Ingkaninan, K. On-line HPLC-MS-DPPH assay for the analysis of phenolic antioxidant compounds in fruit wine: Antidesma thwaitesianum Muell. Food Chem 2010, 118, 147–152. [Google Scholar]
- Nuengchamnong, N.; Krittasilp, K.; Ingkaninan, K. Characterisation of phenolic antioxidants in aqueous extract of Orthosiphon grandiflorus tea by LC-ESI-MS/MS coupled to DPPH assay. Food Chem 2011, 127, 1287–1293. [Google Scholar]
- Shi, S.; Zhao, Y.; Zhou, H.; Zhang, Y.; Jiang, X.; Huang, K. Identification of antioxidants from Taraxacum mongolicum by high-performance liquid chromatography-diode array detection-radical-scavenging detection-electrospray ionization mass spectrometry and nuclear magnetic resonance experiments. J. Chromatogr. A 2008, 1209, 145–152. [Google Scholar]
- Jiang, X.; Shi, S.; Zhang, Y.; Chen, X. Excellent combination of HPLC-RSD-DAD-ESI/MS and HSCCC experiments to screen and identify radical scavengers from Neo-Taraxacum siphonanthun. J. Braz. Chem. Soc 2010, 21, 1524–1529. [Google Scholar]
- Pukalskas, A.; van Beek, T.; de Waard, P. Development of a triple hyphenated HPLC-radical scavenging detection-DAD-SPE-NMR system for the rapid identification of antioxidants in complex plant extracts. J. Chromatogr. A 2005, 1074, 81–88. [Google Scholar]
- Exarchou, V.; Fiamegos, Y.C.; van Beek, T.A.; Nanos, C.; Vervoort, J. Hyphenated chromatographic techniques for the rapid screening and identification of antioxidants in methanolic extracts of pharmaceutically used plants. J. Chromatogr. A 2006, 1112, 293–302. [Google Scholar]
- van der Merwe, J.D.; Joubert, E.; Manley, M.; de Beer, D.; Malherbe, C.J.; Gelderblom, W.C.A. In vitro hepatic biotransformation of aspalathin and nothofagin, dihydrochalcones of rooibos (Aspalathus linearis), and assessment of metabolite antioxidant activity. J. Agric. Food Chem 2010, 58, 2214–2220. [Google Scholar]
- Cano, A.; Alcaraz, O.; Acosta, M.; Arnao, M.B. On-line antioxidant activity determination: Comparison of hydrophilic and lipophilic antioxidant activity using the ABTS•+ assay. Redox Rep 2002, 7, 103–109. [Google Scholar]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med 1999, 26, 1231–1237. [Google Scholar]
- Stewart, A.J.; Mullen, W.; Crozier, A. On-line high-performance liquid chromatography analysis of the antioxidant activity of phenolic compounds in green and black tea. Mol. Nutr. Food Res 2005, 49, 52–60. [Google Scholar]
- He, W.; Liu, X.; Xu, H.; Gong, Y.; Yuan, F.; Gao, Y. On-line HPLC-ABTS screening and HPLC-DAD-MS/MS identification of free radical scavengers in Gardenia (Gardenia jasminoides Ellis) fruit extracts. Food Chem 2010, 123, 521–528. [Google Scholar]
- Li, S.-Y.; Yu, Y.; Li, S.-P. Identification of antioxidants in essential oil of radix Angelicae sinensis using HPLC coupled with DAD-MS and ABTS-based assay. J. Agric. Food Chem 2007, 55, 3358–3362. [Google Scholar]
- Miliauskas, G.; van Beek, T.A.; de Waard, P.; Venskutonis, R.P.; Sudhölter, E.J.R. Identification of radical scavenging compounds in Rhaponticum carthamoides by means of LC-DAD-SPE-NMR. J. Nat. Prod 2005, 68, 168–172. [Google Scholar]
- Capanoglu, E.; Beekwilder, J.; Boyacioglu, D.; Hall, R.; de Vos, R. Changes in antioxidant and metabolite profiles during production of tomato paste. J. Agric. Food Chem 2008, 56, 964–973. [Google Scholar]
- Stalmach, A.; Mullen, W.; Nagai, C.; Crozier, A. On-line HPLC analysis of the antioxidant activity of phenolic compounds in brewed, paper-filtered coffee. Braz. J. Plant Physiol 2006, 18, 253–262. [Google Scholar]
- Raudonis, R.; Bumblauskiene, L.; Jakstas, V.; Pukalskas, A.; Janulis, V. Optimization and validation of post-column assay for screening of radical scavengers in herbal raw materials and herbal preparations. J. Chromatogr. A 2010, 1217, 7690–7698. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of Folin-Ciocalteu reagent. Methods Enzymol 1999, 299, 152–178. [Google Scholar]
- Prieto, P.; Pineda, M.; Aguilar, M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex: Specific application to the determination of vitamin E. Anal. Biochem 1999, 269, 337–341. [Google Scholar]
- Çelik, S.E.; Özyürek, M.; Güçlü, K.; Apak, R. Solvent effects on the antioxidant capacity of lipophilic and hydrophilic antioxidants measured by CUPRAC, ABTS/persulphate and FRAP methods. Talanta 2010, 81, 1300–1309. [Google Scholar]
- Apak, R.; Güçlü, K.; Özyürek, M.; Karademir, S.E. Novel total antioxidant capacity index for dietary polyphenols and vitamins C and E, using their cupric ion reducing capability in the presence of neocuproine: CUPRAC method. J. Agric. Food Chem 2004, 52, 7970–7981. [Google Scholar]
- Apak, R.; Güçlü, K.; Özyürek, M.; Karademir, S.E.; Altun, M. Total antioxidant capacity assay of human serum using copper(II)-neocuproine as chromogenic oxidant: The CUPRAC method. Free Radic. Res 2005, 39, 949–961. [Google Scholar]
- Adcock, J.L.; Francis, P.S.; Barnett, N.W. Acidic potassium permanganate as a chemiluminescence reagent—A review. Anal. Chim. Acta 2007, 601, 36–67. [Google Scholar]
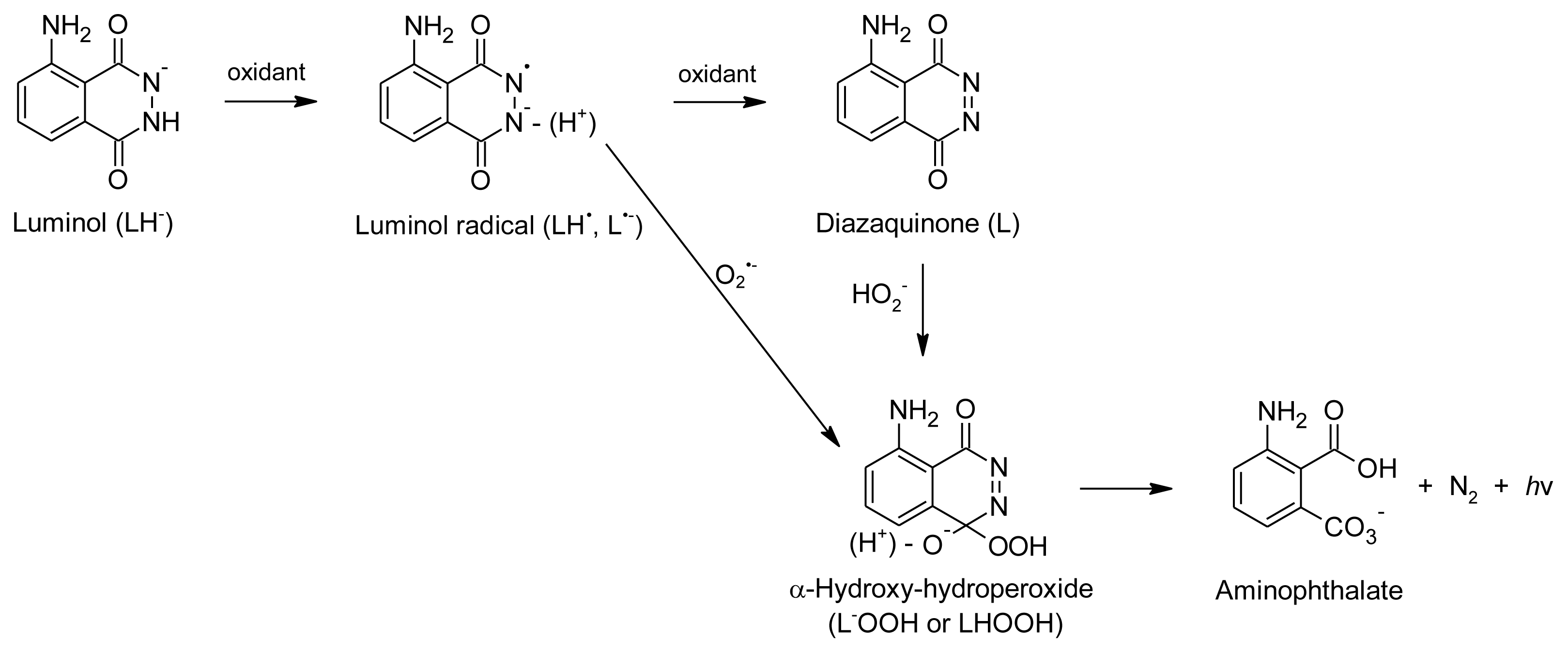
- Merenyi, G.; Lind, J.; Eriksen, T.E. Luminol chemiluminescence: Chemistry, excitation, emitter. J. Biolumin. Chemilumin 1990, 5, 53–56. [Google Scholar]
- Dodeigne, C.; Thunus, L.; Lejeune, R. Chemiluminescence as diagnostic tool. A review. Talanta 2000, 51, 415–439. [Google Scholar]
- Rose, A.L.; Waite, T.D. Chemiluminescence of luminol in the presence of iron(II) and oxygen: Oxidation mechanism and implications for its analytical use. Anal. Chem 2001, 73, 5909–5920. [Google Scholar]
- Gámiz-Gracia, L.; García-Campaña, A.M.; Huertas-Pérez, J.F.; Lara, F.J. Chemiluminescence detection in liquid chromatography: Applications to clinical, pharmaceutical, environmental and food analysis—A review. Anal. Chim. Acta 2009, 640, 7–28. [Google Scholar]
- Dapkevicius, A.; van Beek, T.A.; Niederländer, H.A.G.; de Groot, A. On-line detection of antioxidative activity in high-performance liquid chromatography eluates by chemiluminescence. Anal. Chem 1999, 71, 736–740. [Google Scholar]
- Toyo’oka, T.; Kashiwazaki, T.; Kato, M. On-line screening methods for antioxidants scavenging superoxide anion radical and hydrogen peroxide by liquid chromatography with indirect chemiluminescence detection. Talanta 2003, 60, 467–475. [Google Scholar]
- Ding, X.-P.; Qi, J.; Chang, Y.-X.; Mu, L.-L.; Zhu, D.-N.; Yu, B.-Y. Quality control of flavonoids in Ginkgo biloba leaves by high-performance liquid chromatography with diode array detection and on-line radical scavenging activity detection. J. Chromatogr. A 2009, 1216, 2204–2210. [Google Scholar]
- Ogawa, A.; Arai, H.; Tanizawa, H.; Miyahara, T.; Toyo’oka, T. On-line screening method for antioxidants by liquid chromatography with chemiluminescence detection. Anal. Chim. Acta 1999, 383, 221–230. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Detection | Relevance |
|---|---|---|
| EAD assays | ||
| Acetylcholinesterase inhibitors [6,7,38–41] | UV-Vis, 405 nm [6,7,38] FL, λex 406nm, λem 505 nm [39,40] ESI-MS [41] | Treatment of Alzheimer’s disease, senile dementia, Parkinson’s disease, ataxia and myasthenia gravis |
| α-Glucosidase inhibitors [14,42] | UV-Vis, 405 nm [14,42] | Treatment of diabetes type II |
| Angiotensin 1 converting enzyme [10] | FL, λex 320 nm, λem 420 nm [10] | Treatment of hypertension, cardiac disease, diabetic nephropathy |
| Liver cytochrome P450 ligands [15,43–45] | FL, λex 530 nm, λem 586 nm [15,43] FL, λex 530 nm, λem 590 nm [44] FL, λex 530 nm, λem 580 nm [45] | Cancer prevention |
| Glutathione-S-transferase inhibitors [16,46] | FL, λex 290 nm, λem 465 nm [16,46] | Enhancement of anticancer treatments |
| Serine protease inhibitors [47] | FL, λex 342 nm, λem 440 nm [47] | Treatment of thrombosis |
| HIV-protease inhibitor [48,49] | FL, λex 340 nm, λem 490 nm [48,49] | Treatment of AIDS |
| Phosphodiesterase inhibitors [50] | FL, λex 280 nm, λem 460 nm [50] | Treatment of hypertension, vascular conditions and asthma |
| Kinase/phosphatase inhibitors [51] | FL, λex 425 nm, λem 464 nm [51] | Novel drug targets |
| MAP-kinase inhibitors [52] | FL, λex 355 ± 4 nm, λem 405 ± 5 nm [52] | Treatment of inflammatory diseases |
| RAD assays | ||
| Estrogen receptor ligands [8,11,13,30,53–55] | FL, λex 340 nm, λem 410 nm [8,11,13,30,53,54] FP, λex 485 nm, λem 520 nm [55] | Hormone replacement therapy, chemoprevention, detecting endocrine disruptors |
| Urokinase plasminogen activator receptor ligands [12] | FL, λex 489 nm, λem 520 nm [12] | Important role in angiogenesis, inflammation, wound repair and tumor metastasis |
| Assay | Reaction Mechanism | Detection | Reagent Solution Characteristics | HPLC Mobile Phase Compatibility |
|---|---|---|---|---|
| DPPH• scavenging [5,79,87,88] | H-donation | UV-VIS, 510–520 nm | DPPH• in MeOH or MeOH/buffer (pH 7.6) mixture for RP-HPLC; n-hexane for NP-HPLC | 10–90% organic modifier at pH 3–6 a for RP-HPLC; gradient of n-hexane and isopropanol for NP-HPLC |
| ABTS•+ scavenging [88,89] | e−-transfer | UV-VIS, 410–430, 630–640, 734 nm | ABTS•+ in buffer or MeOH/buffer mixture (pH 7.4or 7.6) for RP-HPLC; MeOH for NP-HPLC | 0–100% organic modifier at pH 3–7.4 (TFA not recommended) for RP-HPLC; gradient of n-hexane and isopropanol for NP-HPLC |
| Galvinoxyl• scavenging [79] | H-donation | UV-VIS, 425 nm | Galvinoxyl• in 100% n-hexane or MTBE | Suitable for NP-HPLC using gradient of n-hexane and MTBE |
| Phosphomolybdate/Folin-Ciocalteau reagent [80–82] | e−-transfer | UV-VIS, 598, 750 nm | Phosphomolybdate/Folin-Ciocalteau reagent in acidic aqueous solution | Suitable for use with most RP-HPLC solvents; <80% organic modifier to prevent precipitation of salts; not suitable for NP-HPLC as reagent not soluble in 100% organic mobile phase |
| CUPRAC reagent [83] | e−-transfer | UV-VIS, 450 nm | Cu(II)-neocuproine in ammonium acetate buffer (pH 7) | Suitable for use with most RP-HPLC solvents; not suitable for NP-HPLC as reagent not soluble in 100% organic mobile phase |
| Acidic KMnO4 reagent [84–86,90] | unknown | CL | KMnO4 and Na polyphosphate or Na hexametaphosphate (enhancer) solution, adjusted to pH 2 or 2.3 with H2SO4 | Acidified aqueous phases combined with MeOH gradients; MeCN not recommended due to CL quenching |
| Assay | Reaction Mechanism | Detection | Reagent Solution Characteristics | HPLC Mobile Phase Compatibility |
|---|---|---|---|---|
| Assays based on H2O2 scavenging | ||||
| HPLC-CL H2O2/MP11/ luminol [125] | Oxidant: H2O2; Catalyst: MP11; Emitter: luminol oxidation product | CL, 425 nm | Reagent 1: MP11 and luminol in 30% methanol/buffer (pH 10); Reagent 2: aqueous H2O2 | No addition of acidifier; acetonitrile content ≥ 30% |
| HPLC-CL H2O2/luminol [126] | Oxidant: H2O2; Emitter: luminol oxidation product | CL | Reagent 1: luminol in 10% methanol/buffer (pH 8); Reagent 2: aqueous H2O2 | Isocratic elution with MeOH/1% H3PO4 (28/71); eluent neutralized before addition of CL reagents |
| HPLC-CL H2O2/EDTA/ luminol [127] | Oxidant: H2O2; Emitter: luminol oxidation product | CL | Reagent 1: luminol and EDTA in buffer (pH 10); Reagent 2: aqueous H2O2 | 0.1% H3PO4 aqueous phase combined with MeCN gradient < 65%; higher acid concentration or MeOH gradient caused baseline drift |
| Assays based on O2•− scavenging | ||||
| HPLC-CL HX/XOD/catalase/ K3Fe(CN)6/ luminol [128] | Oxidant: O2•− (HX/XOD/catalase) Catalyst: K3Fe(CN)6; Emitter: luminol oxidation product | CL | Reagent 1: HX and luminol in 10% methanol/buffer (pH 8); Reagent 2: aqueous K3Fe(CN)6; Reagent 3: XOD and catalase in buffer (pH 8) | No addition of acidifier; MeOH/water gradient |
| HPLC-CL HX/XOD/catalase/ luminol [126] | Oxidant: O2•− (HX/XOD/catalase) Emitter: luminol oxidation product | CL | Reagent 1: HX and luminol in 10% methanol/buffer (pH 8); Reagent 2: XOD and catalase in buffer (pH 8) | Isocratic elution with MeOH/1% H3PO4 (28/71); eluent neutralized before addition of CL reagents |
| HPLC-CL pyrogallol/EDTA/ luminol [127] | Oxidant: O2•− (pyrogallol); Emitter: luminol oxidation product | CL | Reagent 1: luminol and EDTA in buffer (pH 11); Reagent 2: aqueous pyrogallol | 0.1% H3PO4 aqueous phase combined with MeCN gradient < 65%; higher acid concentration or MeOH gradient caused baseline drift |
| HPLC-PAD [9] | Oxidant: O2•− (CYPs/CYP reductase/HRP/ SOD/NADPH); FL-probe: 4-HPAA | FL, λex 320 nm, λem 409 nm | Reagent 1: CYPs, CYP reductase, HRP and SOD in buffer (pH 7.8); Reagent 2: NADPH and 4-HPAA in buffer (pH 7.8) | Make-up flow with reverse gradient added |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Malherbe, C.J.; De Beer, D.; Joubert, E. Development of On-Line High Performance Liquid Chromatography (HPLC)-Biochemical Detection Methods as Tools in the Identification of Bioactives. Int. J. Mol. Sci. 2012, 13, 3101-3133. https://doi.org/10.3390/ijms13033101
Malherbe CJ, De Beer D, Joubert E. Development of On-Line High Performance Liquid Chromatography (HPLC)-Biochemical Detection Methods as Tools in the Identification of Bioactives. International Journal of Molecular Sciences. 2012; 13(3):3101-3133. https://doi.org/10.3390/ijms13033101
Chicago/Turabian StyleMalherbe, Christiaan J., Dalene De Beer, and Elizabeth Joubert. 2012. "Development of On-Line High Performance Liquid Chromatography (HPLC)-Biochemical Detection Methods as Tools in the Identification of Bioactives" International Journal of Molecular Sciences 13, no. 3: 3101-3133. https://doi.org/10.3390/ijms13033101
APA StyleMalherbe, C. J., De Beer, D., & Joubert, E. (2012). Development of On-Line High Performance Liquid Chromatography (HPLC)-Biochemical Detection Methods as Tools in the Identification of Bioactives. International Journal of Molecular Sciences, 13(3), 3101-3133. https://doi.org/10.3390/ijms13033101