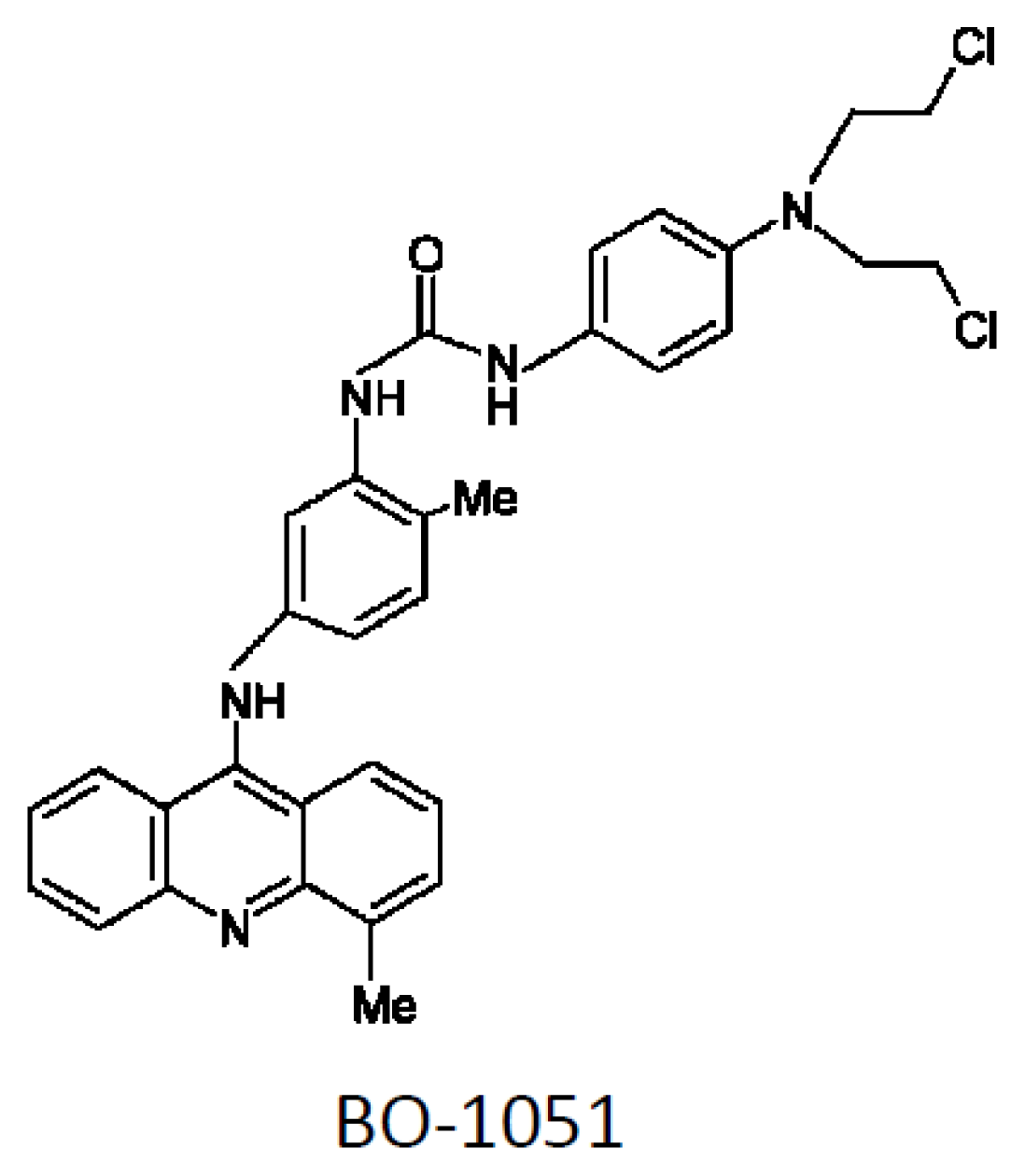
A Combined DNA-Affinic Molecule and N-Mustard Alkylating Agent Has an Anti-Cancer Effect and Induces Autophagy in Oral Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
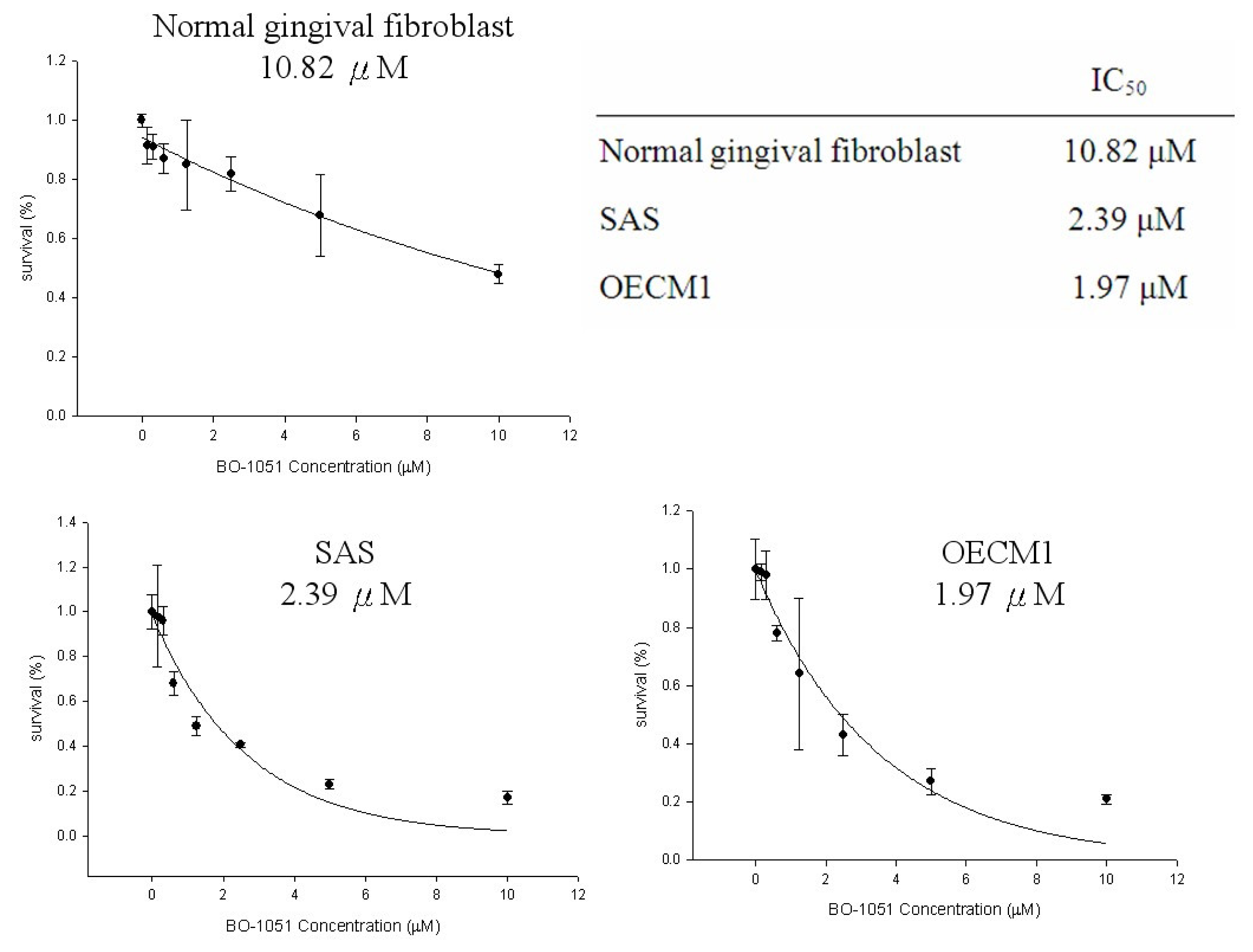
2.1. Cytotoxicity of BO-1051 in Oral Cancer Cell Lines
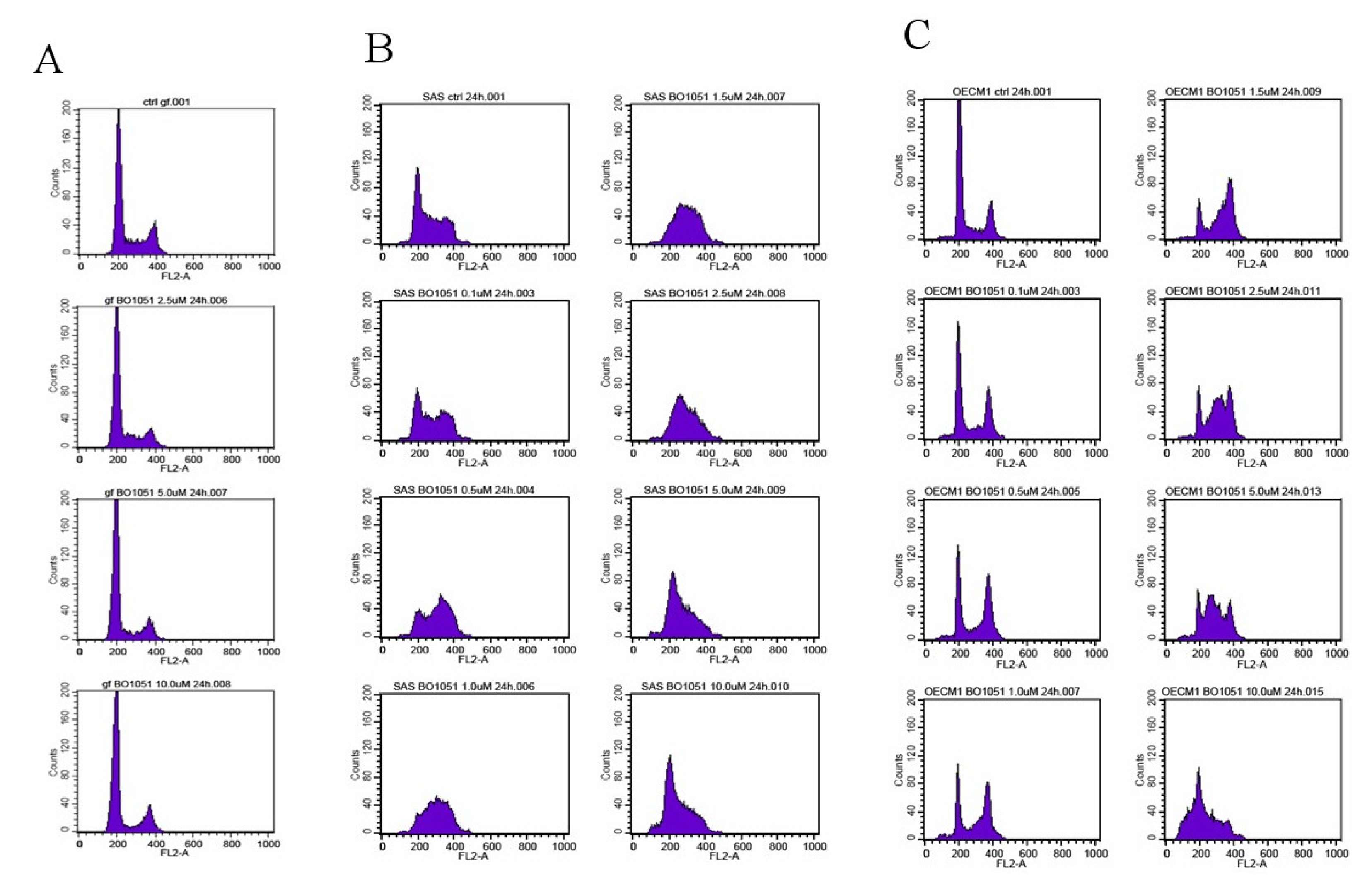
2.2. Diverse Effect of BO-1051 on Cell Cycle in Normal Gingival Fibroblasts and Oral Cancer Cells
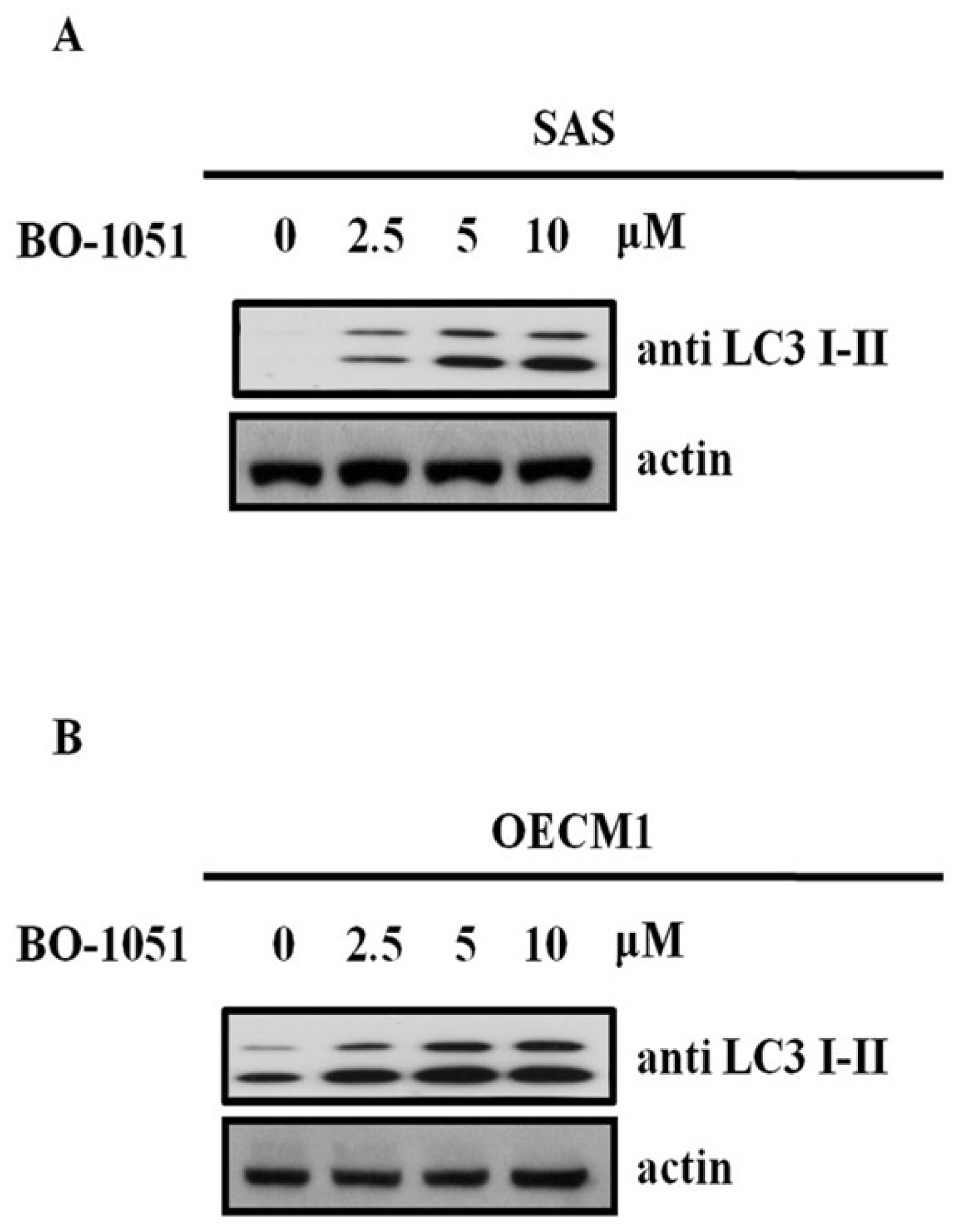
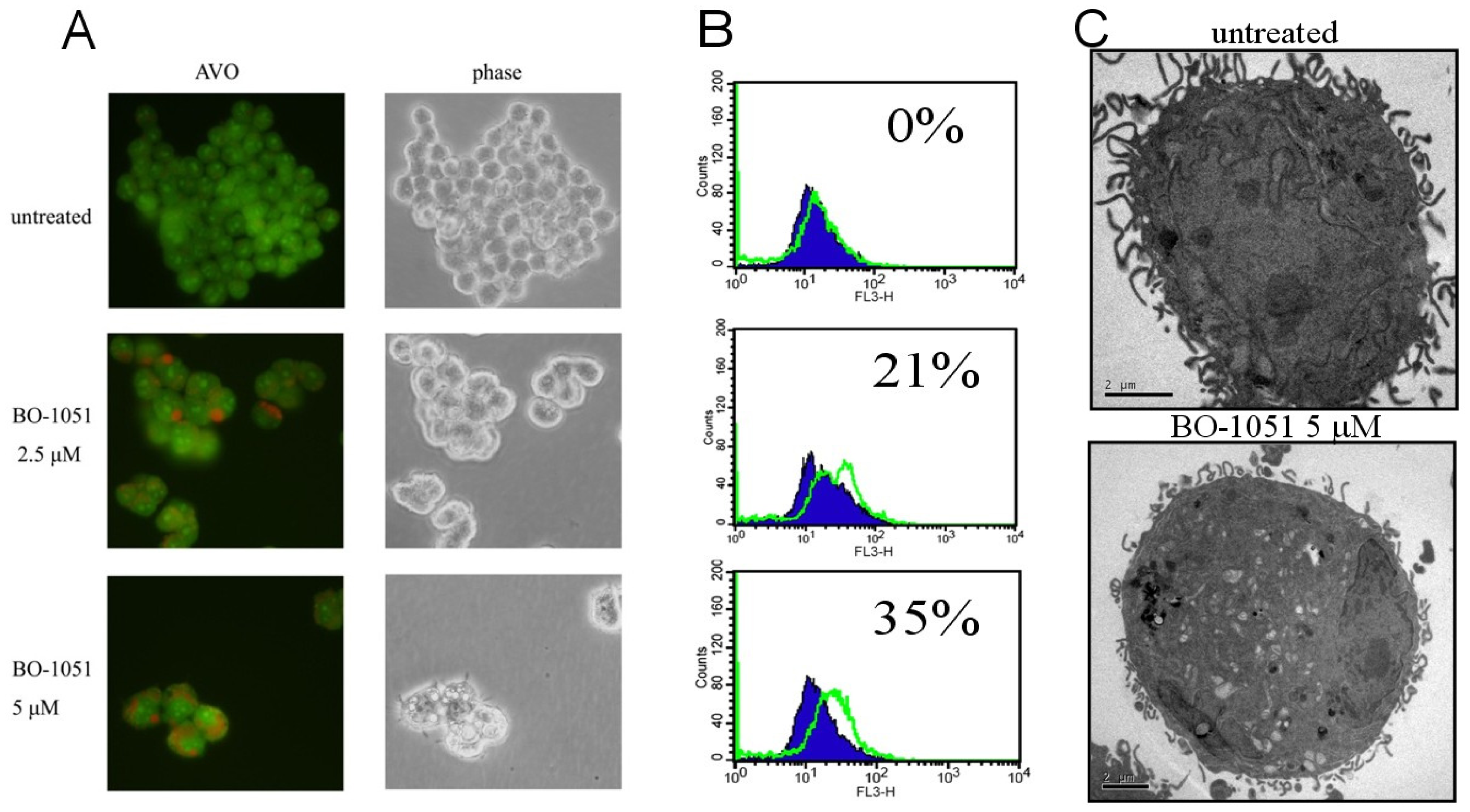
2.3. BO-1051 Induced Autophagy in Oral Cancer Cells
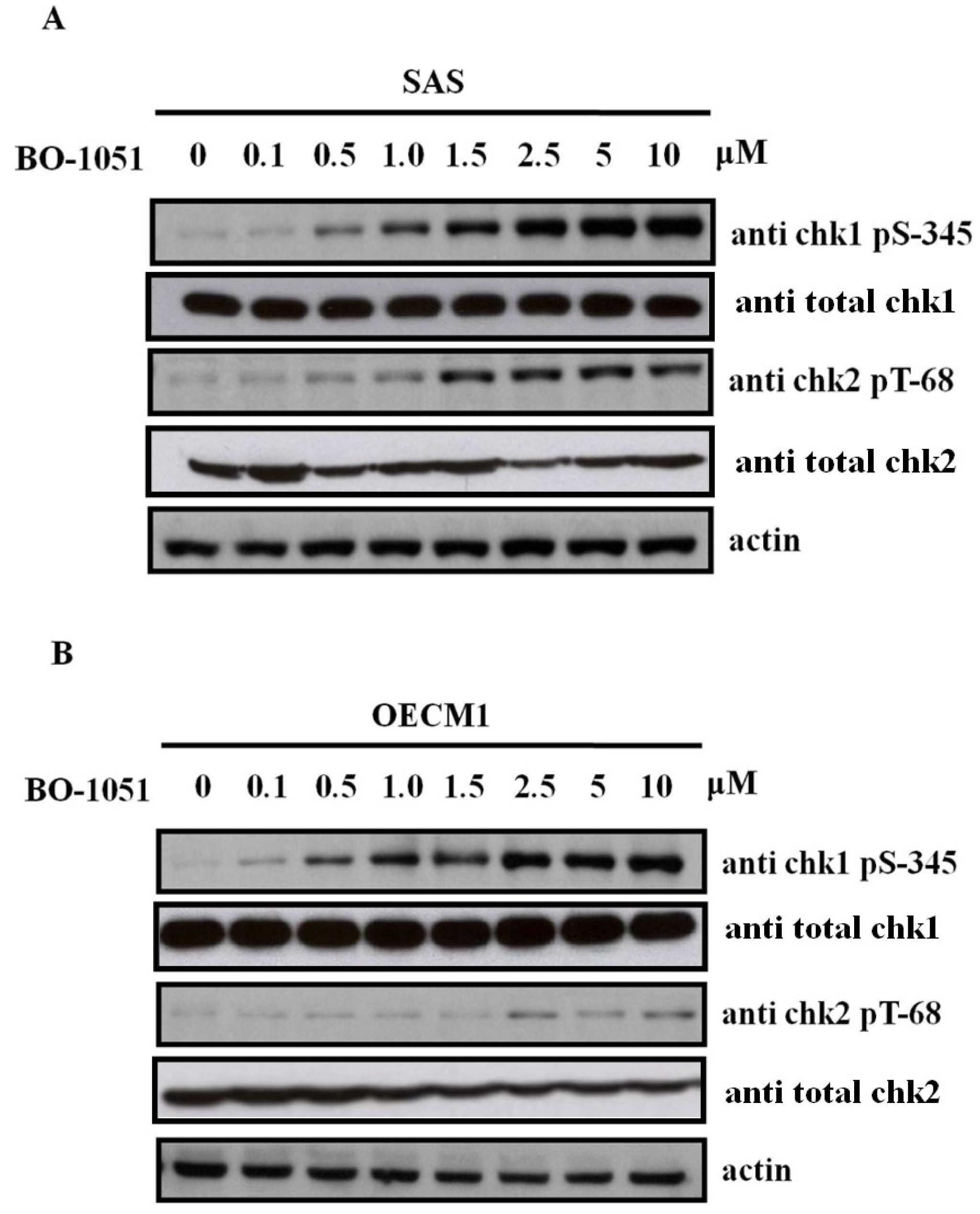
2.4. BO-1051 Induced Checkpoint Kinase Phosphorylation
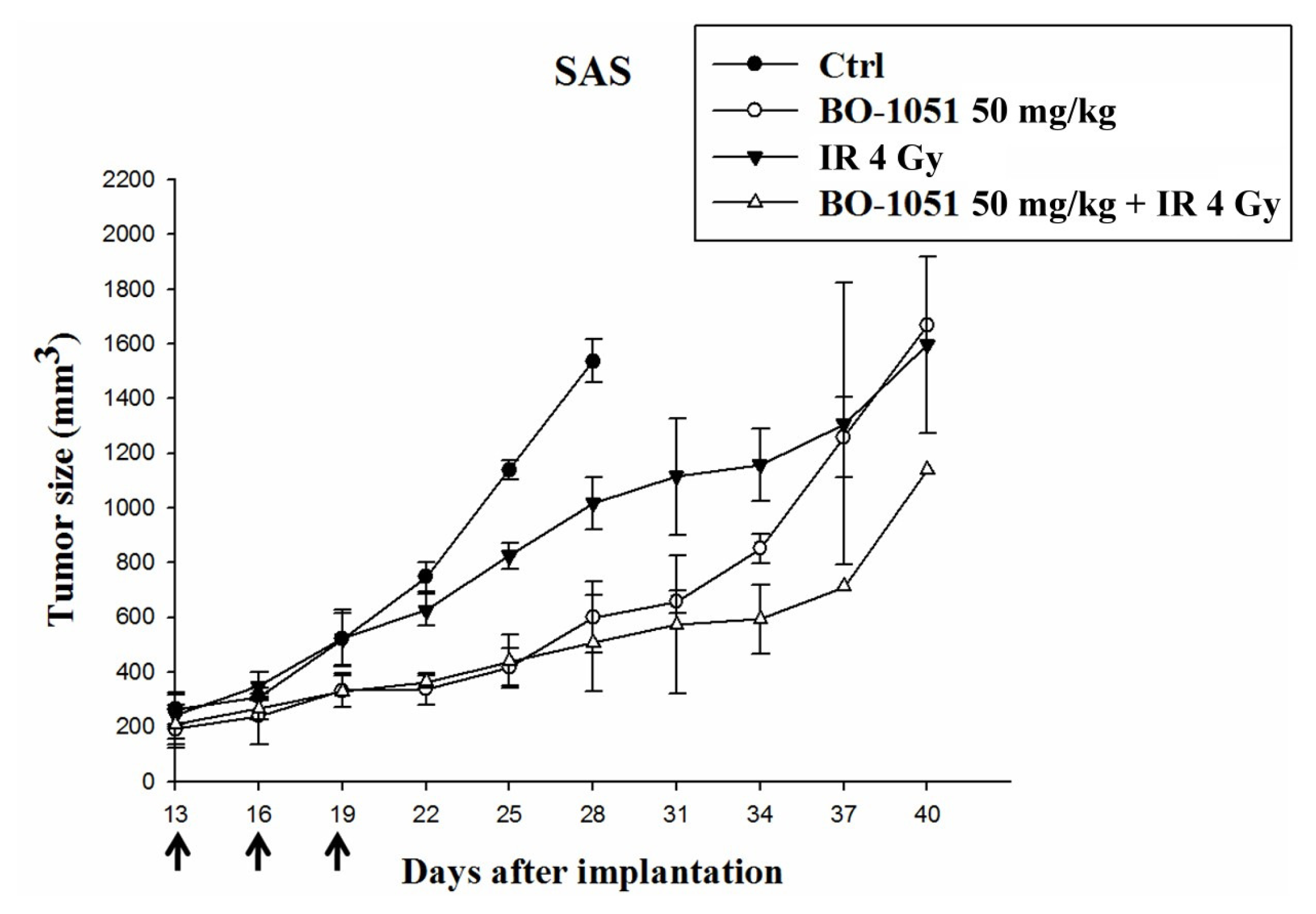
2.5. Tumor Suppressive Effects of BO-1051 on the Xenograft Tumors in Mice
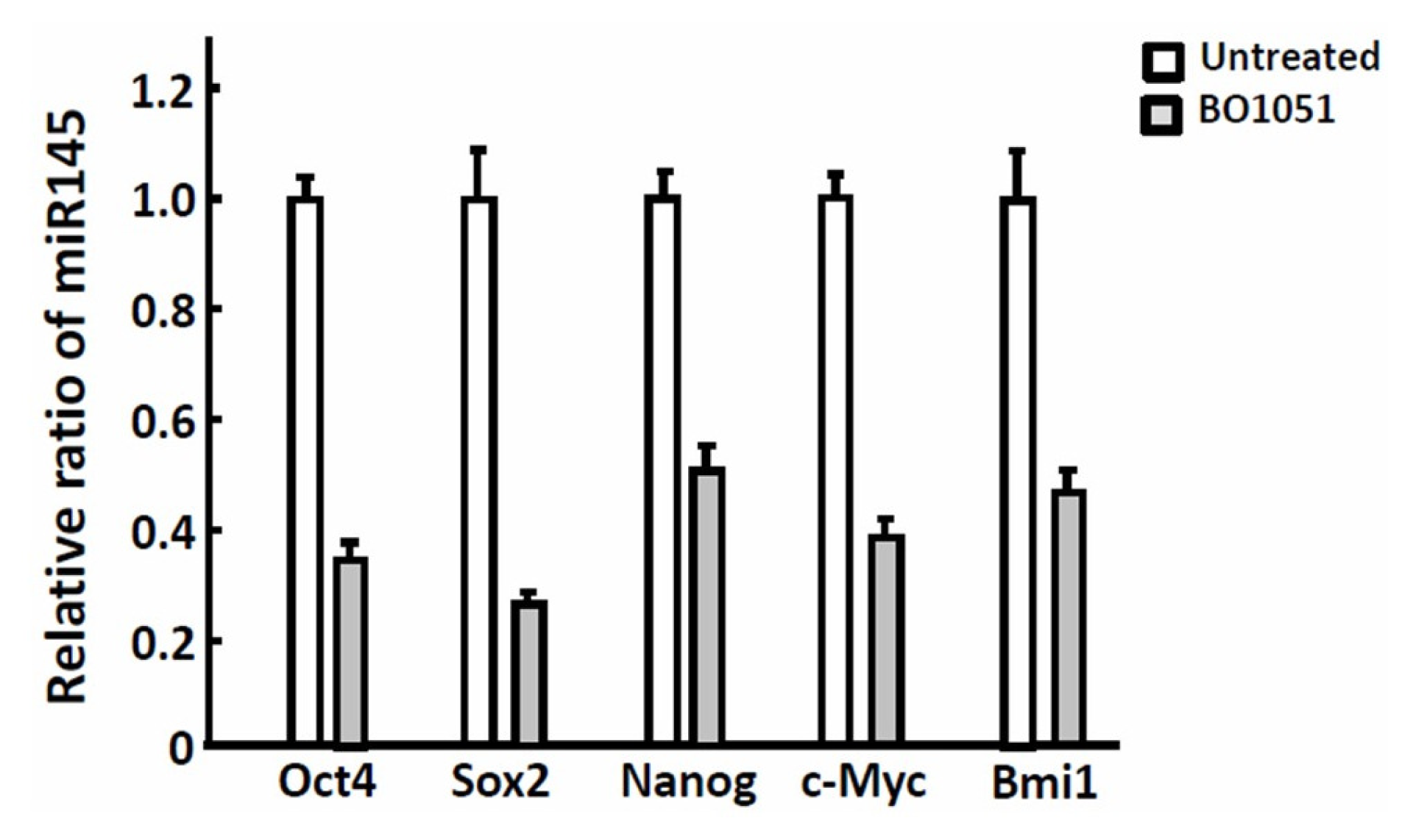
2.6. Discussion
3. Materials and Methods
3.1. MTT Assay
3.2. Cell-Cycle Analysis
3.3. Immunoblot Analysis
3.4. In Vivo Tumorigenesis Model
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin 2005, 55, 74–108. [Google Scholar]
- Seiwert, T.Y.; Cohen, E.E. State-of-the-art management of locally advanced head and neck cancer. Br. J. Cancer 2005, 92, 1341–1348. [Google Scholar]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar]
- Suzukake, K.; Vistica, B.P.; Vistica, D.T. Dechlorination of l-phenylalanine mustard by sensitive and resistant tumor cells and its relationship to intracellular glutathione content. Biochem. Pharmacol 1983, 32, 165–167. [Google Scholar]
- Brendel, M.; Ruhland, A. Relationships between functionality and genetic toxicology of selected DNA-damaging agents. Mutat. Res 1984, 133, 51–85. [Google Scholar]
- Baraldi, P.G.; Romagnoli, R.; Guadix, A.E.; Pineda de las Infantas, M.J.; Gallo, M.A.; Espinosa, A.; Martinez, A.; Bingham, J.P.; Hartley, J.A. Design, synthesis, and biological activity of hybrid compounds between uramustine and DNA minor groove binder distamycin A. J. Med. Chem 2002, 45, 3630–3638. [Google Scholar]
- Gourdie, T.A.; Valu, K.K.; Gravatt, G.L.; Boritzki, T.J.; Baguley, B.C.; Wakelin, L.P.; Wilson, W.R.; Woodgate, P.D.; Denny, W.A. DNA-directed alkylating agents. 1. Structure-activity relationships for acridine-linked aniline mustards: Consequences of varying the reactivity of the mustard. J. Med. Chem 1990, 33, 1177–1186. [Google Scholar]
- McClean, S.; Costelloe, C.; Denny, W.A.; Searcey, M.; Wakelin, L.P. Sequence selectivity, cross-linking efficiency and cytotoxicity of DNA-targeted 4-anilinoquinoline aniline mustards. Anticancer Drug Des 1999, 14, 187–204. [Google Scholar]
- Kapuriya, N.; Kapuriya, K.; Zhang, X.; Chou, T.C.; Kakadiya, R.; Wu, Y.T.; Tsai, T.H.; Chen, Y.T.; Lee, T.C.; Shah, A.; et al. Synthesis and biological activity of stable and potent antitumor agents, aniline nitrogen mustards linked to 9-anilinoacridines via a urea linkage. Bioorg. Med. Chem 2008, 16, 5413–5423. [Google Scholar]
- Elledge, S.J. Cell cycle checkpoints: Preventing an identity crisis. Science 1996, 274, 1664–1672. [Google Scholar]
- Tsai, L.L.; Yu, C.C.; Chang, Y.C.; Yu, C.H.; Chou, M.Y. Markedly increased Oct4 and Nanog expression correlates with cisplatin resistance in oral squamous cell carcinoma. J. Oral Pathol. Med 2011, 40, 621–628. [Google Scholar]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuasen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol 2005, 7, 195–201. [Google Scholar]
- Tomasz, M.; Palom, Y. The mitomycin bioreductive antitumor agents: Cross-linking and alkylation of DNA as the molecular basis of their activity. Pharmacol. Ther 1997, 76, 73–87. [Google Scholar]
- Panasci, L.; Xu, Z.Y.; Bello, V.; Aloyz, R. The role of DNA repair in nitrogen mustard drug resistance. Anticancer Drugs 2002, 13, 211–220. [Google Scholar]
- Chu, P.M.; Chiou, S.H.; Su, T.L.; Lee, Y.J.; Chen, L.H.; Chen, Y.W.; Yen, S.H.; Chen, M.T.; Chen, M.H.; Shih, Y.H.; et al. Enhancement of radiosensitivity in human glioblastoma cells by the DNA N-mustard alkylating agent BO-1051 through augmented and sustained DNA damage response. Radiat. Oncol 2011, 6. [Google Scholar] [CrossRef]
- Chen, L.H.; Loong, C.C.; Su, T.L.; Lee, Y.J.; Chu, P.M.; Tsai, M.L.; Tsai, P.H.; Tu, P.H.; Chi, C.W.; Lee, H.C.; et al. Autophagy inhibition enhances apoptosis triggered by BO-1051, an N-mustard derivative, and involves the ATM signaling pathway. Biochem. Pharmacol 2011, 81, 594–605. [Google Scholar]
- Ferraro, E.; Cecconi, F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem. Biophys 2007, 462, 210–219. [Google Scholar]
- Chang, C.P.; Yang, M.C.; Liu, H.S.; Lin, Y.S.; Lei, H.Y. Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology 2007, 45, 286–296. [Google Scholar]
- Longo, L.; Platini, F.; Scardino, A.; Alabiso, O.; Vasapollo, G.; Tessitore, L. Autophagy inhibition enhances anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol. Cancer Ther 2008, 7, 2476–2485. [Google Scholar]
- Ko, H.; Kim, Y.J.; Park, J.S.; Park, J.H.; Yang, H.O. Autophagy inhibition enhances apoptosis induced by ginsenoside Rk1 in hepatocellular carcinoma cells. Biosci. Biotechnol. Biochem 2009, 73, 2183–2189. [Google Scholar]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004, 11, 448–457. [Google Scholar]
- Tsuboi, Y.; Kurimoto, M.; Nagai, S.; Hayakawa, Y.; Kamiyama, H.; Hayashi, N.; Kitajima, I.; Endo, S. Induction of autophagic cell death and radiosensitization by the pharmacological inhibition of nuclear factor-kappa B activation in human glioma cell lines. J. Neurosurg 2009, 110, 594–604. [Google Scholar]
- Fujiwara, K.; Iwado, E.; Mills, G.B.; Sawaya, R.; Kondo, S.; Kondo, Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int. J. Oncol 2007, 31, 753–760. [Google Scholar]
- Kim, K.W.; Hwang, M.; Moretti, L.; Jaboin, J.J.; Cha, Y.I.; Lu, B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 2008, 4, 659–668. [Google Scholar]








© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lo, W.-L.; Chu, P.-Y.; Lee, T.-H.; Su, T.-L.; Chien, Y.; Chen, Y.-W.; Huang, P.-I.; Tseng, L.-M.; Tu, P.-H.; Kao, S.-Y.; et al. A Combined DNA-Affinic Molecule and N-Mustard Alkylating Agent Has an Anti-Cancer Effect and Induces Autophagy in Oral Cancer Cells. Int. J. Mol. Sci. 2012, 13, 3277-3290. https://doi.org/10.3390/ijms13033277
Lo W-L, Chu P-Y, Lee T-H, Su T-L, Chien Y, Chen Y-W, Huang P-I, Tseng L-M, Tu P-H, Kao S-Y, et al. A Combined DNA-Affinic Molecule and N-Mustard Alkylating Agent Has an Anti-Cancer Effect and Induces Autophagy in Oral Cancer Cells. International Journal of Molecular Sciences. 2012; 13(3):3277-3290. https://doi.org/10.3390/ijms13033277
Chicago/Turabian StyleLo, Wen-Liang, Pen-Yuan Chu, Tsung-Heng Lee, Tsann-Long Su, Yueh Chien, Yi-Wei Chen, Pin-I Huang, Ling-Ming Tseng, Pang-Hsien Tu, Shou-Yen Kao, and et al. 2012. "A Combined DNA-Affinic Molecule and N-Mustard Alkylating Agent Has an Anti-Cancer Effect and Induces Autophagy in Oral Cancer Cells" International Journal of Molecular Sciences 13, no. 3: 3277-3290. https://doi.org/10.3390/ijms13033277