Autophagy in Premature Senescent Cells Is Activated via AMPK Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Autophagy Activity Increases in Stress-Induced Premature Senescent Cells
2.2. Blockage of Autophagy Results in Increased Apoptosis and Impaired Senescence Manifestation During H2O2-Induced Senescent Process
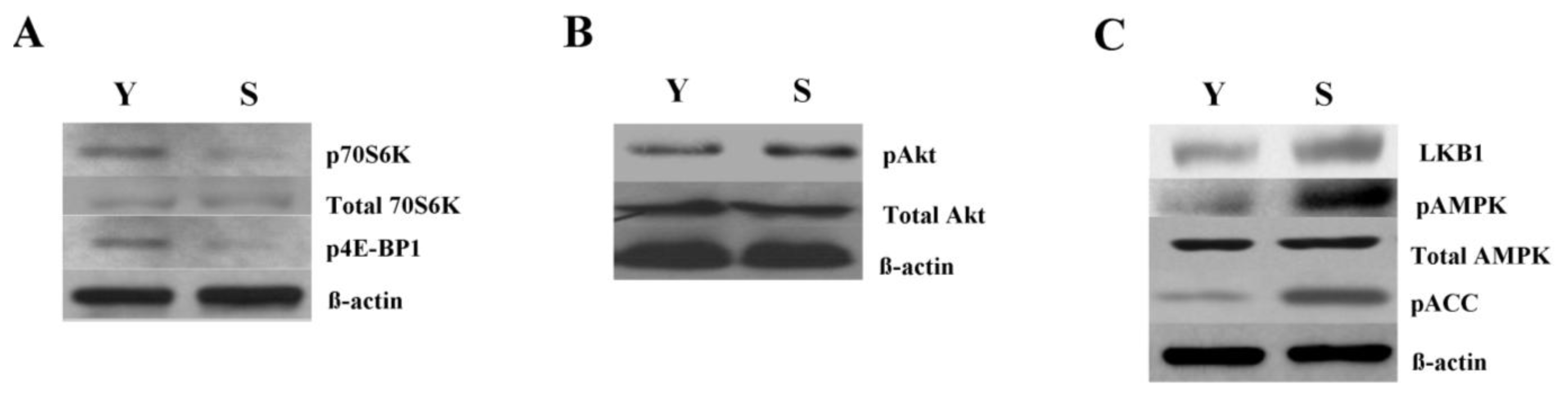
2.3. Increased Autophagy Is Linked to mTOR Inhibition in Senescent Cells
2.4. mTOR Inhibition and Increased Autophagy in Senescent Cells Are Linked to AMPK Activation
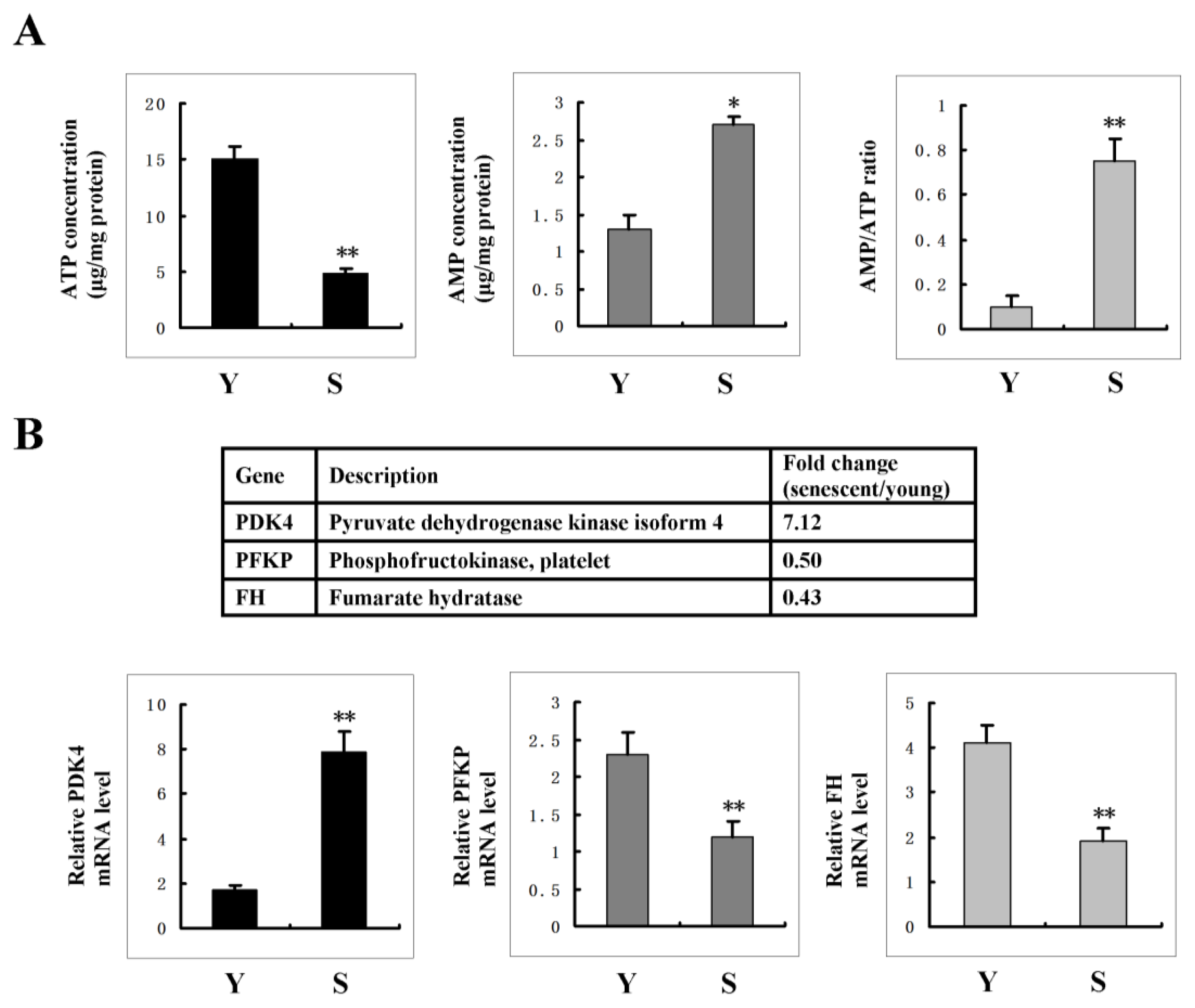
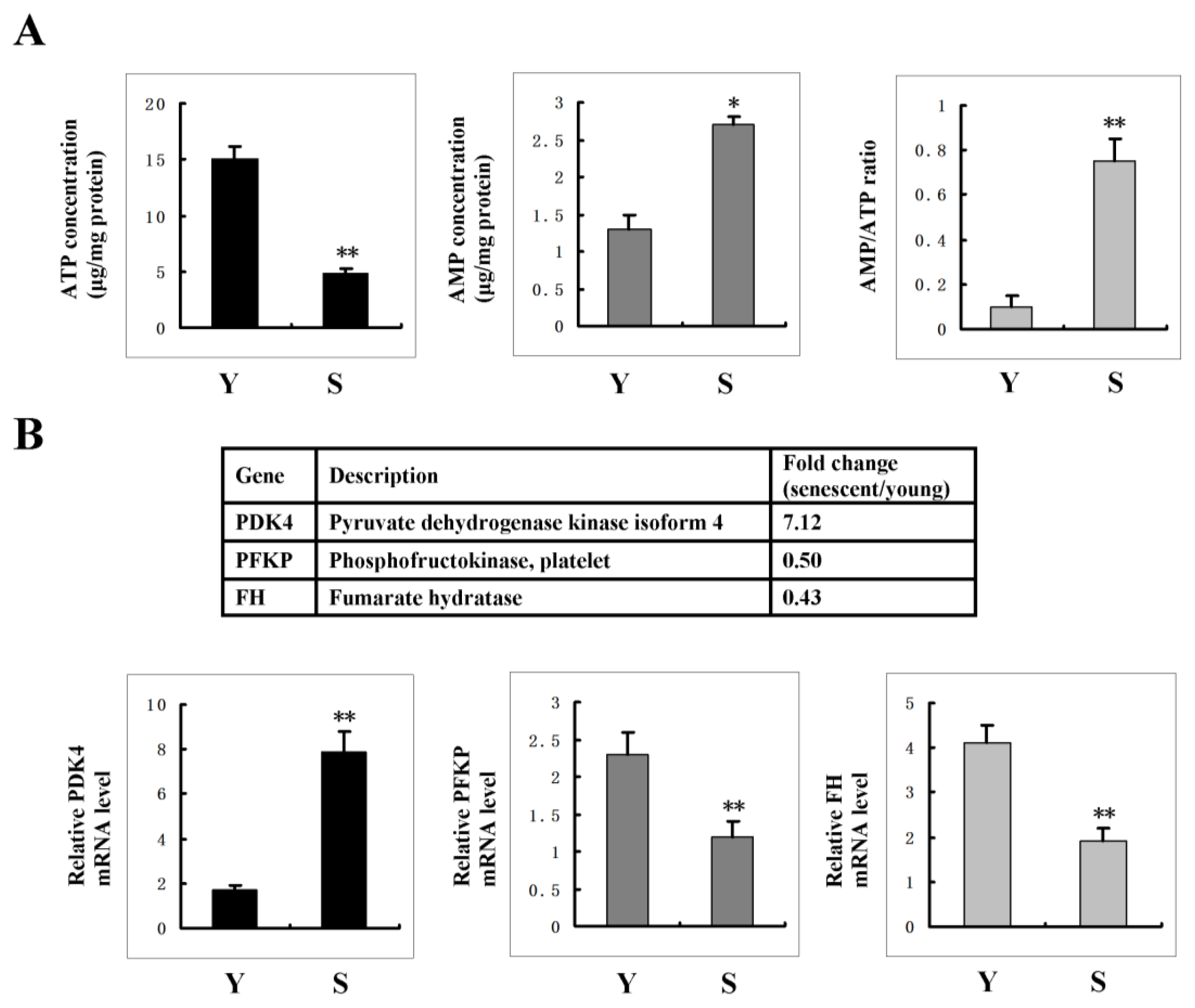
2.5. AMPK Activation Is Linked to the Increase of AMP: ATP Ratio in Senescent Cells
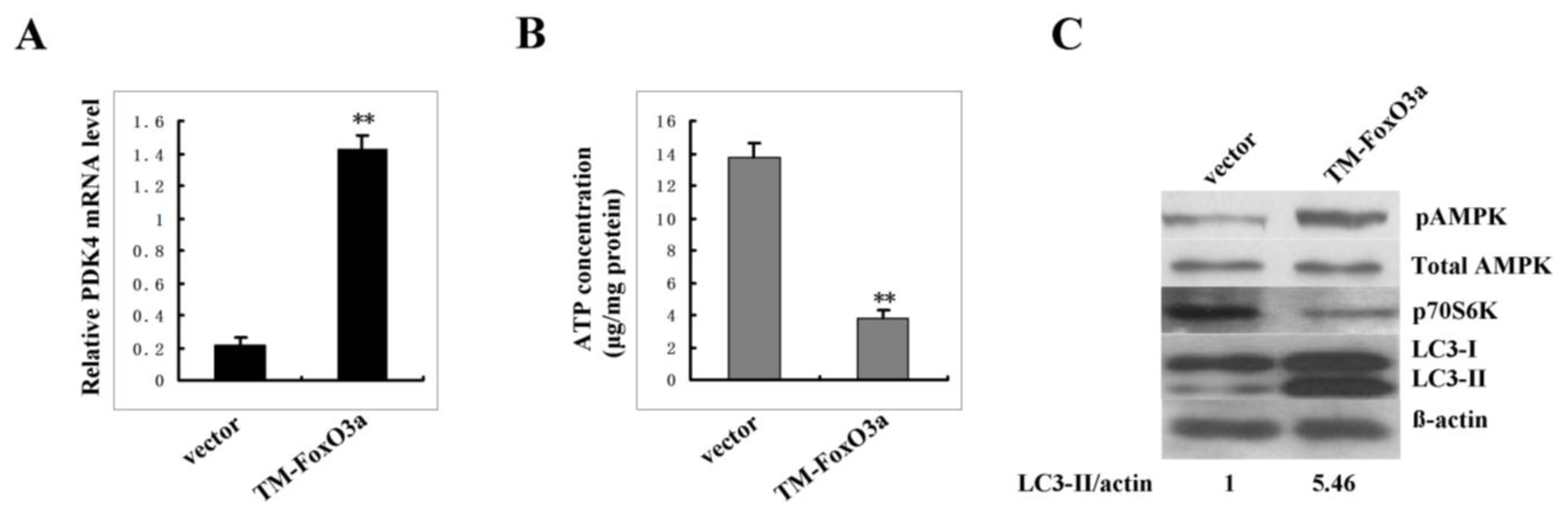
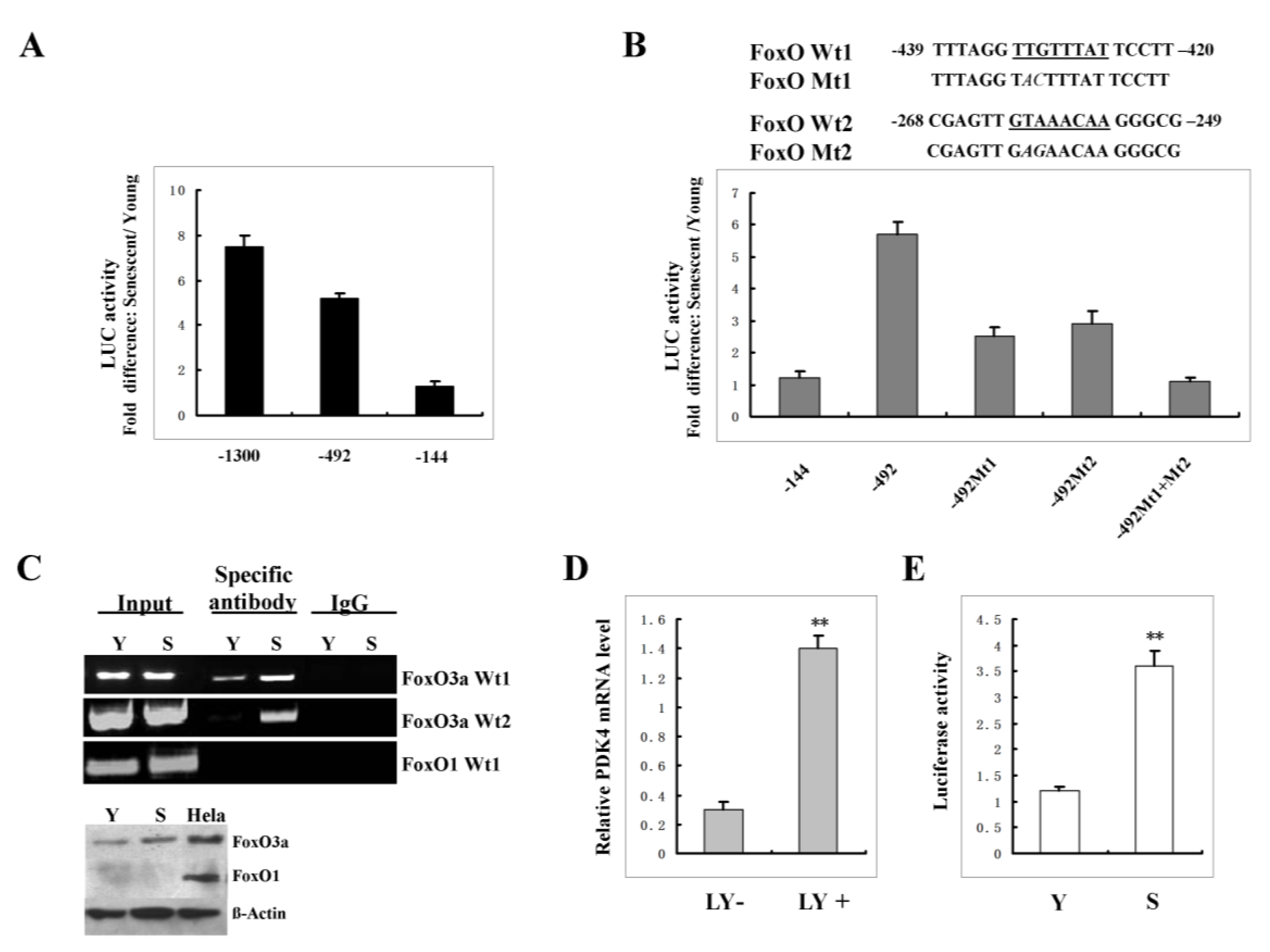
2.6. FoxO3A Activation Transcriptionally Up-Regulates PDK4 Expression and Contributes to the Increase of AMP: ATP Ratio in Senescent Cells
2.7. Constitutively Active FoxO3A Up-Regulates Autophagy Activity in Young Cells
2.8. Knockdown of FoxO3A Inhibits Autophagy Activity in Senescent Process
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Labeling of Autophagic Structures with MDC
4.4. Electron Microscopy
4.5. Immunoblotting
4.6. Flow Cytometry
4.7. Caspase-3 Activity Assay
4.8. Microarray Analysis
4.9. SA-β-gal Staining
4.10. siRNA Transfection
4.11. Plasmid Construct
4.12. Transient Transfection and Luciferase Assay
4.13. Site-Directed Mutagenesis
4.14. Chromatin Immunoprecipitation
4.15. Nucleotide Extraction and AMP/ATP Analysis
4.16. Real-Time PCR
4.17. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Serrano, M.; Blasco, M.A. Putting the stress on senescence. Curr. Opin. Cell Biol 2001, 13, 748–753. [Google Scholar]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res 2003, 63, 2705–2715. [Google Scholar]
- Alexander, K.; Hinds, P.W. Requirement for p27KIP1 in retinoblastoma protein-mediated senescence. Mol. Cell. Biol 2001, 21, 3616–3631. [Google Scholar]
- Sager, R. Senescence as a mode of tumor suppression. Environ. Health Persp 1991, 93, 59–62. [Google Scholar]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001, 11, S27–S31. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; Mcarthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for P53 are developmentally normal but susceptible to spontaneous tumors. Nature 1992, 356, 215–221. [Google Scholar]
- Ghebranious, N.; Donehower, L.A. Mouse models in tumor suppression. Oncogene 1998, 17, 3385–3400. [Google Scholar]
- Wang, C.W.; Klionsky, D.J. The molecular mechanism of autophagy. Mol. Med 2003, 9, 65–76. [Google Scholar]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome c and apoptosis in human prostate cancer cells. Cancer Res 2006, 66, 5828–5835. [Google Scholar]
- Lu, X.F.; Jiang, X.G.; Lu, Y.B.; Bai, J.H.; Mao, Z.B. Characterization of a novel positive transcription regulatory element that differentially regulates the insulin-like growth factor binding protein-3 (IGFBP-3) gene in senescent cells. J. Biol. Chem 2005, 280, 22606–22615. [Google Scholar]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Torok, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol 2004, 6, 1221–1228. [Google Scholar]
- Stroikin, Y.; Dalen, H.; Brunk, U.T.; Terman, A. Testing the “garbage” accumulation theory of ageing: Mitotic activity protects cells from death induced by inhibition of autophagy. Biogerontology 2005, 6, 39–47. [Google Scholar]
- Gerland, L.M.; Peyrol, S.; Lallemand, C.; Branche, R.; Magaud, J.P.; Ffrench, M. Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp. Gerontol 2003, 38, 887–895. [Google Scholar]
- Biederbick, A.; Kern, H.F.; Elsasser, H.P. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol 1995, 66, 3–14. [Google Scholar]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol 2004, 36, 2491–2502. [Google Scholar]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem 2000, 275, 992–998. [Google Scholar]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001, 2, 330–335. [Google Scholar]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase—Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J 2003, 376, 577–586. [Google Scholar]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res 2001, 61, 3443–3449. [Google Scholar]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; Rubinsztein, D.C. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet 2004, 36, 585–595. [Google Scholar]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004, 18, 2893–2904. [Google Scholar] [Green Version]
- Pullen, N.; Thomas, G. The modular phosphorylation and activation of p70s6k. FEBS Lett 1997, 410, 78–82. [Google Scholar]
- Arsham, A.M.; Neufeld, T.P. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol 2006, 18, 589–597. [Google Scholar]
- Hardie, D.G. The AMP-activated protein kinase pathway—new players upstream and downstream. J. Cell Sci 2004, 117, 5479–5487. [Google Scholar]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996, 15, 6541–6551. [Google Scholar]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar]
- Sugden, M.C.; Holness, M.J. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol. Biochem 2006, 112, 139–149. [Google Scholar]
- Kwon, H.S.; Huang, B.; Unterman, T.G.; Harris, R.A. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 2004, 53, 899–910. [Google Scholar]
- Jia, K.; Chen, D.; Riddle, D.L. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Dev. Biol 2004, 131, 3897–3906. [Google Scholar]
- Wu, A.L.; Kim, J.H.; Zhang, C.; Unterman, T.G.; Chen, J. Forkhead box protein O1 negatively regulates skeletal myocyte differentiation through degradation of mammalian target of rapamycin pathway components. Endocrinology 2008, 149, 1407–1414. [Google Scholar]
- Southgate, R.J.; Neill, B.; Prelovsek, O.; El-Osta, A.; Kamei, Y.; Miura, S.; Ezaki, O.; McLoughlin, T.J.; Zhang, W.; Unterman, T.G.; Febbraio, M.A. FOXO1 regulates the expression of 4E-BP1 and inhibits mTOR signaling in mammalian skeletal muscle. J. Biol. Chem 2007, 282, 21176–21186. [Google Scholar]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; di Lisi, R.; Sandri, C.; Zhao, J.; Goldberg, A.L.; Schiaffino, S.; Sandri, M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007, 6, 458–471. [Google Scholar]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007, 6, 472–483. [Google Scholar]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar]
- Wang, Y.; Meng, A.; Zhou, D. Inhibition of phosphatidylinostol 3-kinase uncouples H2O2-induced senescent phenotype and cell cycle arrest in normal human diploid fibroblasts. Exp. Cell Res 2004, 298, 188–196. [Google Scholar]
- Courtois-Cox, S.; Genther Williams, S.M.; Reczek, E.E.; Johnson, B.W.; McGillicuddy, L.T.; Johannessen, C.M.; Hollstein, P.E.; MacCollin, M.; Cichowski, K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006, 10, 459–472. [Google Scholar]
- Mawal-Dewan, M.; Lorenzini, A.; Frisoni, L.; Zhang, H.; Cristofalo, V.J.; Sell, C. Regulation of collagenase expression during replicative senescence in human fibroblasts by Akt-forkhead signaling. J. Biol. Chem 2002, 277, 7857–7864. [Google Scholar]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci 2007, 120, 2479–2487. [Google Scholar]





© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, L.; Xie, B.; Mao, Z. Autophagy in Premature Senescent Cells Is Activated via AMPK Pathway. Int. J. Mol. Sci. 2012, 13, 3563-3582. https://doi.org/10.3390/ijms13033563
Guo L, Xie B, Mao Z. Autophagy in Premature Senescent Cells Is Activated via AMPK Pathway. International Journal of Molecular Sciences. 2012; 13(3):3563-3582. https://doi.org/10.3390/ijms13033563
Chicago/Turabian StyleGuo, Liujing, Bushan Xie, and Zebin Mao. 2012. "Autophagy in Premature Senescent Cells Is Activated via AMPK Pathway" International Journal of Molecular Sciences 13, no. 3: 3563-3582. https://doi.org/10.3390/ijms13033563