Determination of Aniline and Its Derivatives in Environmental Water by Capillary Electrophoresis with On-Line Concentration

Abstract
:1. Introduction
2. Results and Discussion
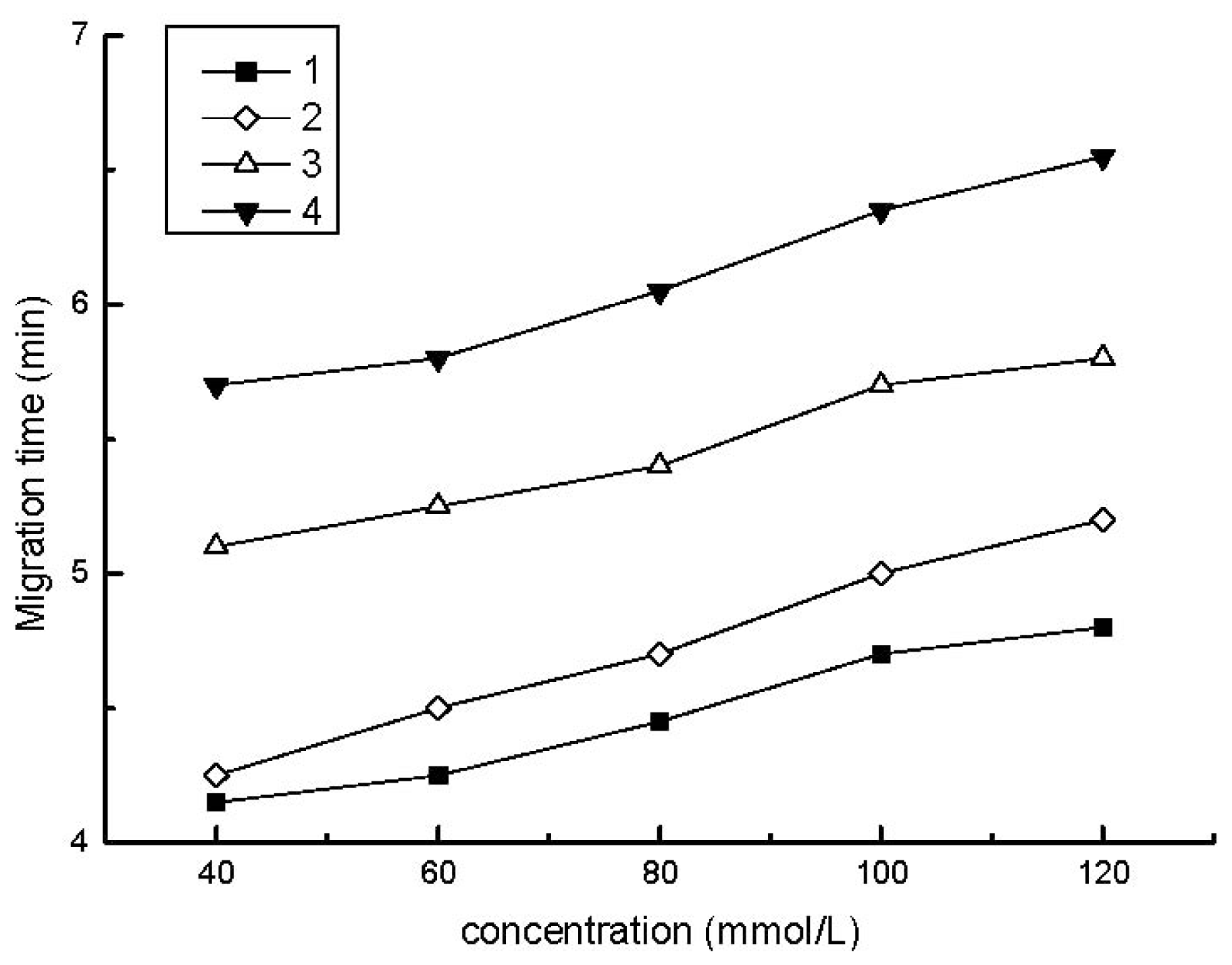
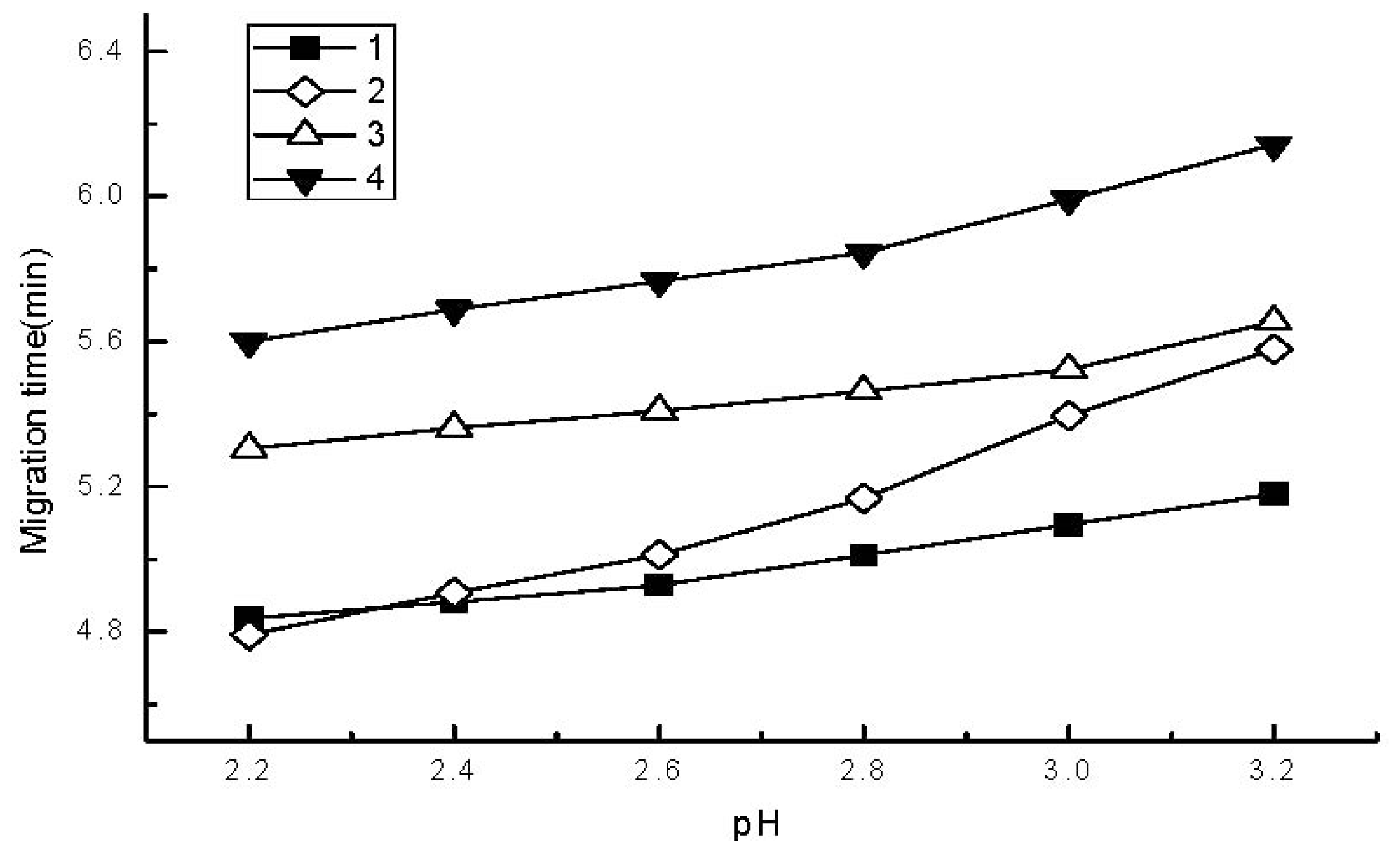
2.1. Development of Separation Parameters
2.2. Optimization of Field-Enhanced Sample Injection
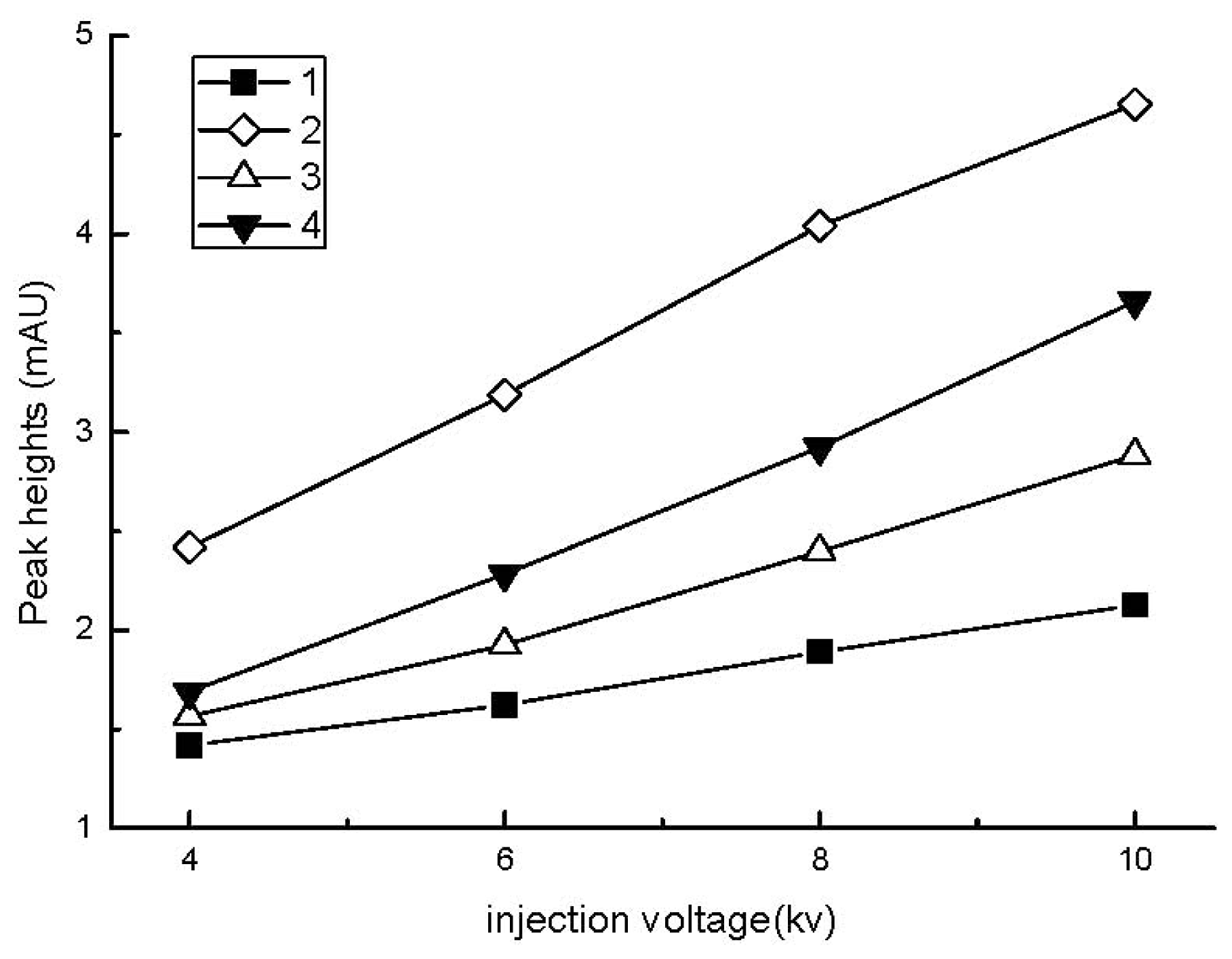
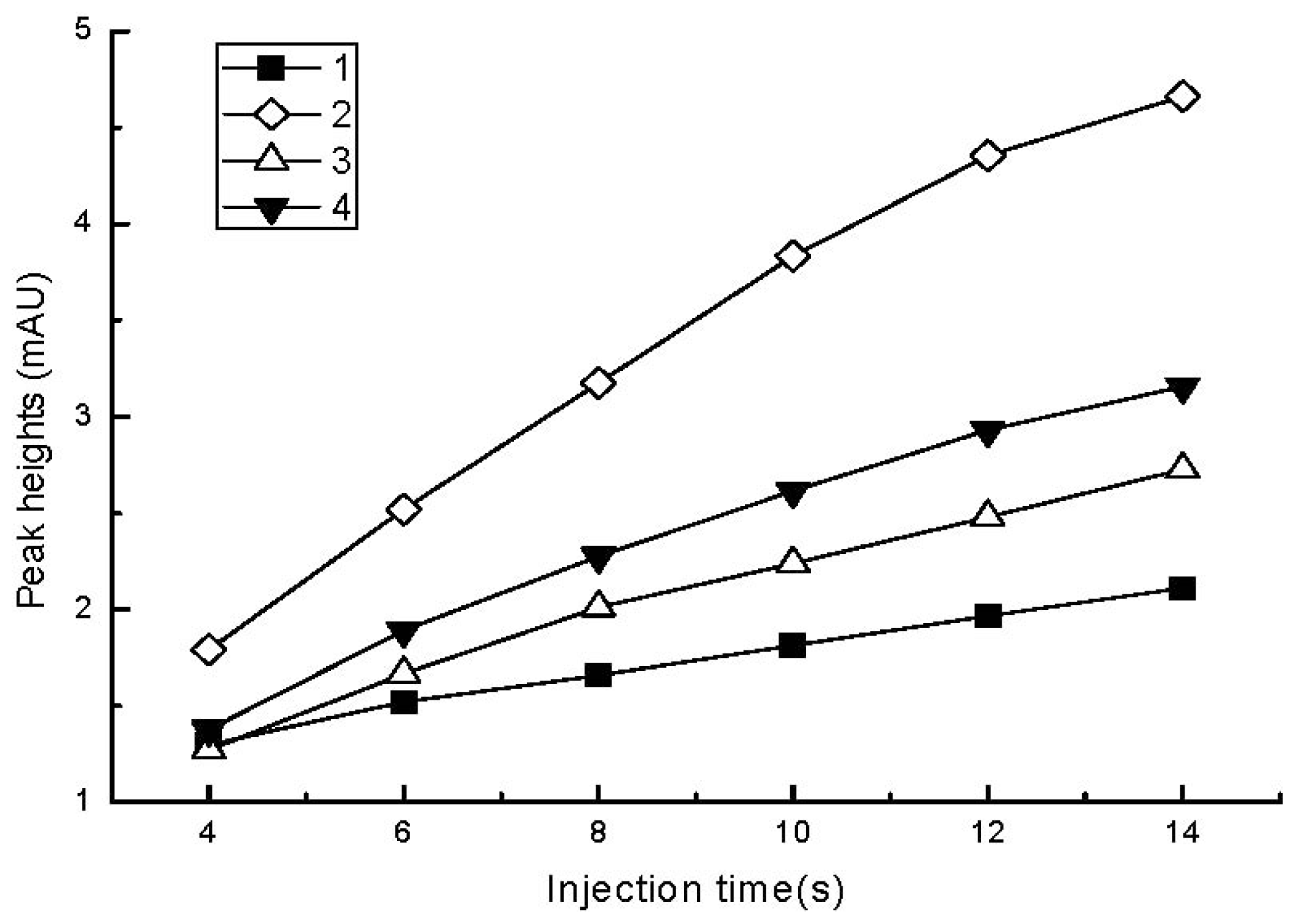
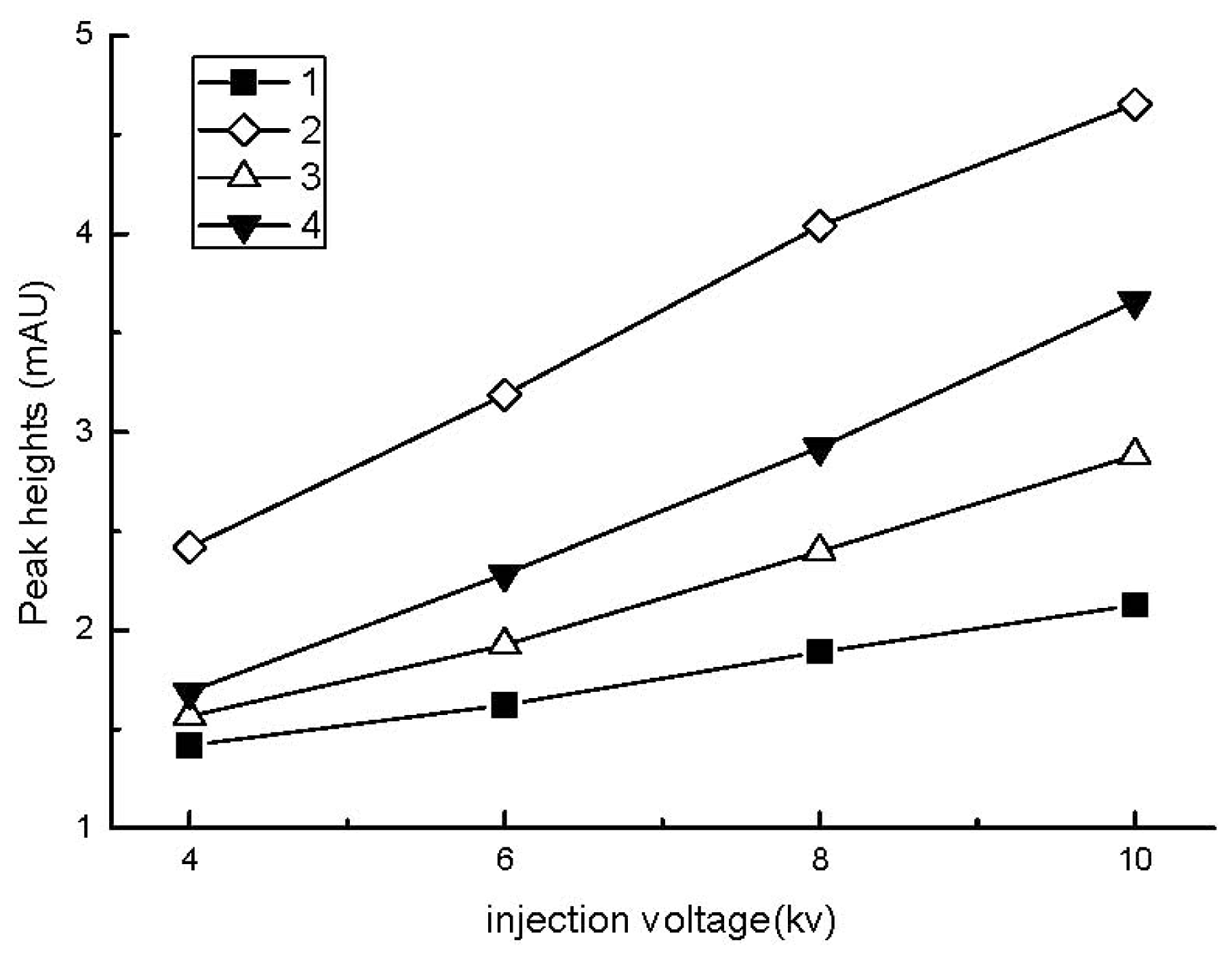
2.2.1. Effect of Sample Injection Time and Voltage
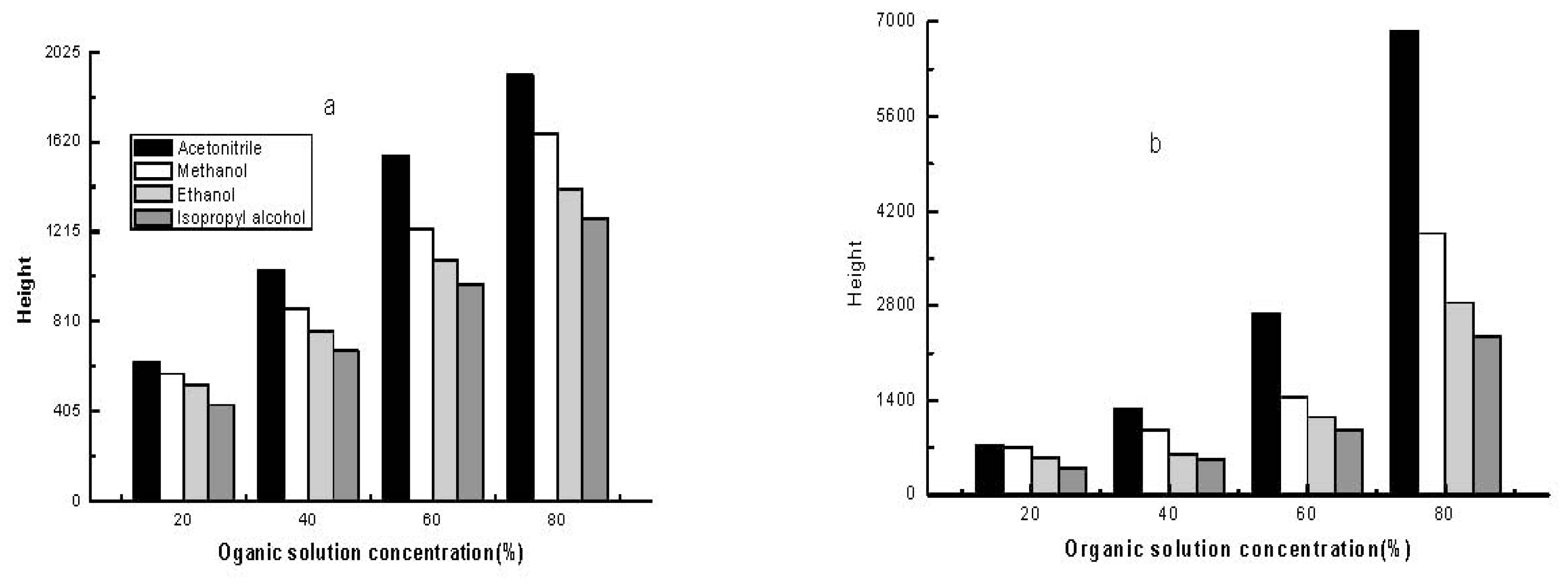
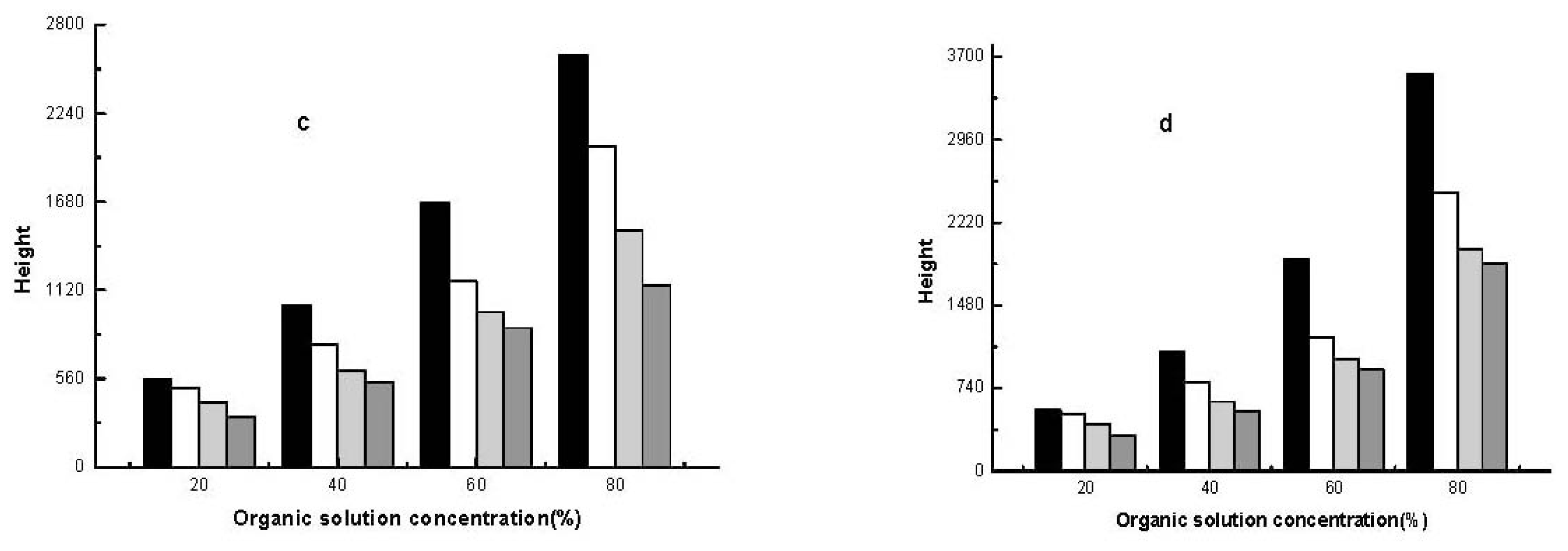
2.2.2. Effect of Different Organic Solvents on Sample Stacking
2.3. Reproducibility, Linearity and Detection Limits
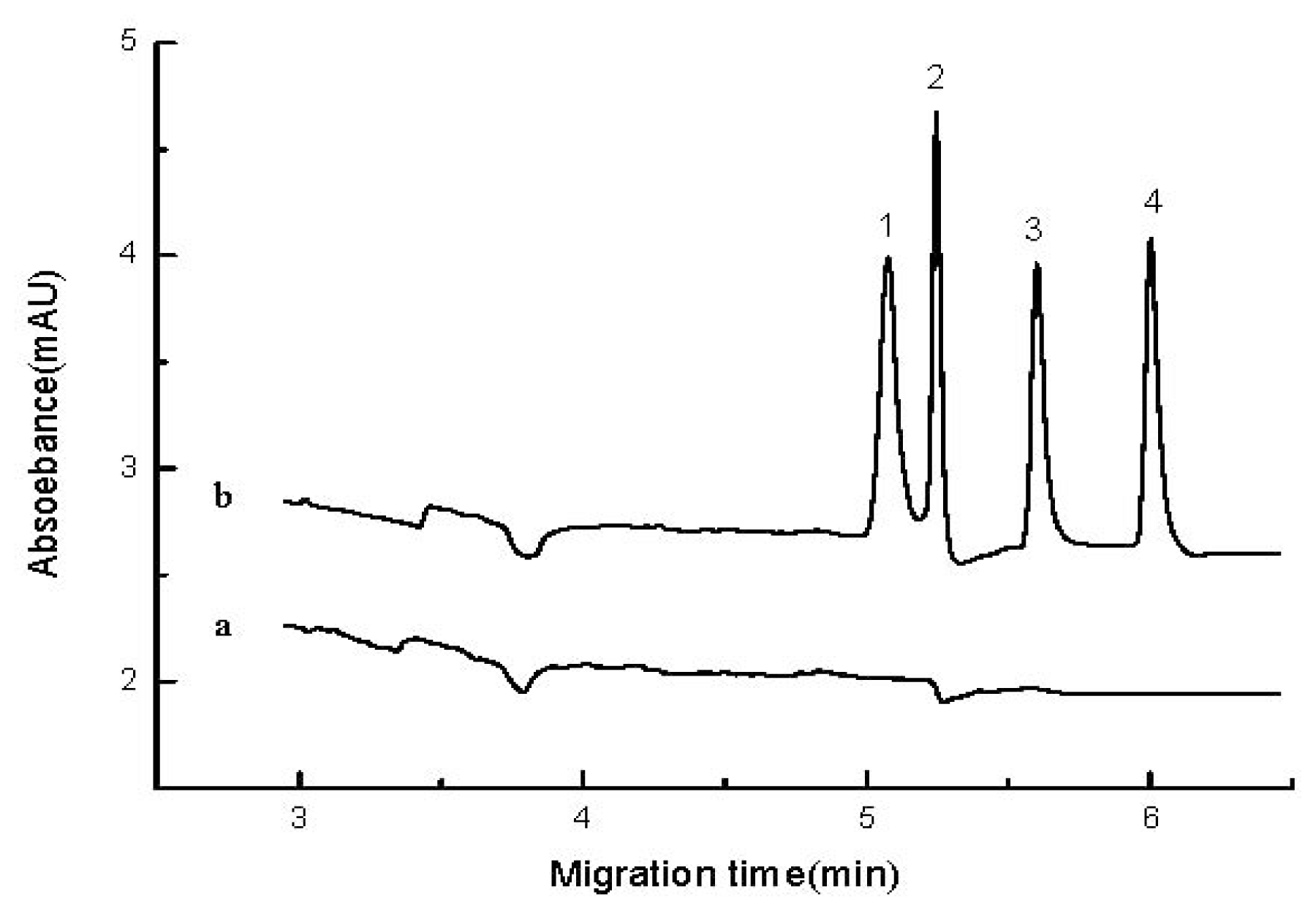
2.4. Real Sample Analysis and Recovery
3. Experimental Section
3.1. Chemicals
3.2. Apparatus
3.3. Experimental Procedure
3.4. Preparation of Real Samples
4. Conclusions
Acknowledgments
References
- Arrhenius, E.; Hultin, T. The effect of 2-aminofluorene and related aromatic amines on the protein and ribonucleic acid metabolism of liver slices. Exp. Cell Res 1961, 22, 476–486. [Google Scholar]
- Scribner, J.D.; Fisk, S.R.; Scribner, N.K. Mechanisms of action of carcinogenic aromatic amines: An investigation using mutagenesis in bacteria. Chem. Biol. Interact 1979, 26, 11–25. [Google Scholar]
- King, C.M.; Land, S.J.; Jones, R.F.; Debiec-Rychter, M.; Lee, M.; Wang, C.Y. Role of acetyltransferases in the metabolism and carcinogenicity of aromatic amines. Mutat. Res 1997, 376, 123–128. [Google Scholar]
- Benigni, R.; Passerini, L. Carcinogenicity of the aromatic amines: From structure-activity relationships to mechanisms of action and risk assessment. Mutat. Res 2002, 511, 191–206. [Google Scholar]
- Zhu, J.; Aikawa, B. Determination of aniline and related mono-aromatic amines in indoor air in selected Canadian residences by a modified thermal desorption GC/MS method. Environ. Int 2004, 30, 135–143. [Google Scholar]
- Reddy-Noone, K.; Jain, A.; Verma, K.K. Liquid-phase micro extraction and GC for the determination of primary secondary and tertiary aromatic amines as their iodo-derivatives. Talanta 2007, 73, 684–691. [Google Scholar]
- Erdemir, S.; Yilmaz, M. Preparation of a new 1,3-alternate-calixarene-bonded HPLC stationary phase for the separation of phenols, aromatic amines and drugs. Talanta 2010, 8, 1240–1246. [Google Scholar]
- Fay, L.B.; Ali, S.; Gross, G.A. Determination of heterocyclic aromatic amines in food products: Automation of the sample preparation method prior to HPLC and HPLC-MS quantification. Mutat. Res 1997, 376, 29–35. [Google Scholar]
- Varney, M.S.; Preson, M.R. Measurement of trace aromatic amines in seawater using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. A 1985, 348, 265–274. [Google Scholar]
- Sun, Y.; Liang, L.; Zhao, X.; Yu, L.; Zhang, J.; Shi, G.; Zhou, T. Determination of aromatic amines in water samples by capillary electrophoresis with amperometric detection. Water Res 2009, 43, 41–46. [Google Scholar]
- Zhang, J.; Wu, X.; Zhang, W.; Xu, L.; Chen, G. A sweeping-micellar electrokinetic chromatography method for direct detection of some aromatic amines in water samples. Electrophoresis 2008, 29, 796–802. [Google Scholar]
- Wakayama, M.; Aoki, N.; Sasaki, H.; Ohsugi, R. Simultaneous analysis of amino acids and carboxylic acids by capillary electrophoresis-mass spectrometry using an acidic electrolyte and uncoated fused-silica capillary. Anal. Chem. 2010, 82, 9967–9976. [Google Scholar]
- Liu, S.; Tian, X.; Chen, X.; Hu, Z. Separation of diastereoisomers of podophyllum lignans by micellar electrokinetic chromatography. J. Chromatogr. A 2002, 959, 263–268. [Google Scholar]
- Terabe, S. Twenty-five years of micellar electrokinetic chromatography. Procedia Chem 2010, 2, 2–8. [Google Scholar]
- Shihabi, Z.K. Stacking in capillary zone electrophoresis. J. Chromatogr. A 2000, 902, 107–117. [Google Scholar]
- Simpson, S.L., Jr; Quirino, J.P.; Terabe, S. On-line sample preconcentration in capillary electrophoresis: Fundamentals and applications. J. Chromatogr. A 2008, 1184, 504–541. [Google Scholar]
- Breadmore, M.C.; Dawod, M.; Quirino, J.P. Recent advances in enhancing the sensitivity of electrophoresis and electrochromatography in capillaries and microchips (2008–2010). Electrophoresis 2011, 32, 127–148. [Google Scholar]
- Kawai, T.; Watanabe, M.; Sueyoshi, K.; Kitagawa, F.; Otsuka, K. Highly sensitive oligosaccharide analysis in capillary electrophoresis using large-volume sample stacking with an electroosmotic flow pump. J. Chromatogr. A 2012, 1232, 52–58. [Google Scholar] [Green Version]
- Liu, S.; Li, Q.; Chen, X.; Hu, Z. Field-amplified sample stacking in capillary electrophoresis for on-column concentration of alkaloids in Sophora flavescens Ait. Electrophoresis 2002, 23, 3392–3397. [Google Scholar]
- Shihabi, Z.K. Organic solvent high-field amplified stacking for basic compounds in capillary electrophoresis. J. Chromatogr. A 2005, 1066, 205–210. [Google Scholar]
- Shihabi, Z.K. Field amplified injection in the presence of salts for capillary electrophoresis. J. Chromatogr. A 1999, 853, 3–9. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Regression Equation | Correlation Coefficient | Linear Range (ng/mL) | Detection Limit (ng/mL) |
|---|---|---|---|---|
| Aniline | y = 40.8x + 50.061 | 0.9983 | 1~50 | 0.29 |
| y = 5.1371x + 1149.9 | 0.9995 | 50~1000 | ||
| o-Tolidine; o-Toluidine | y = 30.588x + 324.09 | 0.9996 | 5~800 | 0.39 |
| y = 27.016x + 16.356 | 0.9999 | 5~50 | 0.44 | |
| y = 9.9569x + 907.81 | 0.9993 | 50~800 | ||
| 3-Aminophenol | y = 27.895x + 49.048 | 0.9997 | 5~50 | 0.43 |
| y = 12.142x + 769.55 | 0.9994 | 50~1000 | ||
| Compound | Analytes Spiked (ng/mL) | Measured Amount (ng/mL) | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| Aniline | 20 | 20.8 | 4.93 | 104 |
| 300 | 278.2 | 3.60 | 93 | |
| o-Tolidine | 20 | 19.2 | 1.69 | 96 |
| 300 | 290.8 | 5.85 | 97 | |
| o-Toluidine | 20 | 19.4 | 4.37 | 97 |
| 300 | 279.8 | 4.11 | 93 | |
| m-Aminophenol | 20 | 18.9 | 2.79 | 95 |
| 300 | 294.5 | 5.17 | 98 | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, S.; Wang, W.; Chen, J.; Sun, J. Determination of Aniline and Its Derivatives in Environmental Water by Capillary Electrophoresis with On-Line Concentration. Int. J. Mol. Sci. 2012, 13, 6863-6872. https://doi.org/10.3390/ijms13066863
Liu S, Wang W, Chen J, Sun J. Determination of Aniline and Its Derivatives in Environmental Water by Capillary Electrophoresis with On-Line Concentration. International Journal of Molecular Sciences. 2012; 13(6):6863-6872. https://doi.org/10.3390/ijms13066863
Chicago/Turabian StyleLiu, Shuhui, Wenjun Wang, Jie Chen, and Jianzhi Sun. 2012. "Determination of Aniline and Its Derivatives in Environmental Water by Capillary Electrophoresis with On-Line Concentration" International Journal of Molecular Sciences 13, no. 6: 6863-6872. https://doi.org/10.3390/ijms13066863
APA StyleLiu, S., Wang, W., Chen, J., & Sun, J. (2012). Determination of Aniline and Its Derivatives in Environmental Water by Capillary Electrophoresis with On-Line Concentration. International Journal of Molecular Sciences, 13(6), 6863-6872. https://doi.org/10.3390/ijms13066863