The Tricyclodecan-9-yl-xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. Flow Cytometry Analyses
2.4. Protein Extraction and Western Blotting Analyses
2.5. Determination of SMS and GCS Activities in Jurkat Cells
2.6. Ceramide Measurement
2.7. Fluorogenic DEVD Cleavage Enzyme Assays
2.8. Morphological Analysis
2.9. Statistical Analysis
3. Results
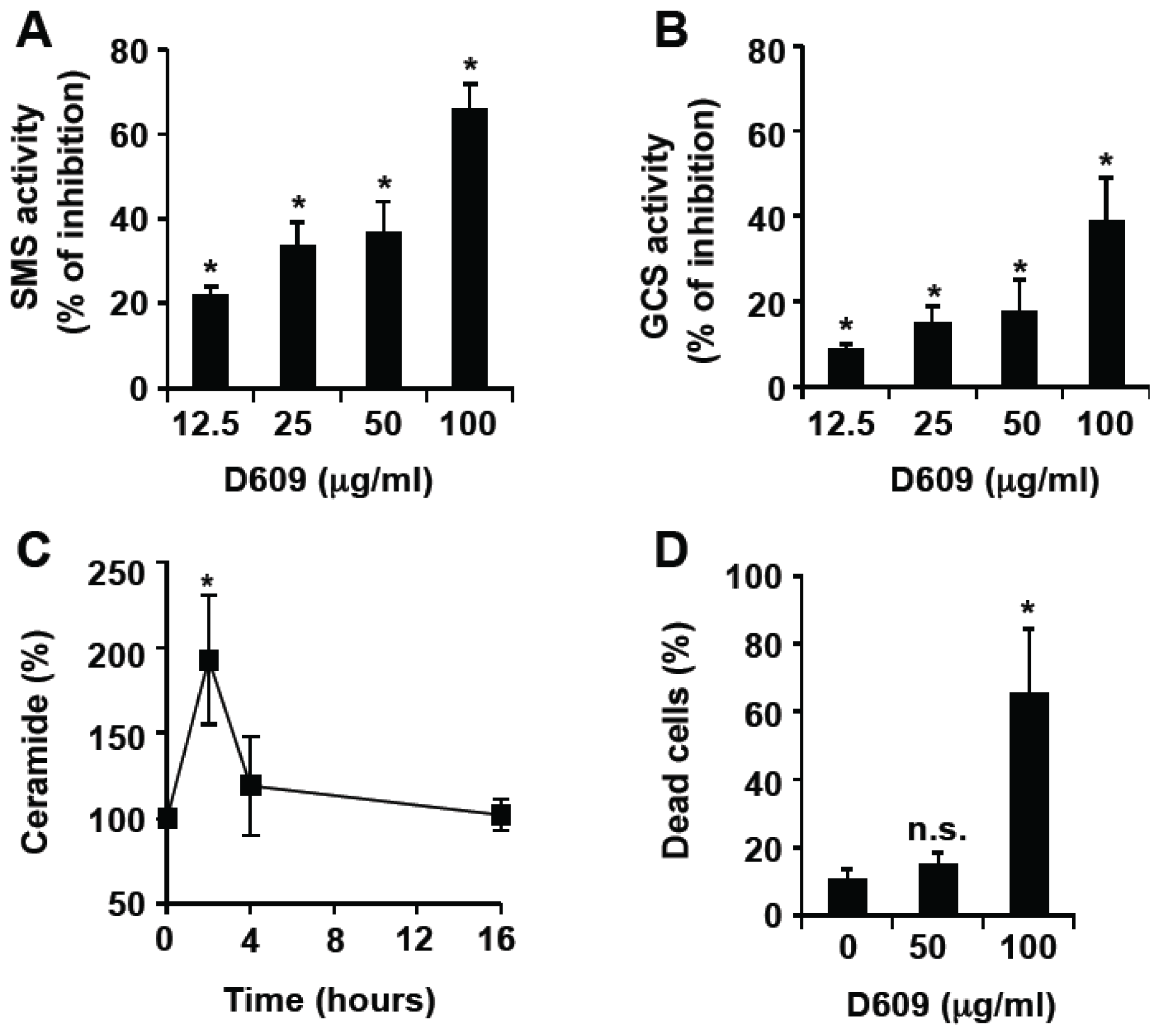
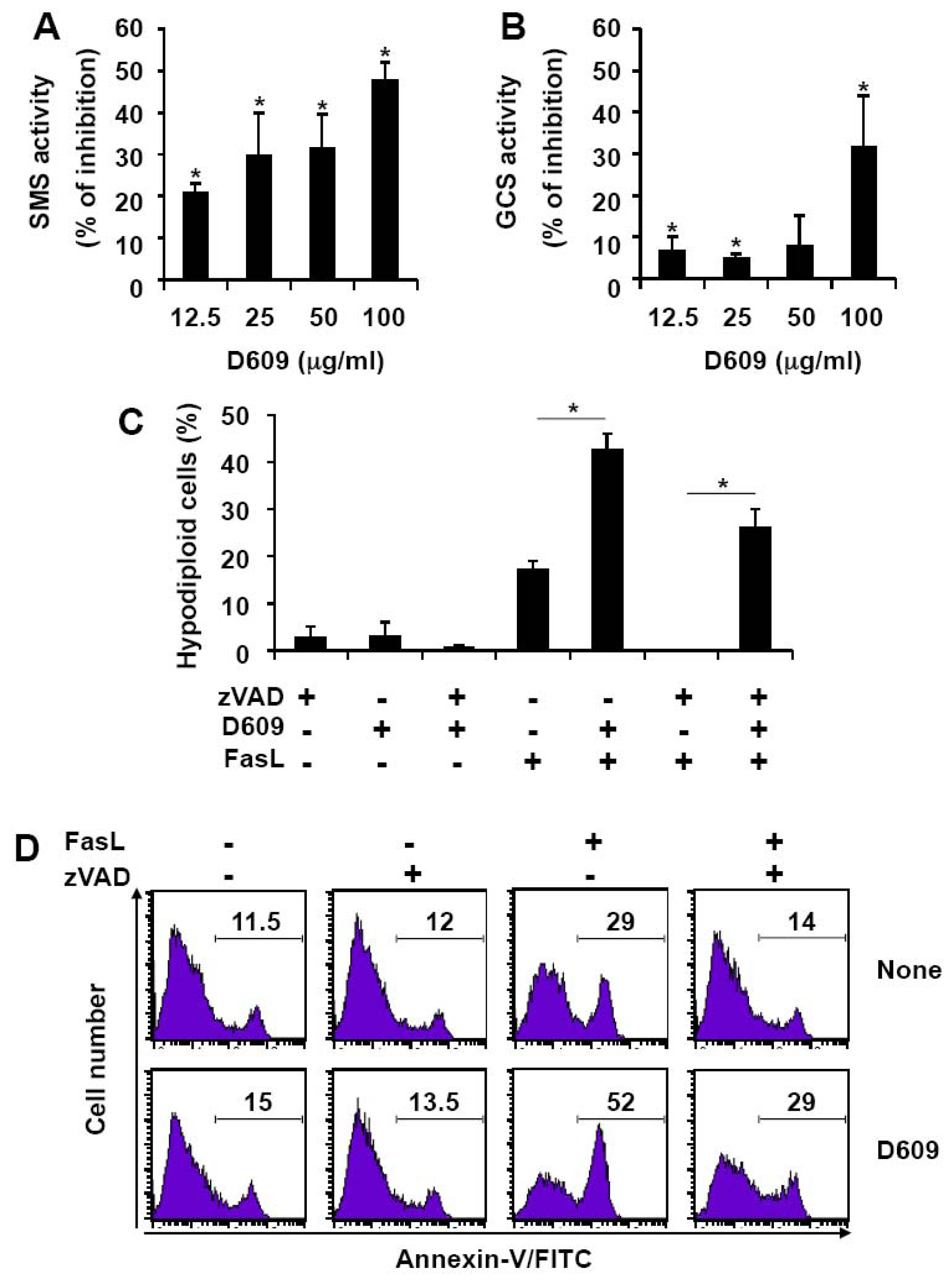
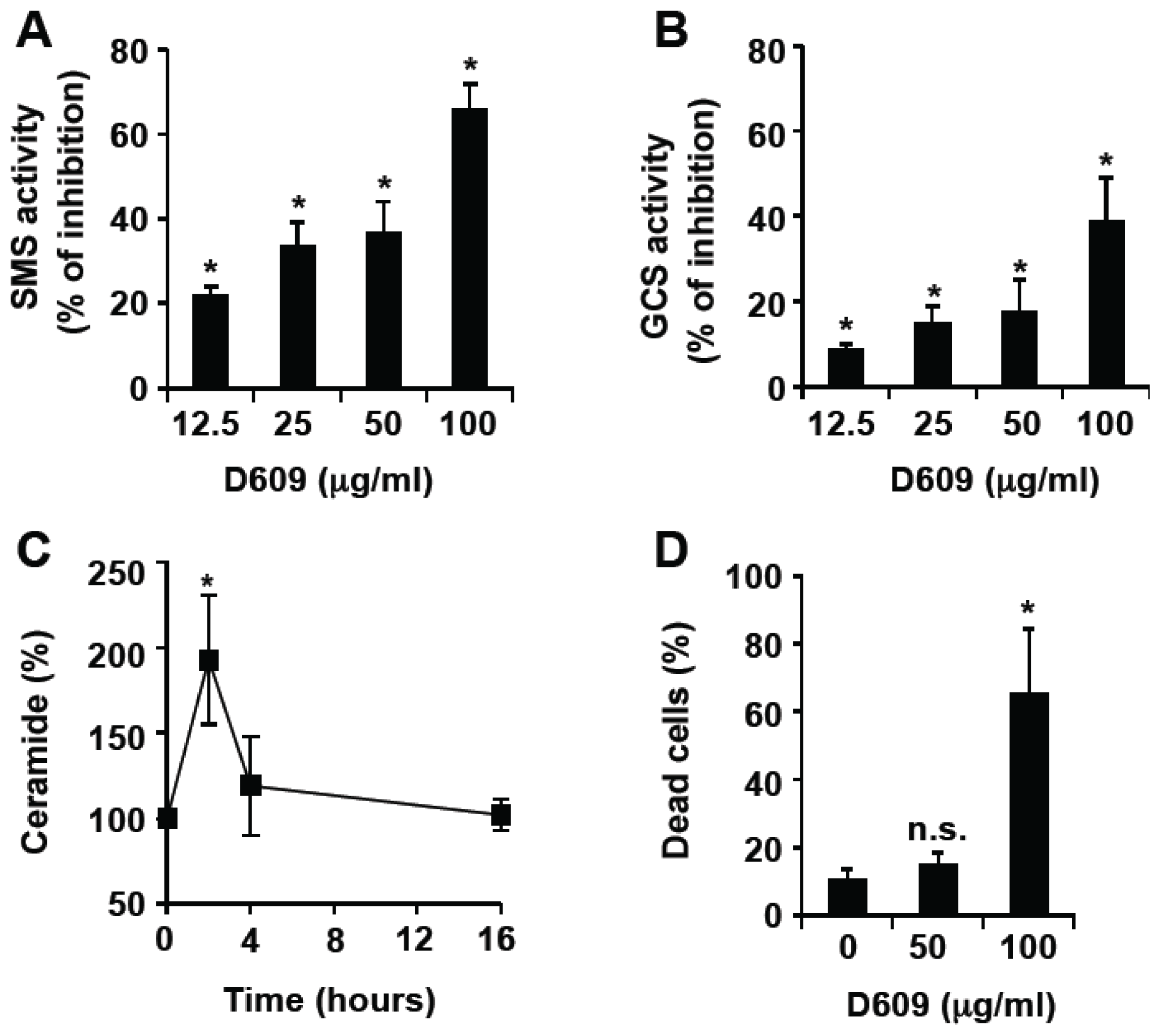
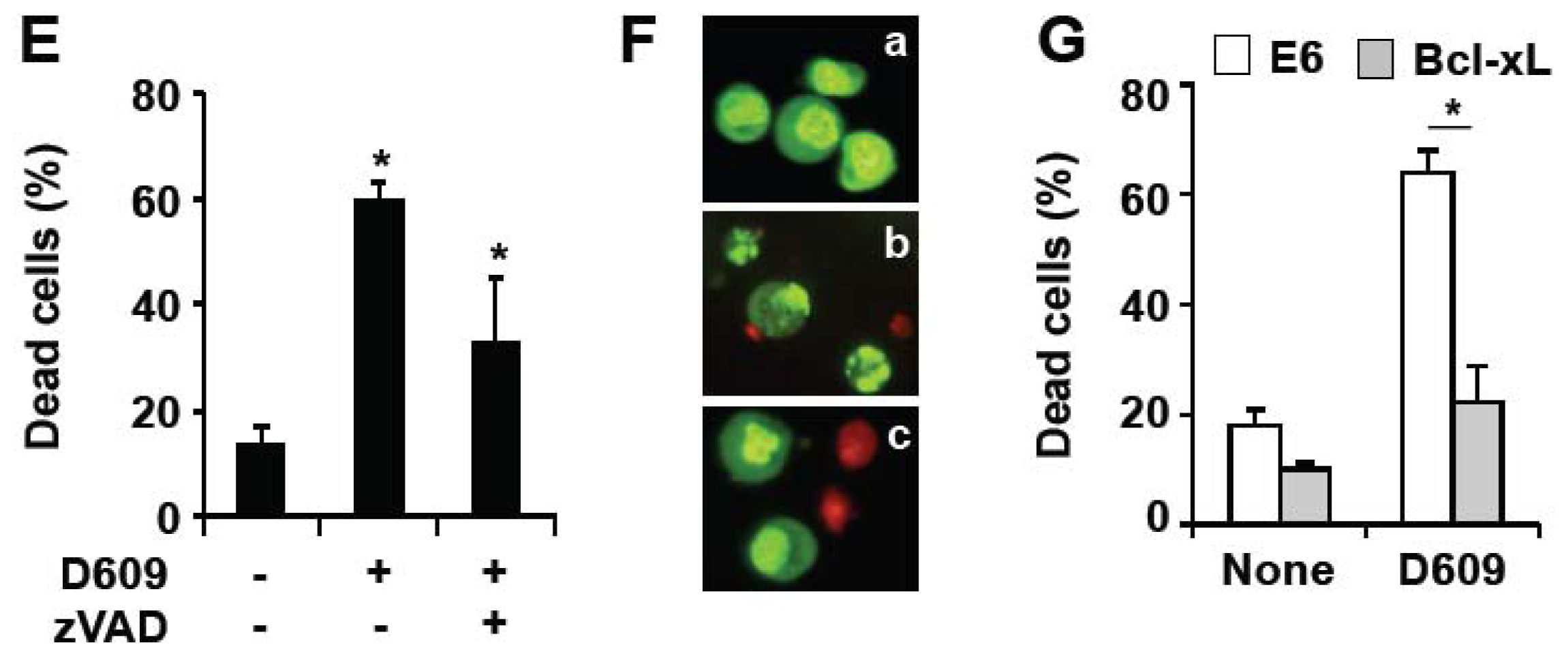
3.1. Inhibition of SMS and GCS Activities by D609 Triggers Ceramide Increase and Cell Death in Jurkat Cells
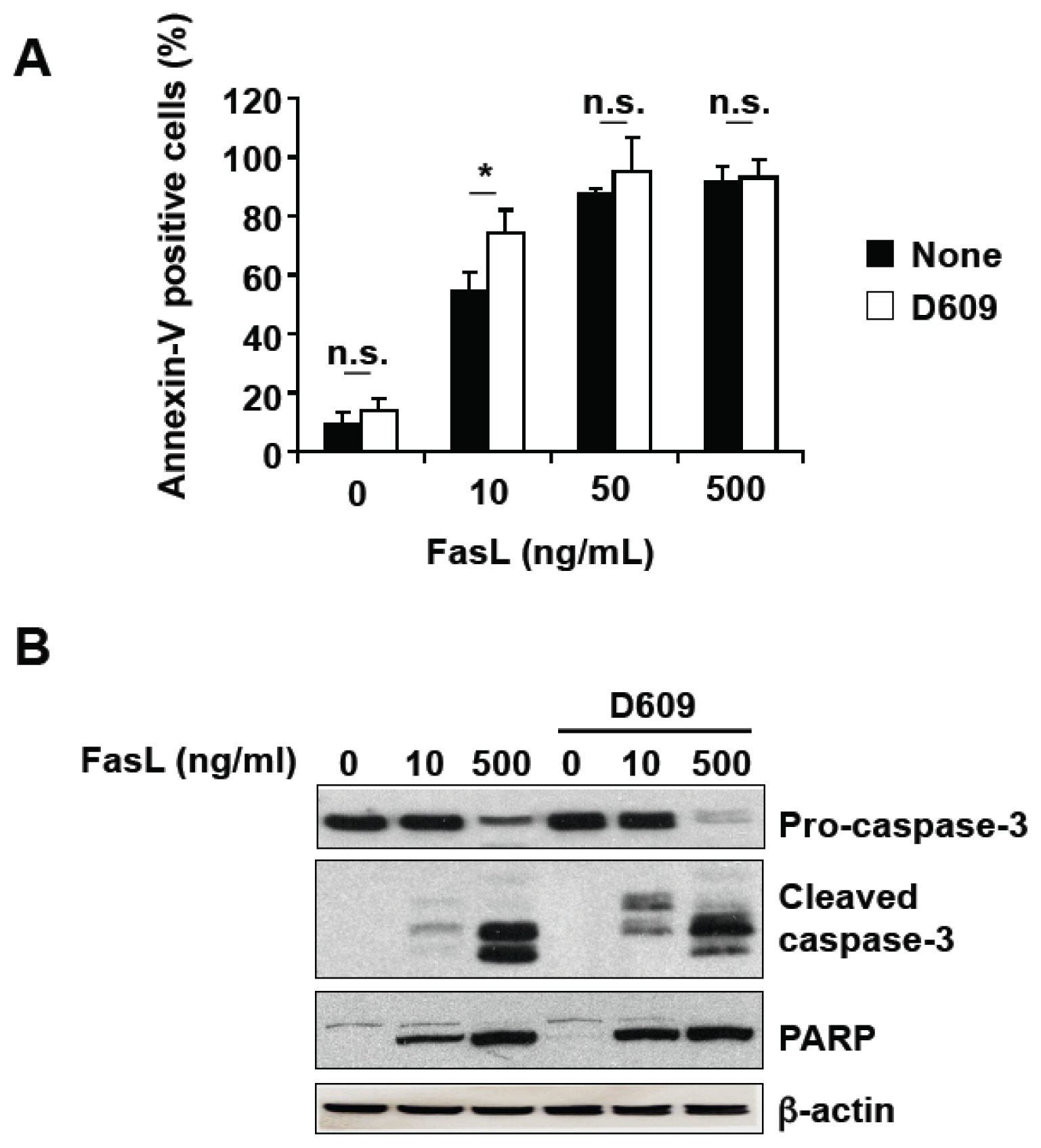
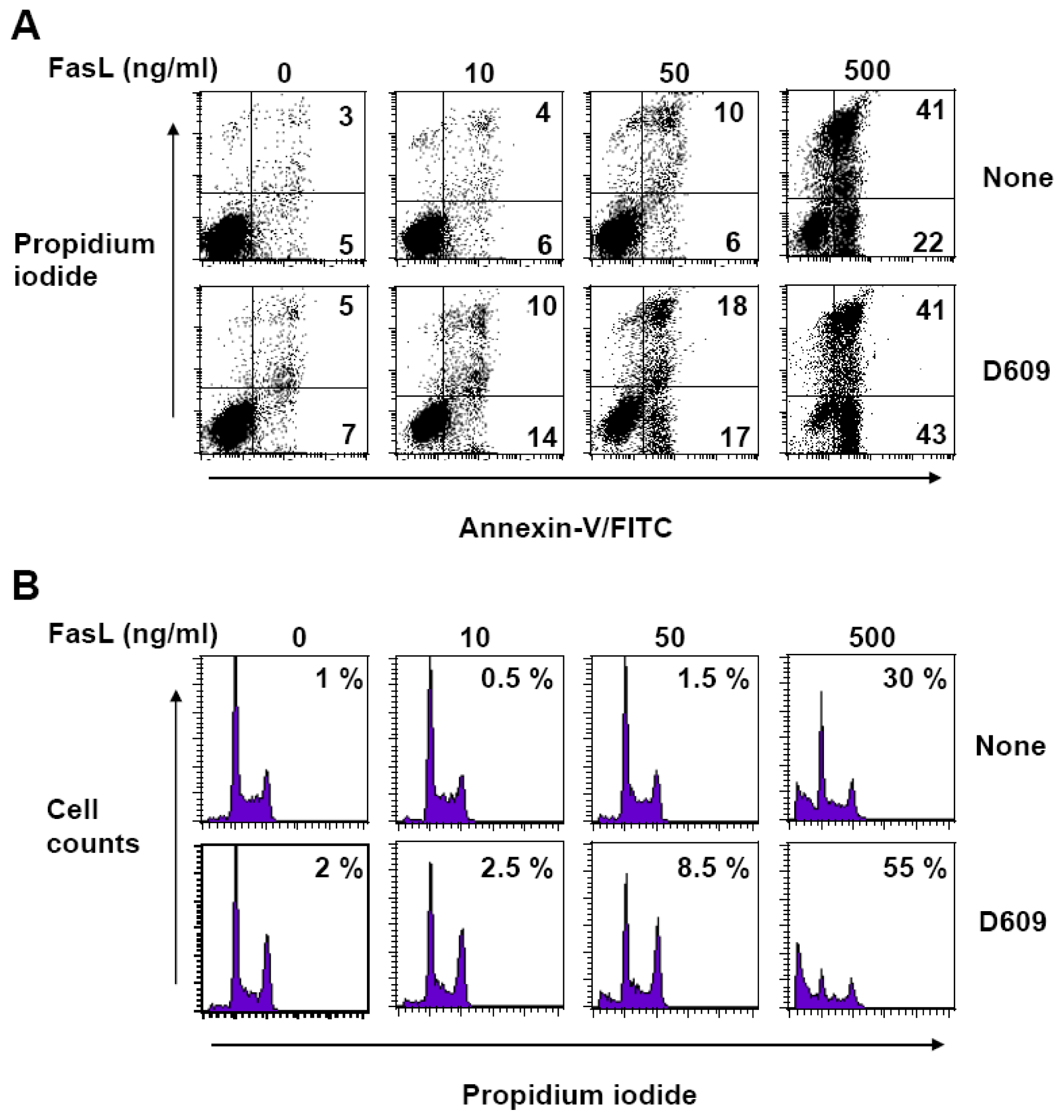
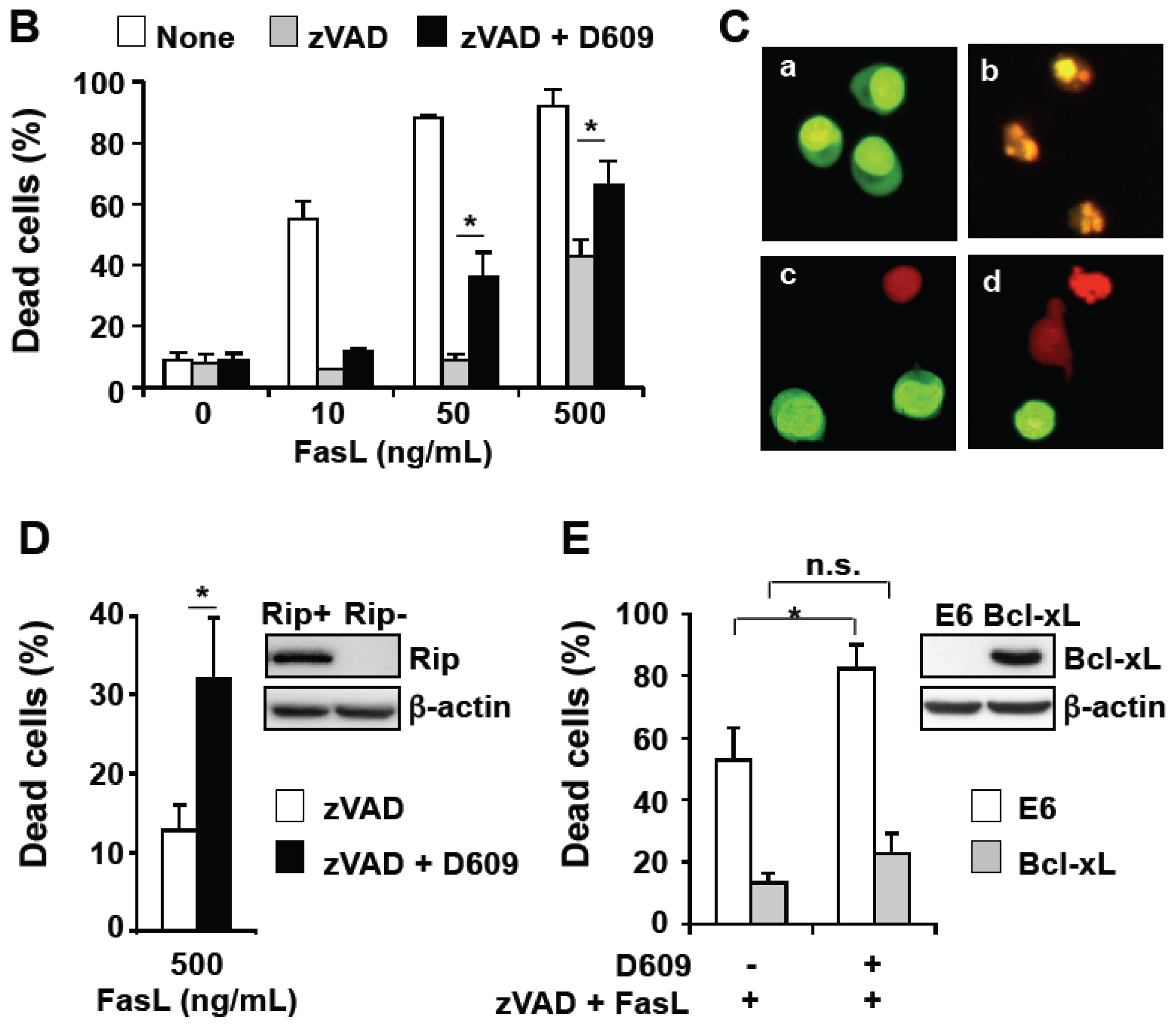
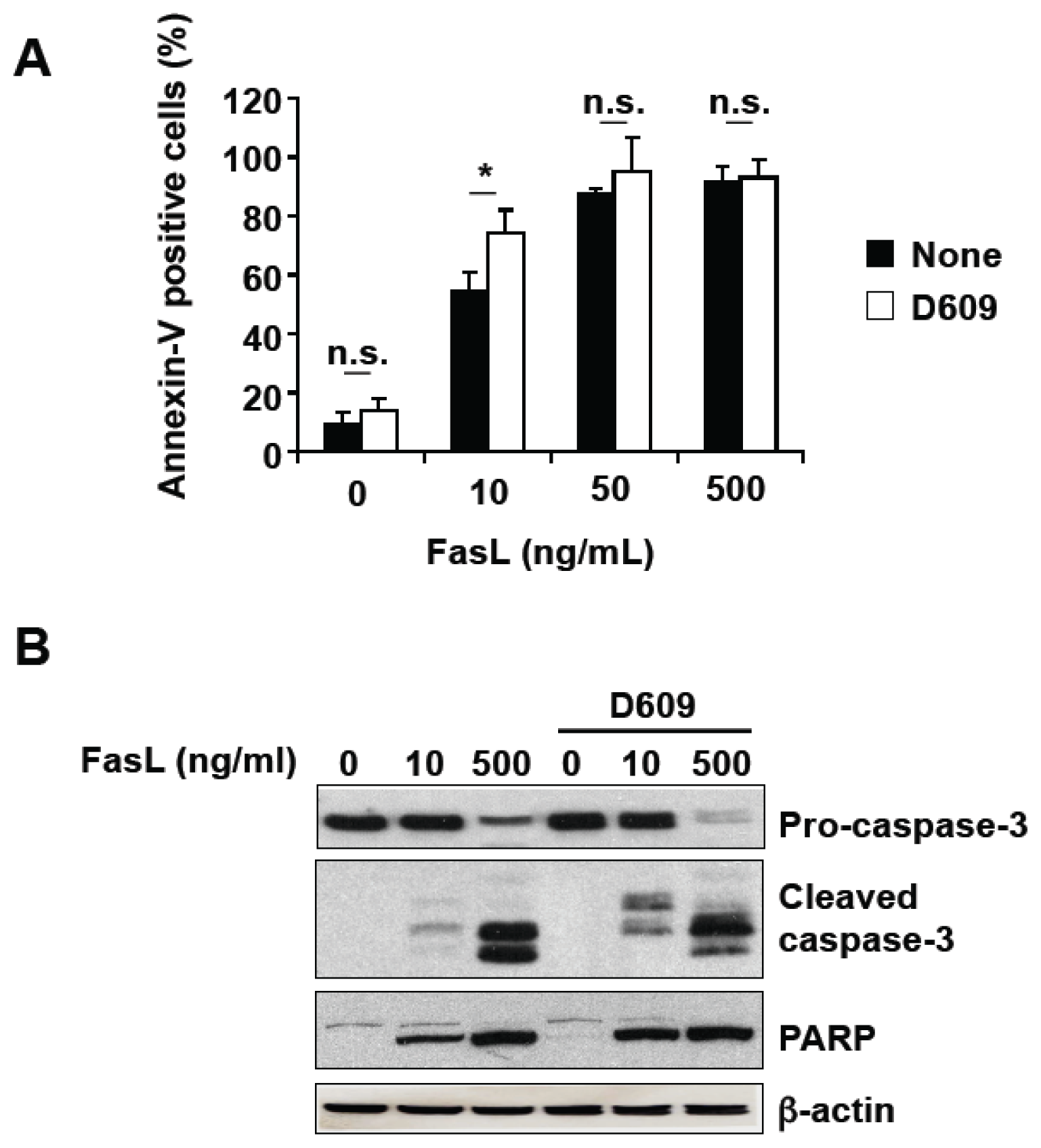
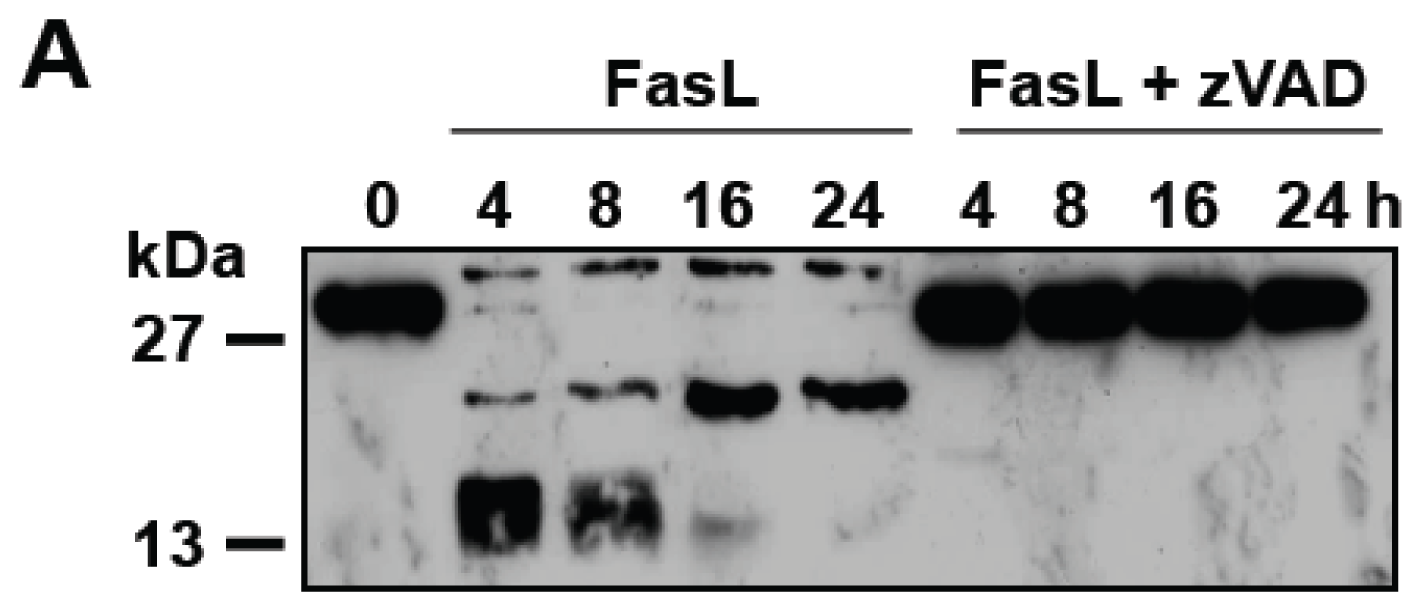
3.2. D609 Stimulates FasL-Induced Caspase Activation and Apoptosis in Jurkat Cells
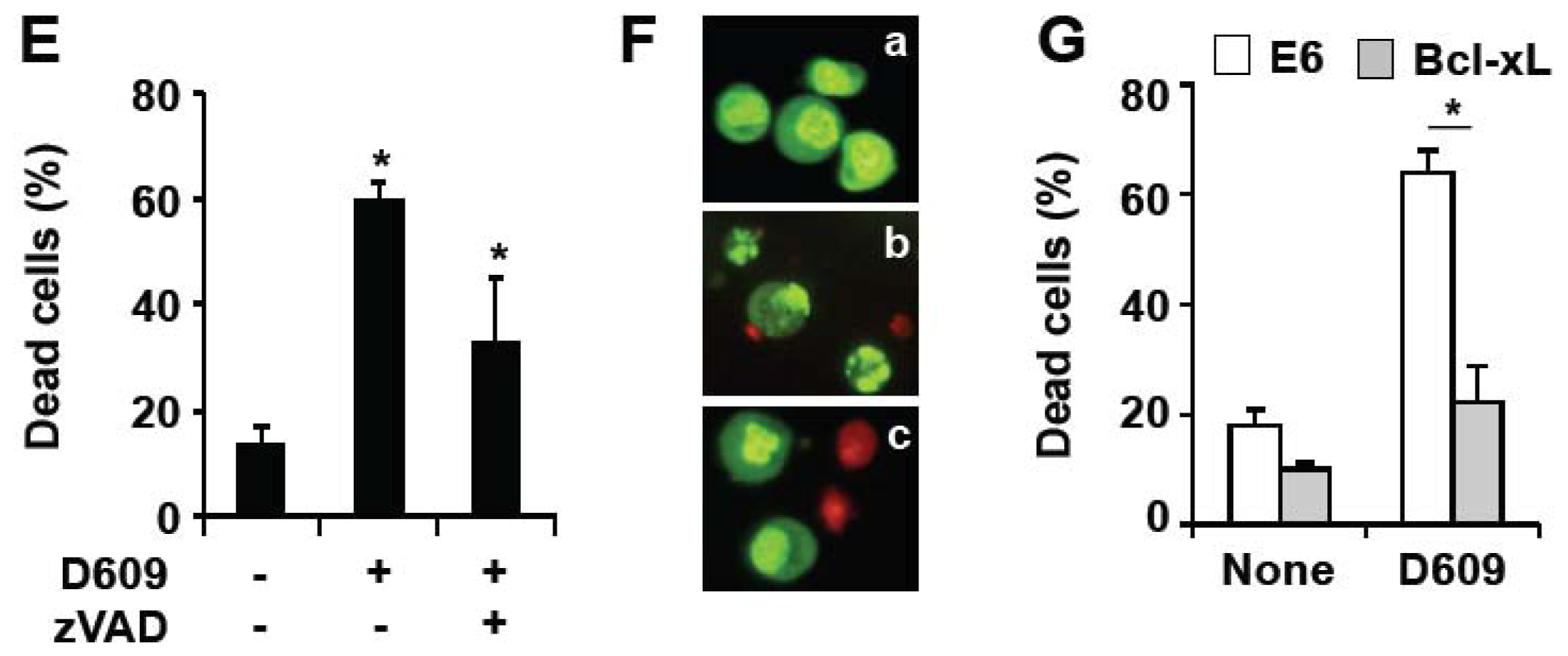
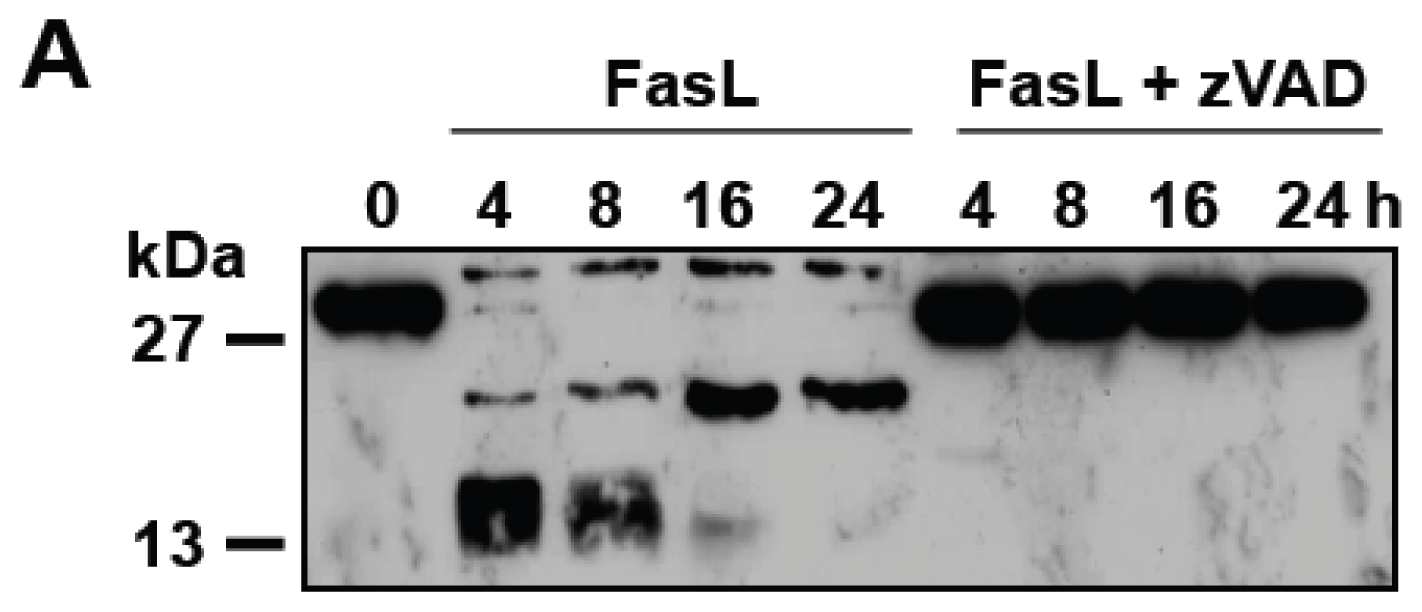
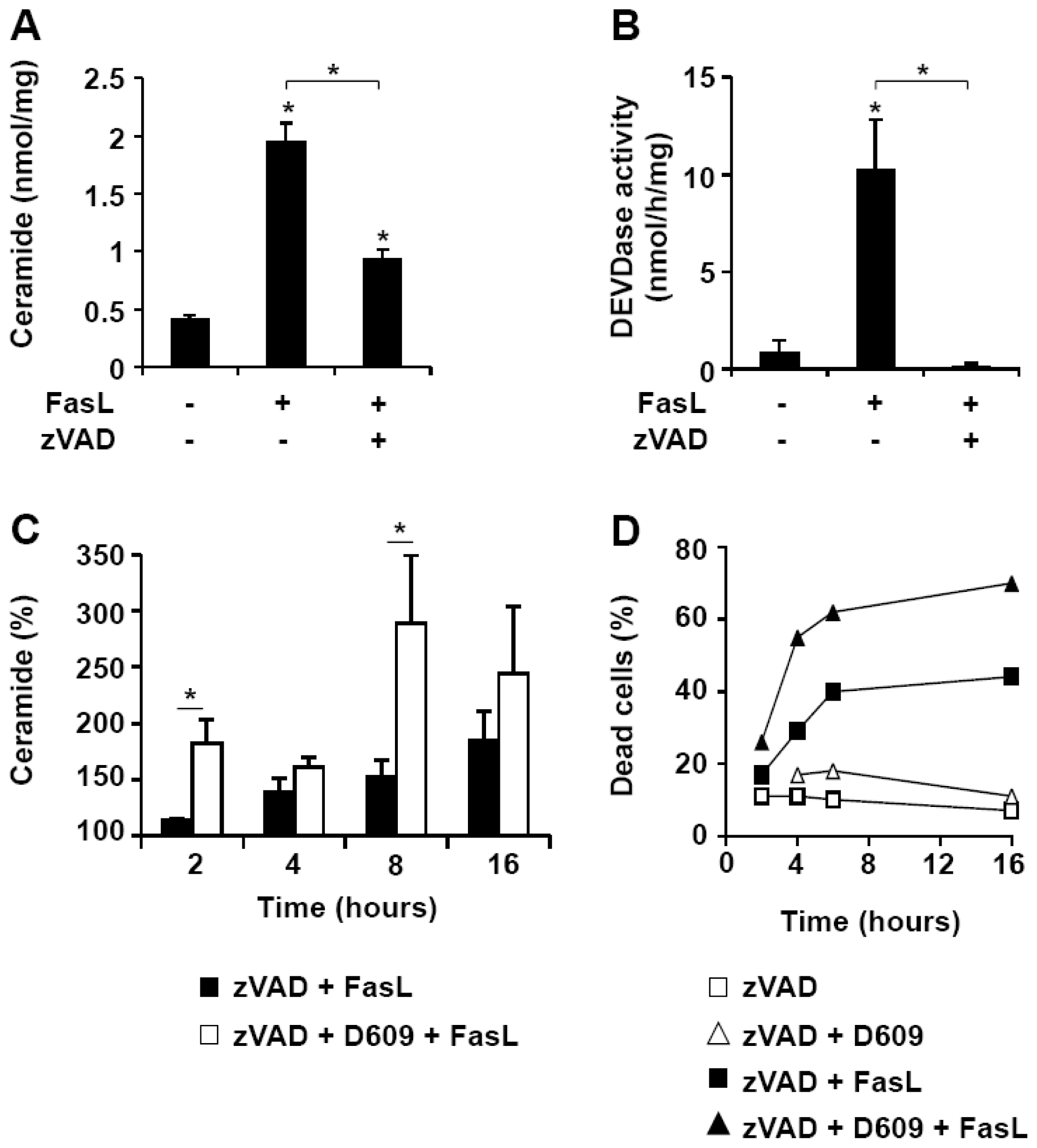
3.3. D609 Enhances FasL-Induced Caspase-Independent Cell Death and Overcomes Caspase-8 and RIP Deficiency
3.4. D609 Increases FasL-Induced Caspase-Independent Ceramide Production
3.5. D609 Enhances FasL-Induced Cell Death in T Lymphocytes
4. Discussion
Acknowledgments
References
- Sauer, G.; Amtmann, E.; Melber, K.; Knapp, A.; Muller, K.; Hummel, K.; Scherm, A. DNA and RNA virus species are inhibited by xanthates, a class of antiviral compounds with unique properties. Proc. Natl. Acad. Sci. USA 1984, 81, 3263–3267. [Google Scholar]
- Amtmann, E. The antiviral, antitumoural xanthate D609 is a competitive inhibitor of phosphatidylcholine-specific phospholipase C. Drugs Exp. Clin. Res 1996, 22, 287–294. [Google Scholar]
- Amtmann, E.; Sauer, G. Selective killing of tumor cells by xanthates. Cancer Lett 1987, 35, 237–244. [Google Scholar]
- Machleidt, T.; Kramer, B.; Adam, D.; Neumann, B.; Schutze, S.; Wiegmann, K.; Kronke, M. Function of the p55 tumor necrosis factor receptor “death domain” mediated by phosphatidylcholine-specific phospholipase C. J. Exp. Med 1996, 184, 725–733. [Google Scholar]
- Adibhatla, R.M.; Hatcher, J.F.; Gusain, A. Tricyclodecan-9-yl-xanthogenate (D609) mechanism of actions: A mini-review of literature. Neurochem. Res 2012, 37, 671–679. [Google Scholar]
- Goebeler, M.; Gillitzer, R.; Kilian, K.; Utzel, K.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Multiple signaling pathways regulate NF-κB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood 2001, 97, 46–55. [Google Scholar]
- Mas, V.M.; Hernandez, H.; Plo, I.; Bezombes, C.; Maestre, N.; Quillet-Mary, A.; Filomenko, R.; Demur, C.; Jaffrezou, J.P.; Laurent, G. Protein kinase Czeta mediated Raf-1/extracellular-regulated kinase activation by daunorubicin. Blood 2003, 101, 1543–1550. [Google Scholar]
- Marchetti, M.C.; Di Marco, B.; Cifone, G.; Migliorati, G.; Riccardi, C. Dexamethasone-induced apoptosis of thymocytes: Role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood 2003, 101, 585–593. [Google Scholar]
- Luft, T.; Rodionova, E.; Maraskovsky, E.; Kirsch, M.; Hess, M.; Buchholtz, C.; Goerner, M.; Schnurr, M.; Skoda, R.; Ho, A.D. Adaptive functional differentiation of dendritic cells: Integrating the network of extra- and intracellular signals. Blood 2006, 107, 4763–4769. [Google Scholar]
- Schutze, S.; Potthoff, K.; Machleidt, T.; Berkovic, D.; Wiegmann, K.; Kronke, M. TNF activates NF-κB by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 1992, 71, 765–776. [Google Scholar]
- Cifone, M.G.; de Maria, R.; Roncaioli, P.; Rippo, M.R.; Azuma, M.; Lanier, L.L.; Santoni, A.; Testi, R. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J. Exp. Med 1994, 180, 1547–1552. [Google Scholar]
- Wiegmann, K.; Schutze, S.; Machleidt, T.; Witte, D.; Kronke, M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 1994, 78, 1005–1015. [Google Scholar]
- Cifone, M.G.; Roncaioli, P.; de Maria, R.; Camarda, G.; Santoni, A.; Ruberti, G.; Testi, R. Multiple pathways originate at the Fas/APO-1 (CD95) receptor: Sequential involvement of phosphatidylcholine-specific phospholipase C and acidic sphingomyelinase in the propagation of the apoptotic signal. EMBO J 1995, 14, 5859–5868. [Google Scholar]
- Cifone, M.G.; Migliorati, G.; Parroni, R.; Marchetti, C.; Millimaggi, D.; Santoni, A.; Riccardi, C. Dexamethasone-induced thymocyte apoptosis: Apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood 1999, 93, 2282–2296. [Google Scholar]
- Luberto, C.; Hannun, Y.A. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J. Biol. Chem 1998, 273, 14550–14559. [Google Scholar]
- Watanabe, M.; Kitano, T.; Kondo, T.; Yabu, T.; Taguchi, Y.; Tashima, M.; Umehara, H.; Domae, N.; Uchiyama, T.; Okazaki, T. Increase of nuclear ceramide through caspase-3-dependent regulation of the “sphingomyelin cycle” in Fas-induced apoptosis. Cancer Res 2004, 64, 1000–1007. [Google Scholar]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.; Holthuis, J.C. Identification of a family of animal sphingomyelin synthases. EMBO J 2004, 23, 33–44. [Google Scholar]
- Meng, A.; Luberto, C.; Meier, P.; Bai, A.; Yang, X.; Hannun, Y.A.; Zhou, D. Sphingomyelin synthase as a potential target for D609-induced apoptosis in U937 human monocytic leukemia cells. Exp. Cell Res 2004, 292, 385–392. [Google Scholar]
- Zhou, D.; Lauderback, C.M.; Yu, T.; Brown, S.A.; Butterfield, D.A.; Thompson, J.S. D609 inhibits ionizing radiation-induced oxidative damage by acting as a potent antioxidant. J. Pharmacol. Exp. Ther 2001, 298, 103–109. [Google Scholar]
- Gusain, A.; Hatcher, J.F.; Adibhatla, R.M.; Wesley, U.V.; Dempsey, R.J. Anti-proliferative effects of tricyclodecan-9-yl-xanthogenate (D609) involve ceramide and cell cycle inhibition. Mol. Neurobiol 2012, 45, 455–464. [Google Scholar]
- Barcelo-Coblijn, G.; Martin, M.L.; de Almeida, R.F.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Luth, A.; Kleuser, B.; Halver, J.E.; Escriba, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar]
- Thon, L.; Mohlig, H.; Mathieu, S.; Lange, A.; Bulanova, E.; Winoto-Morbach, S.; Schutze, S.; Bulfone-Paus, S.; Adam, D. Ceramide mediates caspase-independent programmed cell death. FASEB J 2005, 19, 1945–1956. [Google Scholar]
- Porn-Ares, M.I.; Chow, S.C.; Slotte, J.P.; Orrenius, S. Induction of apoptosis and potentiation of TNF- and Fas-mediated apoptosis in U937 cells by the xanthogenate compound D609. Exp. Cell Res 1997, 235, 48–54. [Google Scholar]
- Zhang, L.; Shimizu, S.; Tsujimoto, Y. Two distinct Fas-activated signaling pathways revealed by an antitumor drug D609. Oncogene 2005, 24, 2954–2962. [Google Scholar]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998, 17, 1675–1687. [Google Scholar]
- Muzio, M.; Salvesen, G.S.; Dixit, V.M. FLICE induced apoptosis in a cell-free system. Cleavage of caspase zymogens. J. Biol. Chem 1997, 272, 2952–2956. [Google Scholar]
- Vincenz, C.; Dixit, V.M. Fas-associated death domain protein interleukin-1beta-converting enzyme 2 (FLICE2), an ICE/Ced-3 homologue, is proximally involved in CD95- and p55-mediated death signaling. J. Biol. Chem 1997, 272, 6578–6583. [Google Scholar]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar]
- Milhas, D.; Cuvillier, O.; Therville, N.; Clave, P.; Thomsen, M.; Levade, T.; Benoist, H.; Segui, B. Caspase-10 triggers bid cleavage and caspase cascade activation in FasL-induced apoptosis. J. Biol. Chem 2005, 280, 19836–19842. [Google Scholar]
- Fischer, U.; Stroh, C.; Schulze-Osthoff, K. Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 2006, 25, 152–159. [Google Scholar]
- Lee, K.H.; Feig, C.; Tchikov, V.; Schickel, R.; Hallas, C.; Schutze, S.; Peter, M.E.; Chan, A.C. The role of receptor internalization in CD95 signaling. EMBO J 2006, 25, 1009–1023. [Google Scholar]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol 2000, 1, 489–495. [Google Scholar]
- Segui, B.; Andrieu-Abadie, N.; Jaffrezou, J.P.; Benoist, H.; Levade, T. Sphingolipids as modulators of cancer cell death: potential therapeutic targets. Biochim. Biophys. Acta 2006, 1758, 2104–2120. [Google Scholar]
- Lafont, E.; Dupont, R.; Andrieu-Abadie, N.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; Segui, B. Ordering of ceramide formation and caspase-9 activation in CD95L-induced Jurkat leukemia T cell apoptosis. Biochim. Biophys Acta 2012, 1821, 684–693. [Google Scholar]
- Lafont, E.; Milhas, D.; Carpentier, S.; Garcia, V.; Jin, Z.X.; Umehara, H.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; Segui, B. Caspase-mediated inhibition of sphingomyelin synthesis is involved in FasL-triggered cell death. Cell Death Differ 2010, 17, 642–654. [Google Scholar]
- Miyaji, M.; Jin, Z.X.; Yamaoka, S.; Amakawa, R.; Fukuhara, S.; Sato, S.B.; Kobayashi, T.; Domae, N.; Mimori, T.; Bloom, E.T.; Okazaki, T.; Umehara, H. Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J. Exp. Med 2005, 202, 249–259. [Google Scholar]
- Lafont, E.; Kitatani, K.; Okazaki, T.; Segui, B. Regulation of death and growth signals at the plasma membrane by sphingomyelin synthesis: Implications for hematological malignancies. Recent Pat. Anticancer Drug Discov 2011, 6, 324–333. [Google Scholar]
- Shimizu, M.; Fontana, A.; Takeda, Y.; Yagita, H.; Yoshimoto, T.; Matsuzawa, A. Induction of antitumor immunity with Fas/APO-1 ligand (CD95L)-transfected neuroblastoma neuro-2a cells. J. Immunol 1999, 162, 7350–7357. [Google Scholar]
- Juo, P.; Kuo, C.J.; Yuan, J.; Blenis, J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol 1998, 8, 1001–1008. [Google Scholar]
- Ting, A.T.; Pimentel-Muinos, F.X.; Seed, B. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J 1996, 15, 6189–6196. [Google Scholar]
- Boise, L.H.; Thompson, C.B. Bcl-x(L) can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc. Natl. Acad. Sci. USA 1997, 94, 3759–3764. [Google Scholar]
- Segui, B.; Allen-Baume, V.; Cockcroft, S. Phosphatidylinositol transfer protein beta displays minimal sphingomyelin transfer activity and is not required for biosynthesis and trafficking of sphingomyelin. Biochem. J 2002, 366, 23–34. [Google Scholar]
- Van Veldhoven, P.P.; Matthews, T.J.; Bolognesi, D.P.; Bell, R.M. Changes in bioactive lipids, alkylacylglycerol and ceramide, occur in HIV-infected cells. Biochem. Biophys. Res. Commun 1992, 187, 209–216. [Google Scholar]
- Cuvillier, O.; Edsall, L.; Spiegel, S. Involvement of sphingosine in mitochondria-dependent Fas-induced apoptosis of type II Jurkat T cells. J. Biol. Chem 2000, 275, 15691–15700. [Google Scholar]
- Segui, B.; Andrieu-Abadie, N.; Adam-Klages, S.; Meilhac, O.; Kreder, D.; Garcia, V.; Bruno, A.P.; Jaffrezou, J.P.; Salvayre, R.; Kronke, M.; Levade, T. CD40 signals apoptosis through FAN-regulated activation of the sphingomyelin-ceramide pathway. J. Biol. Chem 1999, 274, 37251–37258. [Google Scholar]
- Lafont, E.; Milhas, D.; Teissie, J.; Therville, N.; Andrieu-Abadie, N.; Levade, T.; Benoist, H.; Segui, B. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Matsumura, H.; Shimizu, Y.; Ohsawa, Y.; Kawahara, A.; Uchiyama, Y.; Nagata, S. Necrotic death pathway in Fas receptor signaling. J. Cell Biol 2000, 151, 1247–1256. [Google Scholar]
- Hetz, C.A.; Hunn, M.; Rojas, P.; Torres, V.; Leyton, L.; Quest, A.F. Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: Onset of necrosis is associated with delayed ceramide increase. J. Cell Sci 2002, 115, 4671–4683. [Google Scholar]
- Juo, P.; Woo, M.S.; Kuo, C.J.; Signorelli, P.; Biemann, H.P.; Hannun, Y.A.; Blenis, J. FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ 1999, 10, 797–804. [Google Scholar]
- Kiss, Z.; Tomono, M. Compound D609 inhibits phorbol ester-stimulated phospholipase D activity and phospholipase C-mediated phosphatidylethanolamine hydrolysis. Biochim. Biophys. Acta 1995, 1259, 105–108. [Google Scholar]
- Perry, R.J.; Ridgway, N.D. The role of de novo ceramide synthesis in the mechanism of action of the tricyclic xanthate D609. J. Lipid Res 2004, 45, 164–173. [Google Scholar]
- Ségui, B.; Bezombes, C.; Uro-Coste, E.; Medin, J.A.; Andrieu-Abadie, N.; Auge, N.; Brouchet, A.; Laurent, G.; Salvayre, R.; Jaffrezou, J.P.; Levade, T. Stress-induced apoptosis is not mediated by endolysosomal ceramide. FASEB J 2000, 14, 36–47. [Google Scholar]
- Birbes, H.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J 2001, 15, 2669–2679. [Google Scholar]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism—A strategy for overcoming drug resistance. J. Natl. Cancer Inst 2001, 93, 347–357. [Google Scholar]
- Tepper, A.D.; Diks, S.H.; van Blitterswijk, W.J.; Borst, J. Glucosylceramide synthase does not attenuate the ceramide pool accumulating during apoptosis induced by CD95 or anti-cancer regimens. J. Biol. Chem 2000, 275, 34810–34817. [Google Scholar]
- Bidere, N.; Su, H.C.; Lenardo, M.J. Genetic disorders of programmed cell death in the immune system. Annu. Rev. Immunol 2006, 24, 321–352. [Google Scholar]
- Bai, A.; Meier, G.P.; Wang, Y.; Luberto, C.; Hannun, Y.A.; Zhou, D. Prodrug modification increases potassium tricyclo[5.2.1.0(2,6)]-decan-8-yl dithiocarbonate (D609) chemical stability and cytotoxicity against U937 leukemia cells. J. Pharmacol. Exp. Ther 2004, 309, 1051– 1059. [Google Scholar]




© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Milhas, D.; Andrieu-Abadie, N.; Levade, T.; Benoist, H.; Ségui, B. The Tricyclodecan-9-yl-xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes. Int. J. Mol. Sci. 2012, 13, 8834-8852. https://doi.org/10.3390/ijms13078834
Milhas D, Andrieu-Abadie N, Levade T, Benoist H, Ségui B. The Tricyclodecan-9-yl-xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes. International Journal of Molecular Sciences. 2012; 13(7):8834-8852. https://doi.org/10.3390/ijms13078834
Chicago/Turabian StyleMilhas, Delphine, Nathalie Andrieu-Abadie, Thierry Levade, Hervé Benoist, and Bruno Ségui. 2012. "The Tricyclodecan-9-yl-xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes" International Journal of Molecular Sciences 13, no. 7: 8834-8852. https://doi.org/10.3390/ijms13078834
APA StyleMilhas, D., Andrieu-Abadie, N., Levade, T., Benoist, H., & Ségui, B. (2012). The Tricyclodecan-9-yl-xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes. International Journal of Molecular Sciences, 13(7), 8834-8852. https://doi.org/10.3390/ijms13078834