Effects of a Buried Cysteine-To-Serine Mutation on Yeast Triosephosphate Isomerase Structure and Stability

 ,
,
Abstract
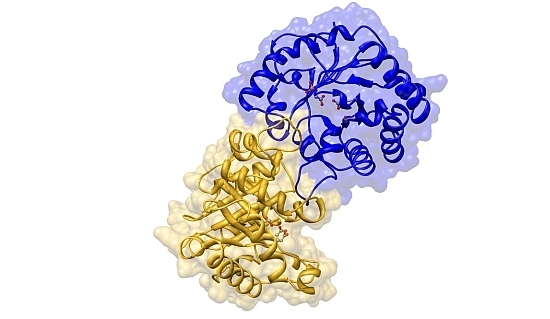
:
1. Introduction
2. Results and Discussion
2.1. Enzyme Preparation, and Crystallization
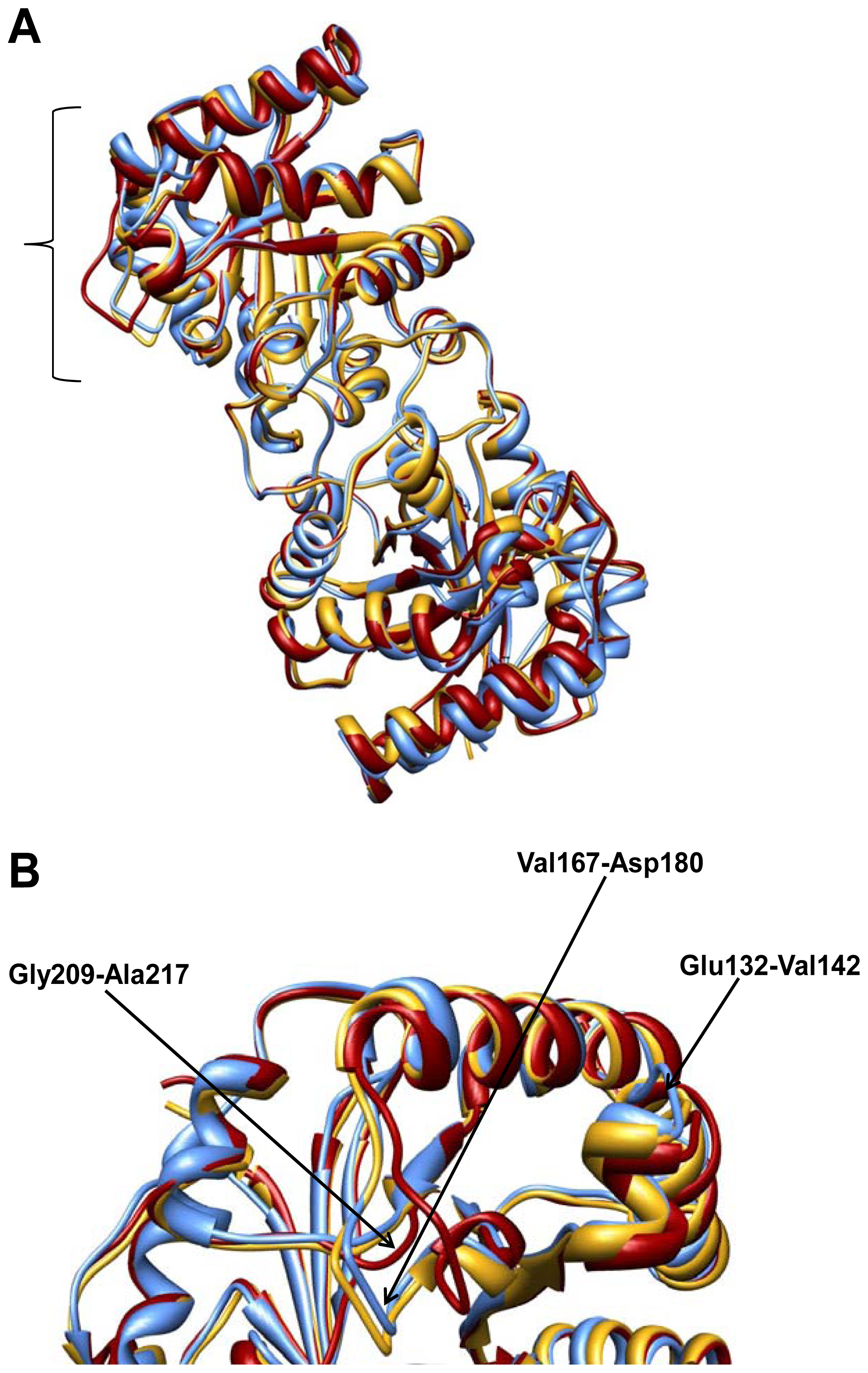
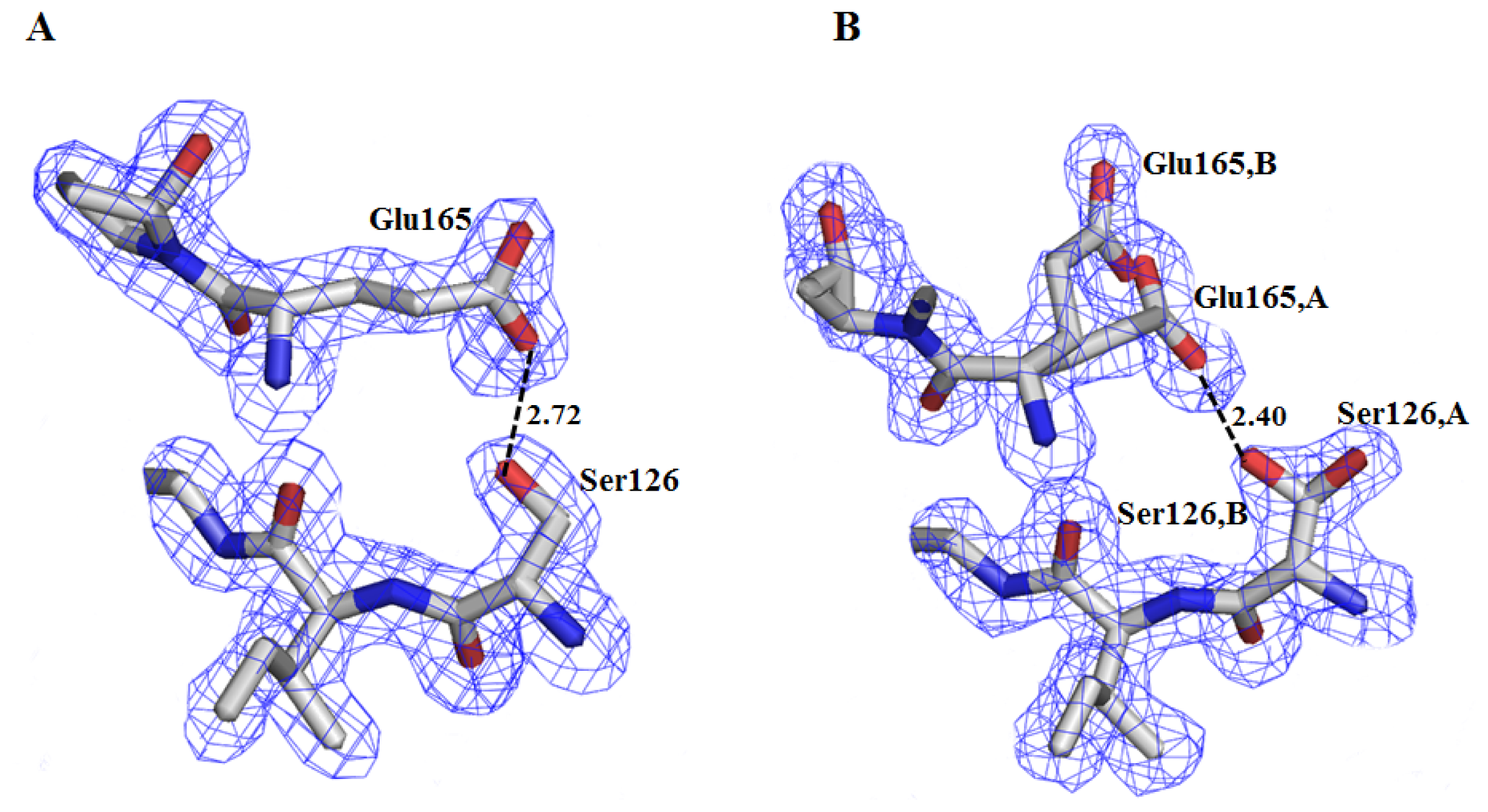
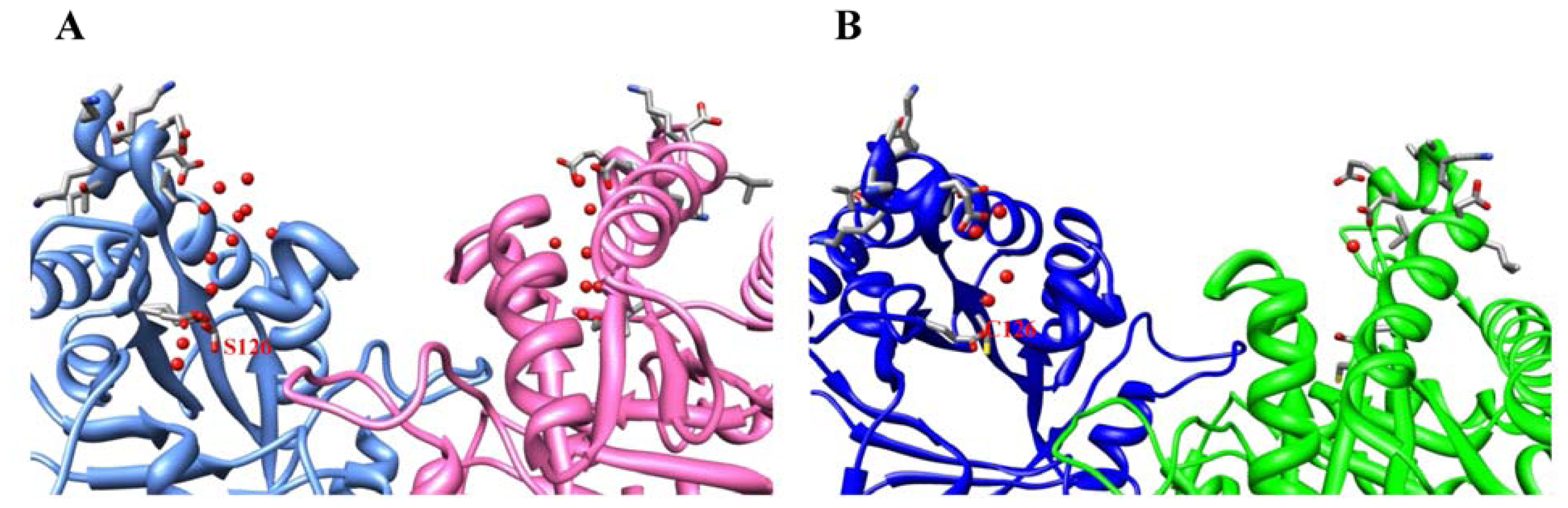
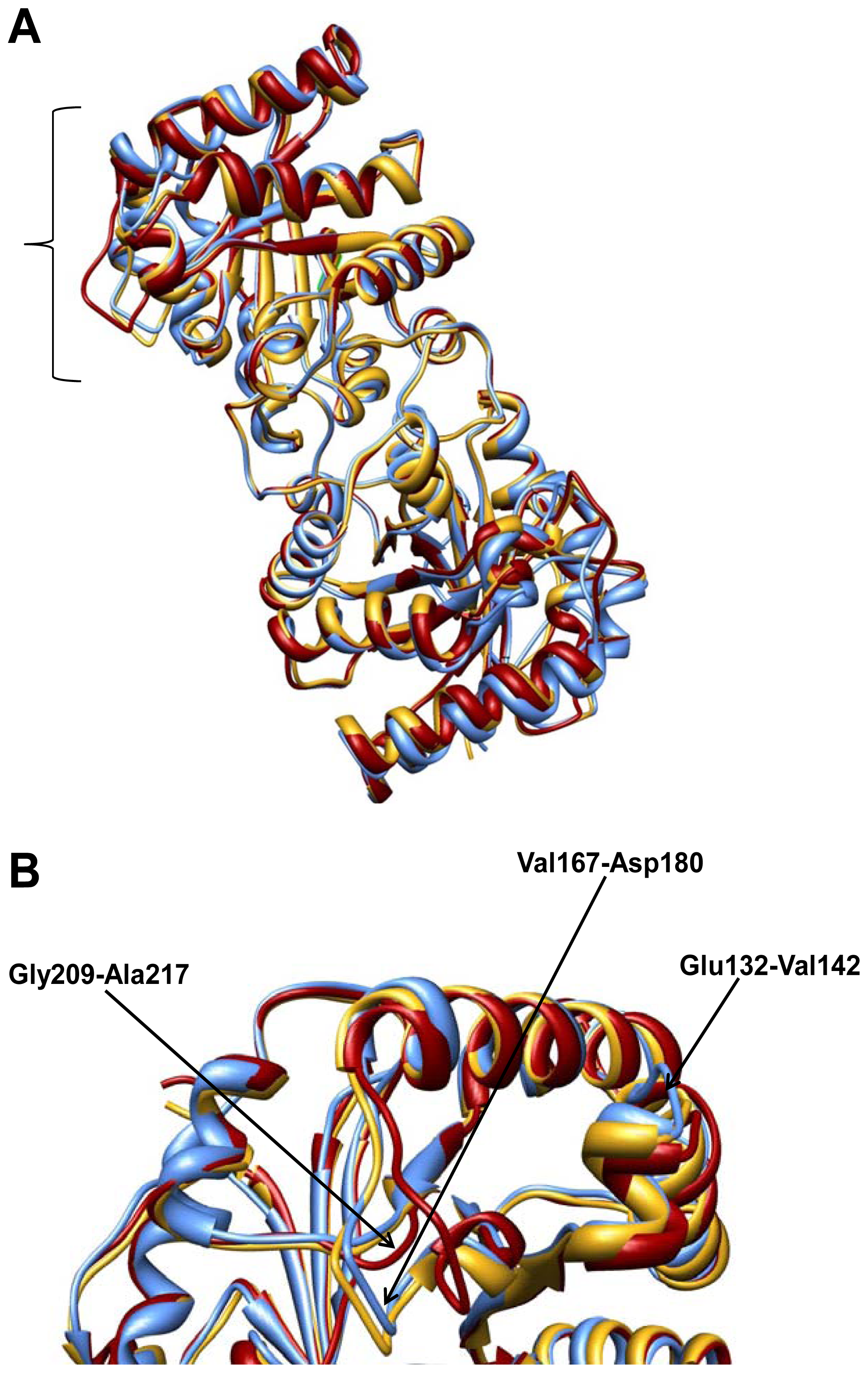
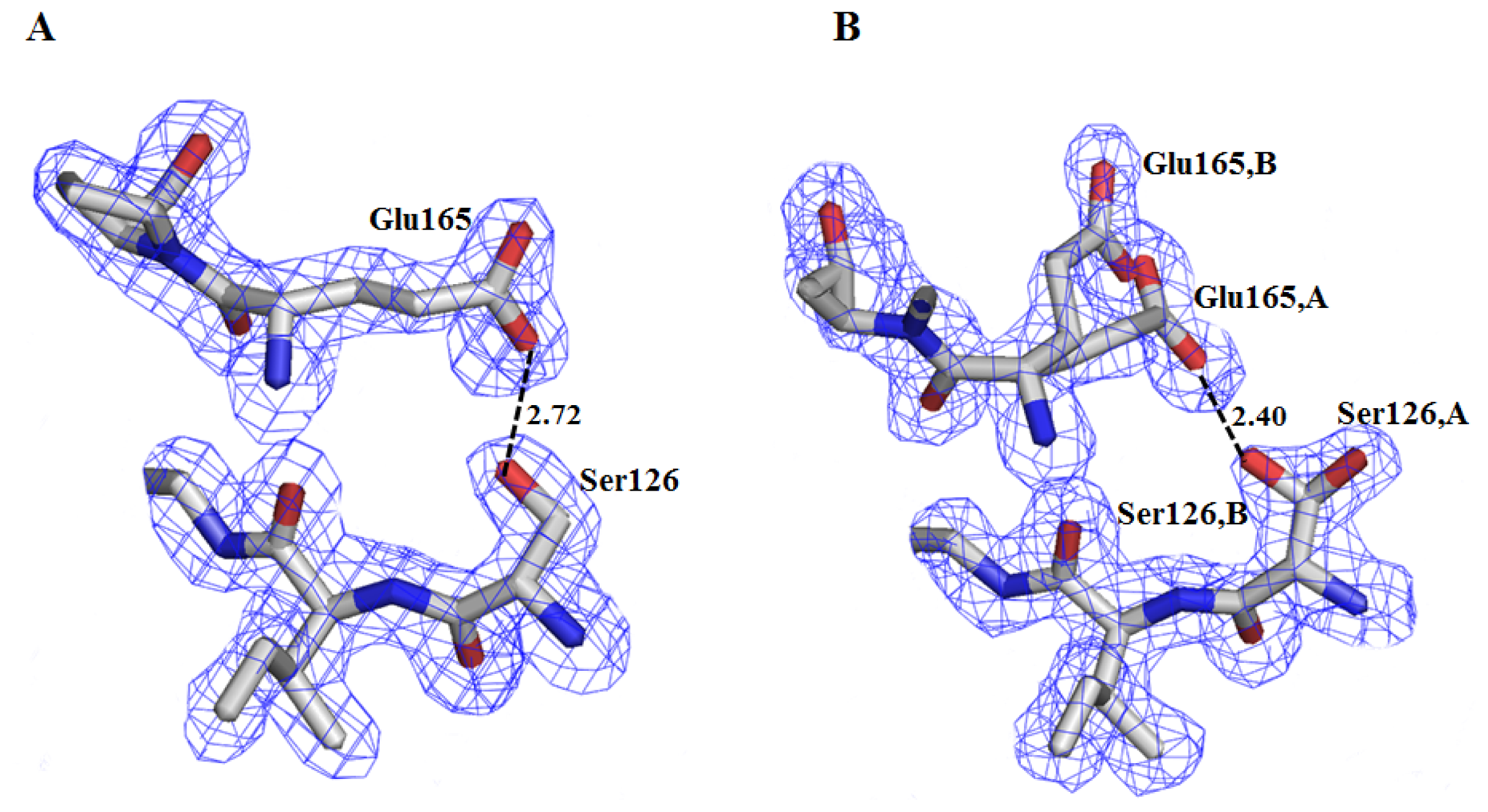
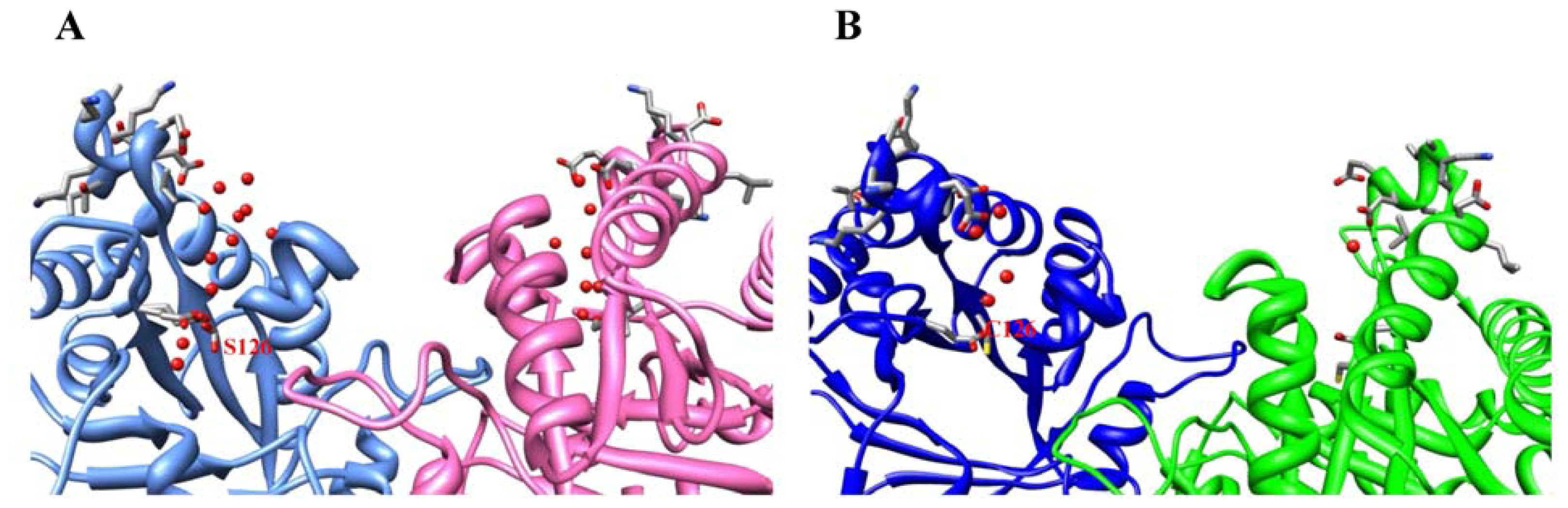
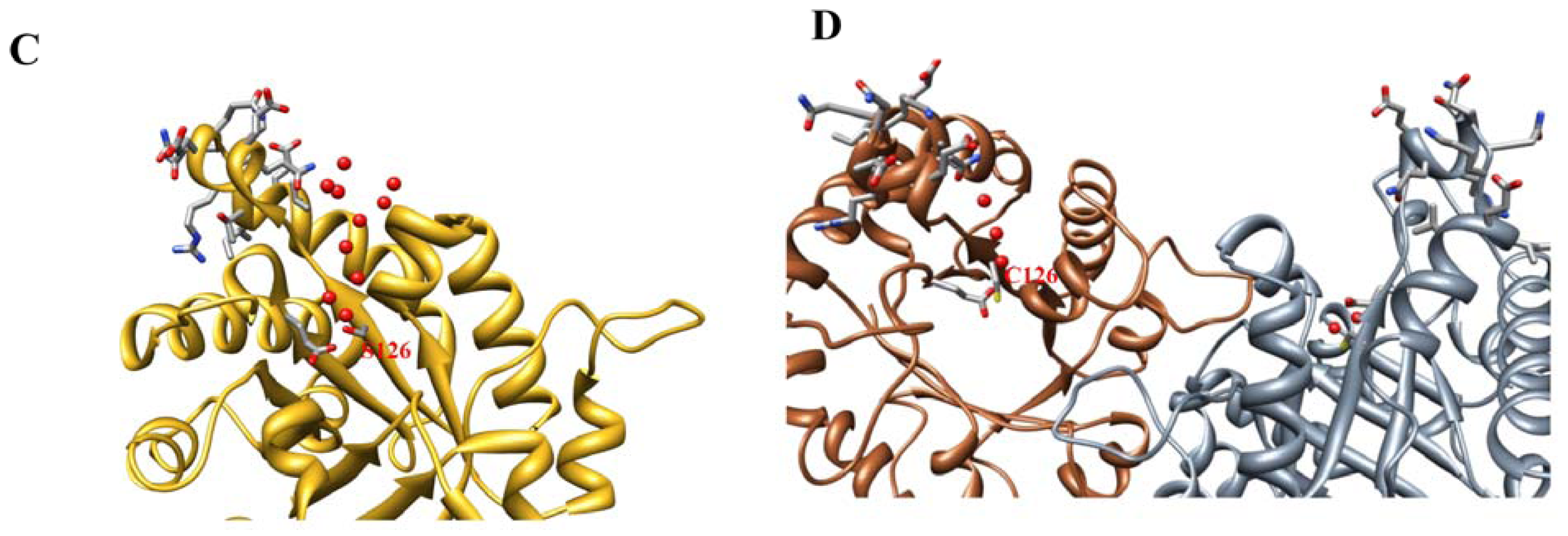
2.2. Crystal Structure Overview
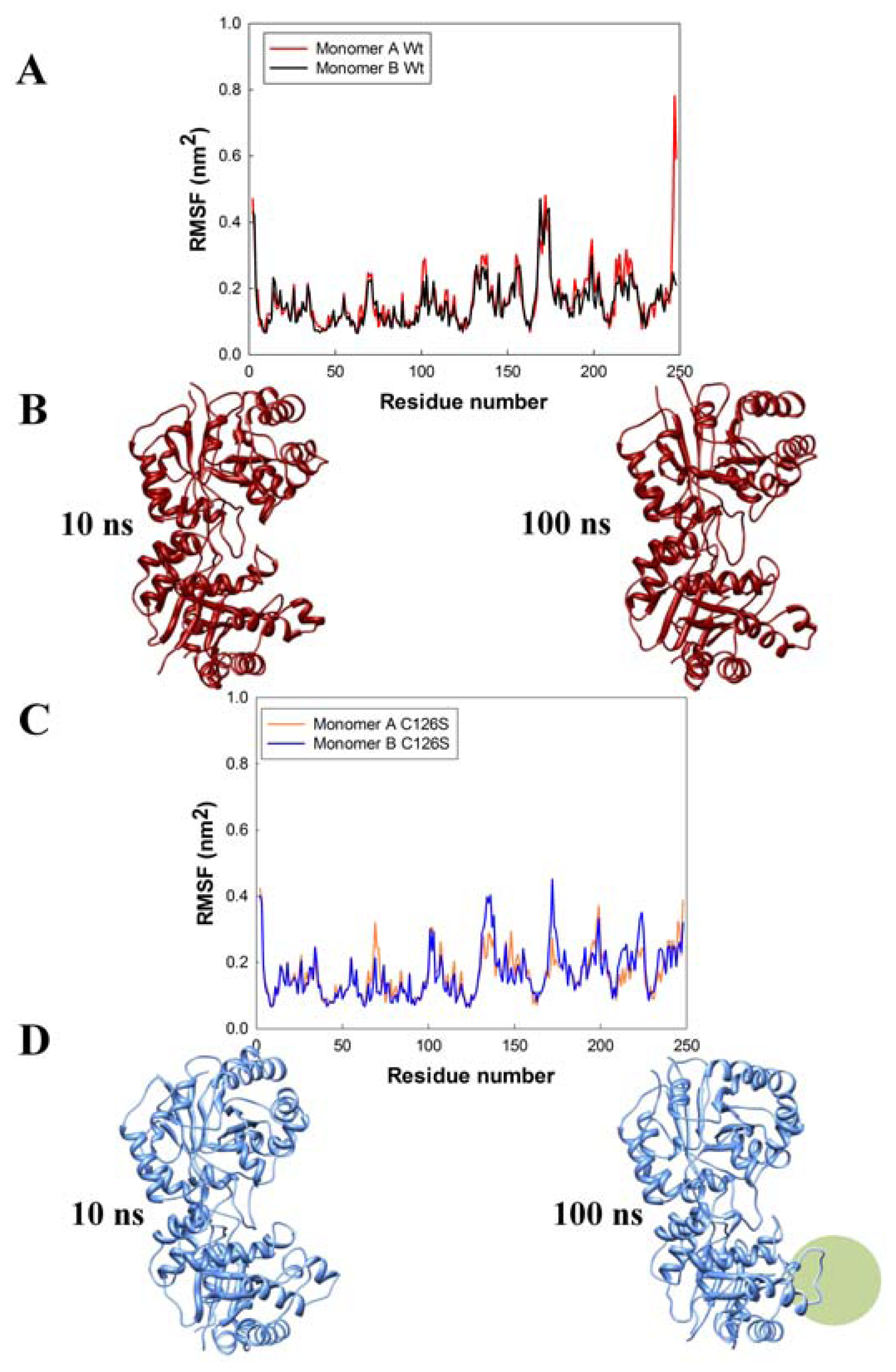
2.3. Molecular Dynamics
3. Experimental Section
3.1. Protein Expression, Purification and Crystallization
3.2. Structural Determination and Refinement
3.3. Molecular Dynamics
4. Conclusions
Supplementary Materials
ijms-13-10010-s001.pdfAcknowledgments
References
- González-Mondragón, E.; Zubillaga, R.A.; Saavedra, E.; Chánez-Cárdenas, M.A.; Pérez-Montfort, R.; Hernández-Arana, A. Conserved cysteine 126 in triosephosphate isomerase is required not for enzymatic activity but for proper folding and stability. Biochemistry 2004, 43, 3255–3263. [Google Scholar]
- Jeffery, C.J.; Gloss, L.M.; Petsko, G.A.; Ringe, D. The role of residues outside the active site: Structural basis for function of C191 mutants of E. coli aspartate aminotransferase. Protein Eng 2000, 13, 105–112. [Google Scholar]
- Lolis, E.; Alber, T.; Davenport, R.C.; Rose, D.; Hartman, F.C.; Petsko, G.A. Structure of yeast triosephosphate isomerase at 1.9-A resolution. Biochemistry 1990, 29, 6609–6618. [Google Scholar]
- Lolis, E.; Petsko, G.A. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-A resolution: Implications for catalysis. Biochemistry 1990, 29, 6619–6625. [Google Scholar]
- Cruces-Ángeles, M.E.; Cabrera, N.; Pérez-Montfort, R.; Reyes-López, C.A.; Hernández-Arana, A. Thermodynamic and kinetic destabilization of triosephosphate isomerase resulting from the mutation of conserved and non-conserved cysteines. Protein Pept. Lett 2011, 18, 1290–1298. [Google Scholar]
- Benítez-Cardoza, C.G.; Rojo-Domínguez, A.; Hernández-Arana, A. Temperature-Induced denaturation and renaturation of triosephosphate isomerase from Saccharomyces cerevisiae: Evidence of dimerization coupled to refolding of the thermally unfolded protein. Biochemistry 2001, 40, 9049–9058. [Google Scholar]
- Samanta, M.; Banerjee, M.; Murthy, M.R.; Balaram, H.; Balaram, P. Probing the role of the fully conserved Cys126 in triosephosphate isomerase by site-specific mutagenesis-distal effects on dimer stability. FEBS J 2011, 278, 1932–1943. [Google Scholar]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol 1968, 33, 491–497. [Google Scholar]
- Parthasarathy, S.; Eaazhisai, K.; Balaram, H.; Balaram, P.; Murthy, M.R. Structure of Plasmodium falciparum triose-phosphate isomerase-2-phosphoglycerate complex at 1.1-A resolution. J. Biol. Chem 2003, 278, 52461–52470. [Google Scholar]
- Miller, S.; Janin, L.; Lesk, A.M.; Chotia, C. Interior and surface of monomeric proteins. J. Mol. Biol 1987, 196, 641–656. [Google Scholar]
- Vázquez-Contreras, E.; Zubillaga, R.A.; Mendoza-Hernández, G.; Costas, M.; Fernández-Velasco, D.A. Equilibrium unfolding of yeast triosephosphate isomerase: A monomeric intermediate in guanidine-HCl and two-state behavior in urea. Protein Pept. Lett 2000, 7, 57–64. [Google Scholar]
- Norton, I.L.; Hartman, F.C. Haloacetol phosphates. A comparative study of the active sites of yeast and muscle triosephosphate isomerase. Biochemistry 1972, 11, 4435–4441. [Google Scholar]
- Rozacky, E.E.; Sawyer, T.H.; Barton, R.A.; Gracy, R.W. Studies of human triosephosphate isomerase. I. Isolation and properties of the enzyme from erythrocytes. Arch. Biochem. Biophys 1971, 146, 312–320. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr 2010, 66, 125–132. [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Biol. Crystallogr. 1994, 50, 760–763.
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr 2006, 62, 72–82. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst 2007, 40, 658–674. [Google Scholar]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.-W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr 2002, 58, 1948–1954. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-Building tools for molecular graphics. Acta Crystallogr 2004, 60, 2126–2132. [Google Scholar]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom structure validation for macromolecular crystallography. Acta Crystallogr 2010, 66, 12–21. [Google Scholar]
- Cohen, G.H. ALIGN: A program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Cryst 1997, 30, 1160–1161. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA; p. 2002.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data collection | |
| Space group | P212121 |
| Cell dimensions (Å) | a = 46.92 b = 61.44 c = 160.24 α = β = γ = 90.00 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.9791 |
| Resolution limit (Å) | 40.49–1.86 (1.93–1.86) |
| Reflection collected | 251,929 (30,945) |
| Unique reflections a | 39,548 (5593) |
| R bmerge | 10.6 (44.3) |
| Mean I/σI | 11.4 (3.6) |
| Completeness (%) | 99.7 (98.6) |
| Redundancy | 6.4 (5.5) |
| Wilson B-factor (Å2) | 21.45 |
| Refinement | |
| R cwork/Rfree (%) | 17.47 (22.21)/21.20 (25.20) |
| No. of atoms of protein/solvent | 3786/318 |
| No. of residues of | |
| SO4/PO4/glycerol/Na/PGA/ | 2/3/12/1/1 |
| Average B-value (Å2) | 13.6 |
| Macromolecule | 12.7 |
| Solvent | 18.1 |
| R.m.s.d. from ideal | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.29 |
| Ramachandran statistics of ϕ/ψ angles (%) | |
| Most favored | 98 |
| Additionally allowed | 2 |
| PDB code | 4FF7 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hernández-Santoyo, A.; Domínguez-Ramírez, L.; Reyes-López, C.A.; González-Mondragón, E.; Hernández-Arana, A.; Rodríguez-Romero, A. Effects of a Buried Cysteine-To-Serine Mutation on Yeast Triosephosphate Isomerase Structure and Stability. Int. J. Mol. Sci. 2012, 13, 10010-10021. https://doi.org/10.3390/ijms130810010
Hernández-Santoyo A, Domínguez-Ramírez L, Reyes-López CA, González-Mondragón E, Hernández-Arana A, Rodríguez-Romero A. Effects of a Buried Cysteine-To-Serine Mutation on Yeast Triosephosphate Isomerase Structure and Stability. International Journal of Molecular Sciences. 2012; 13(8):10010-10021. https://doi.org/10.3390/ijms130810010
Chicago/Turabian StyleHernández-Santoyo, Alejandra, Lenin Domínguez-Ramírez, César A. Reyes-López, Edith González-Mondragón, Andrés Hernández-Arana, and Adela Rodríguez-Romero. 2012. "Effects of a Buried Cysteine-To-Serine Mutation on Yeast Triosephosphate Isomerase Structure and Stability" International Journal of Molecular Sciences 13, no. 8: 10010-10021. https://doi.org/10.3390/ijms130810010