Macromolecule-Assisted de novo Protein Folding

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Uncovered Aspects of the Anfinsen Postulate and Molecular Chaperones
2.1. Anfinsen Postulate
2.2. Molecular Chaperones
3. Macromolecule Linkage-Mediated Folding Helper Systems in Vivo
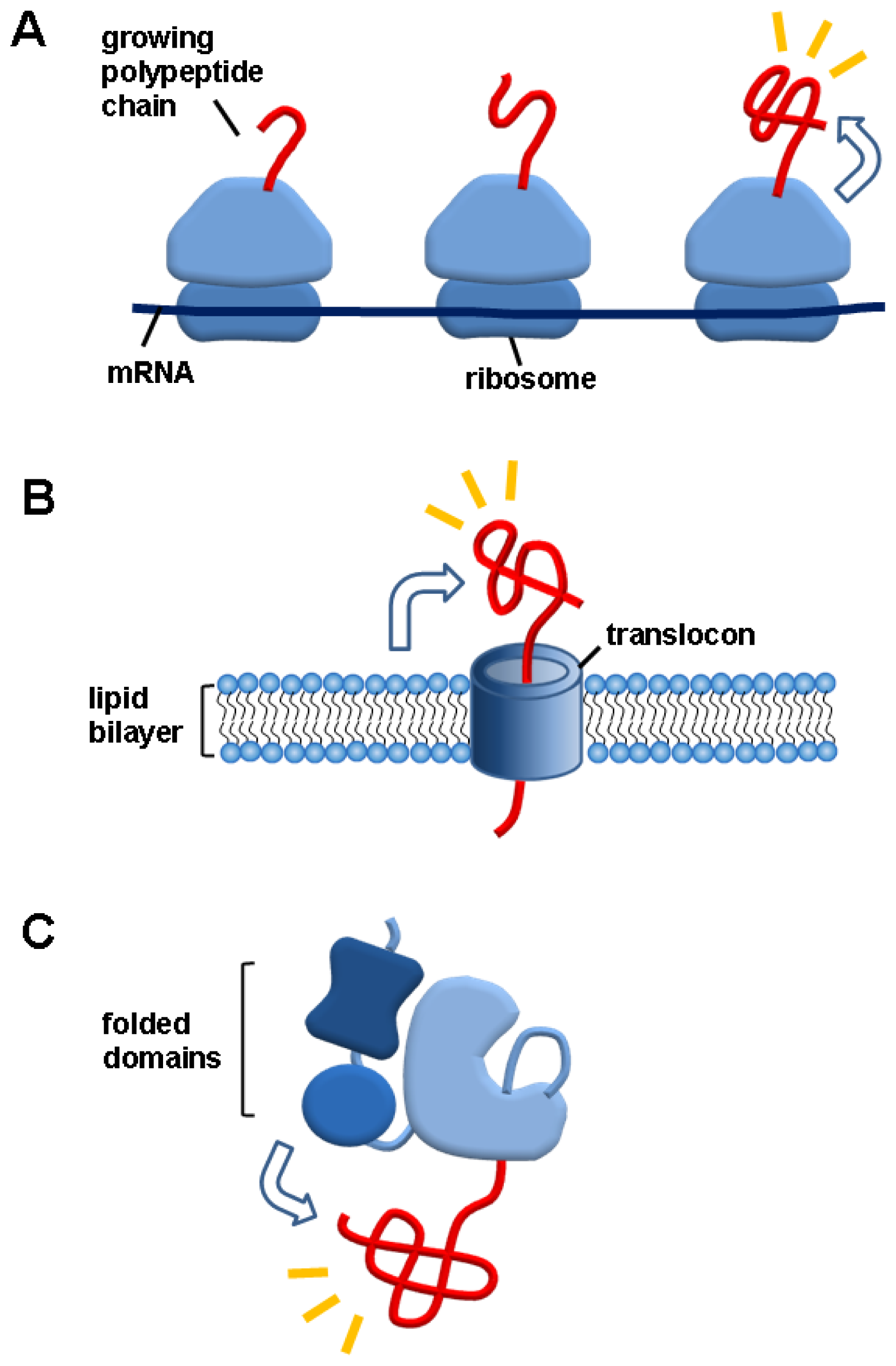
3.1. Ribosomes
3.2. Lipid Bilayers
3.3. Folded Domains in Multidomain Proteins
3.4. Salient Features of the Macromolecule Linkage-Mediated Folding Helper Systems
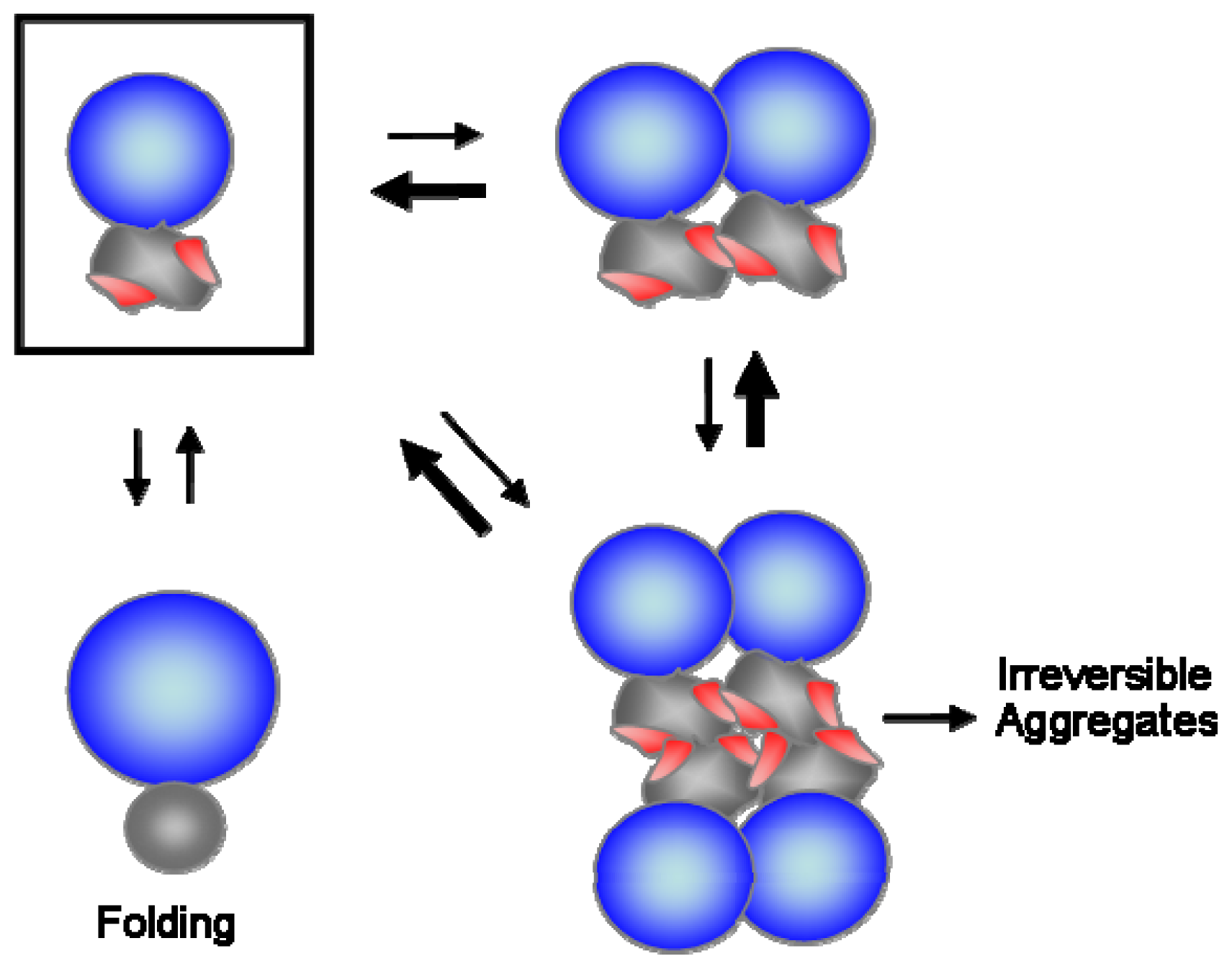
4. Hydrophobic Interaction-Based Stabilizing Mechanisms of Molecular Chaperones
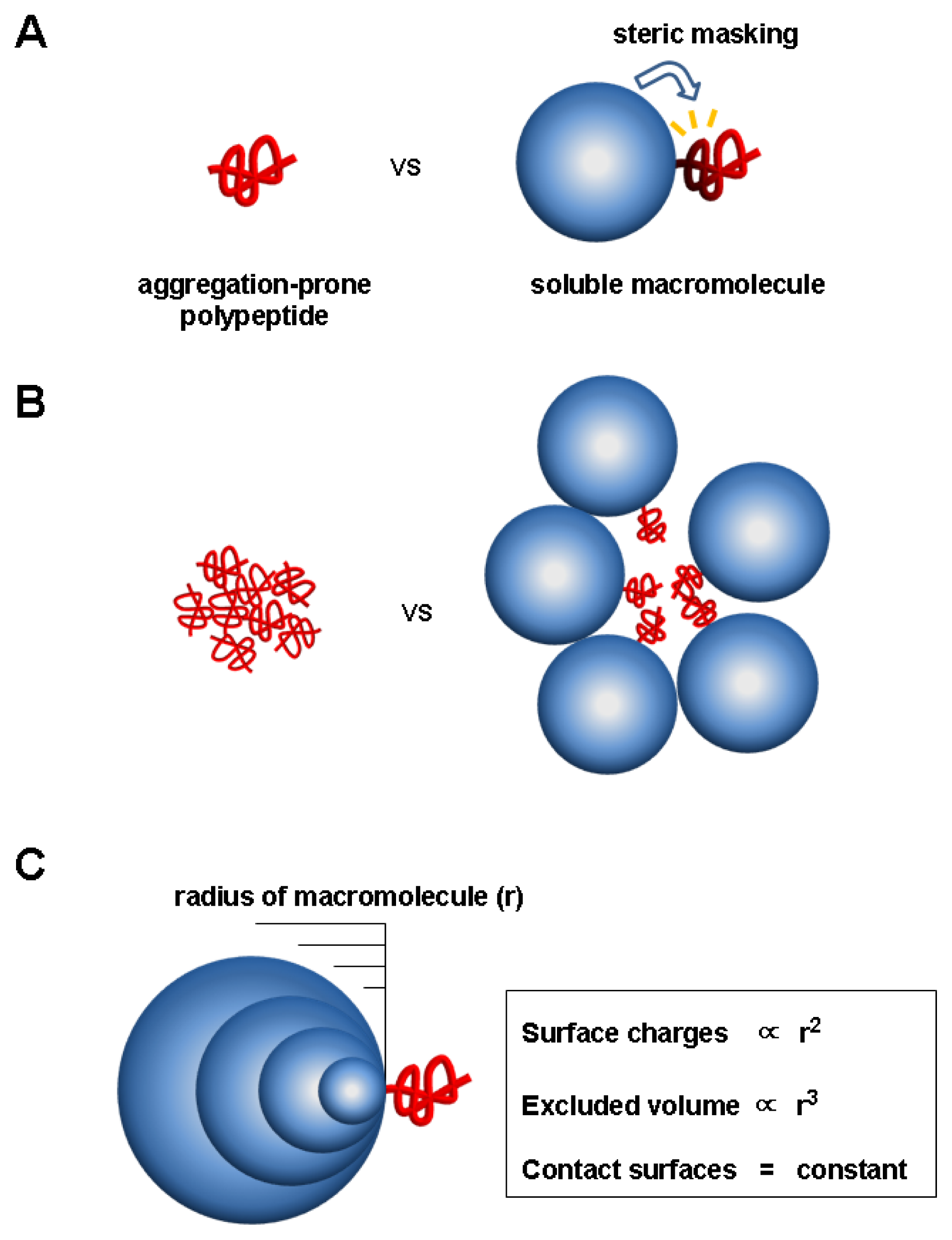
5. Charge and Steric Hindrance as Stabilizing Factors
5.1. Evidence for Charge and Steric Hindrance as Stabilizing Factors
5.2. Unique Features of the Charge and Steric Factors
5.3. Significance of the Charge and Steric Factors of Macromolecules in Chaperoning Functions
6. Conclusions and Perspectives
Acknowledgements
References
- Hemmingsen, S.M.; Woolford, C.; van der Vies, S.M.; Tilly, K.; Dennis, D.T.; Georgopoulos, C.P.; Hendrix, R.W.; Ellis, R.J. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature 1988, 333, 330–334. [Google Scholar]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar]
- Choi, S.I.; Ryu, K.; Seong, B.L. RNA-mediated chaperone type for de novo protein folding. RNA Biol 2009, 6, 21–24. [Google Scholar]
- Creighton, T.E. Protein Folding; W.H. Freeman and Company: New York, NY, USA, 1992; pp. 405–454. [Google Scholar]
- Kolb, V.A.; Makeyev, E.V.; Spirin, A.S. Folding of firefly luciferase during translation in a cell-free system. EMBO J 1994, 13, 3631–3637. [Google Scholar]
- Fedorov, A.N.; Baldwin, T.O. Cotranslational protein folding. J. Biol. Chem 1997, 272, 32715–32718. [Google Scholar]
- Kramer, G.; Ramachandiran, V.; Hardesty, B. Cotranslational folding-omnia mea mecum porto? Int. J. Biochem. Cell. Biol 2001, 33, 541–553. [Google Scholar]
- Cabrita, L.D.; Dobson, C.M.; Christodoulou, J. Protein folding on the ribosome. Curr. Opin. Struct. Biol 2010, 20, 33–45. [Google Scholar]
- Zhang, G.; Hubalewska, M.; Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol 2009, 16, 274–280. [Google Scholar]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar]
- Lilie, H.; Lang, K.; Rudolph, R.; Buchner, J. Prolyl isomerases catalyze antibody folding in vitro. Protein Sci 1993, 2, 1490–1496. [Google Scholar]
- Gruber, C.W.; Cemazar, M.; Heras, B.; Martin, J.L.; Craik, D.J. Protein disulfide isomerase: The structure of oxidative folding. Trends Biochem. Sci 2006, 31, 455–464. [Google Scholar]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol 2008, 18, 756–764. [Google Scholar]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: Intrinsically disordered proteins in Archaea. BMC Syst. Biol 2010, 4, S1. [Google Scholar]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol 2009, 5, 789–796. [Google Scholar]
- Chakraborty, K.; Chatila, M.; Sinha, J.; Shi, Q.; Poschner, B.C.; Sikor, M.; Jiang, G.; Lamb, D.C.; Hartl, F.U.; Hayer-Hartl, M. Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell 2010, 142, 112–122. [Google Scholar]
- Ellis, R.J. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol 2001, 11, 114–119. [Google Scholar]
- Szabo, A.; Langer, T.; Schröder, H.; Flanagan, J.; Bukau, B.; Hartl, F.U. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. USA 1994, 91, 10345–10349. [Google Scholar]
- Buchberger, A.; Schröder, H.; Hesterkamp, T.; Schönfeld, H.J.; Bukau, B. Substrate shuttling between the DnaK and GroEL systems indicates a chaperone network promoting protein folding. J. Mol. Biol 1996, 261, 328–333. [Google Scholar]
- Hesterkamp, T.; Bukau, B. Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E. coli. EMBO J 1998, 17, 4818–4828. [Google Scholar]
- Deuerling, E.; Schulze-Specking, A.; Tomoyasu, T.; Mogk, A.; Bukau, B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature 1999, 400, 693–696. [Google Scholar]
- Houry, W.A.; Frishman, D.; Eckerskorn, C.; Lottspeich, F.; Hartl, F.U. Identification of in vivo substrates of the chaperonin GroEL. Nature 1999, 402, 147–154. [Google Scholar]
- Koll, H.; Guiard, B.; Rassow, J.; Ostermann, J.; Horwich, A.L.; Neupert, W.; Hartl, F.U. Antifolding activity of hsp60 couples protein import into the mitochondrial matrix with export to the intermembrane space. Cell 1992, 68, 1163–1175. [Google Scholar]
- Kandror, O.; Busconi, L.; Sherman, M.; Goldberg, A.L. Rapid degradation of an abnormal protein in Escherichia coli involves the chaperones GroEL and GroES. J. Biol. Chem 1994, 269, 23575–23582. [Google Scholar]
- De Los Rios, P.; Ben-Zvi, A.; Slutsky, O.; Azem, A.; Goloubinoff, P. Hsp70 chaperones accelerate protein translocation and the unfolding of stable protein aggregates by entropic pulling. Proc. Natl. Acad. Sci. USA 2006, 103, 6166–6171. [Google Scholar]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol 2010, 11, 515–528. [Google Scholar]
- Frydman, J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem 2001, 70, 603–647. [Google Scholar]
- Vorderwülbecke, S.; Kramer, G.; Merz, F.; Kurz, T.A.; Rauch, T.; Zachmann-Brand, B.; Bukau, B.; Deuerling, E. Low temperature or GroEL/ES overproduction permits growth of Escherichia coli cells lacking trigger factor and DnaK. FEBS Lett 2004, 559, 181–187. [Google Scholar]
- Ullers, R.S.; Luirink, J.; Harms, N.; Schwager, F.; Georgopoulos, C.; Genevaux, P. SecB is a bona fide generalized chaperone in Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 7583–7588. [Google Scholar]
- Masters, M.; Blakely, G.; Coulson, A.; McLennan, N.; Yerko, V.; Acord, J. Protein folding in Escherichia coli: The chaperonin GroE and its substrates. Res. Microbiol 2009, 160, 267–277. [Google Scholar]
- Wong, P.; Houry, W.A. Chaperone networks in bacteria: Analysis of protein homeostasis in minimal cells. J. Struct. Biol 2004, 146, 79–89. [Google Scholar]
- Horwich, A.L.; Low, K.B.; Fenton, W.A.; Hirshfield, I.N.; Furtak, K. Folding in vivo of bacterial cytoplasmic proteins: Role of GroEL. Cell 1993, 74, 909–917. [Google Scholar]
- Chapman, E.; Farr, G.W.; Usaite, R.; Furtak, K.; Fenton, W.A.; Chaudhuri, T.K.; Hondorp, E.R.; Matthews, R.G.; Wolf, S.G.; Yates, J.R.; et al. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc. Natl. Acad. Sci. USA 2006, 103, 15800–15805. [Google Scholar]
- Sewell, B.T.; Best, R.B.; Chen, S.; Roseman, A.M.; Farr, G.W.; Horwich, A.L.; Saibil, H.R. A mutant chaperonin with rearranged inter-ring electrostatic contacts and temperature-sensitive dissociation. Nat. Struct. Mol. Biol 2004, 11, 1128–1133. [Google Scholar]
- Sot, B.; von Germar, F.; Mäntele, W.; Valpuesta, J.M.; Taneva, S.G.; Muga, A. Ionic interactions at both inter-ring contact sites of GroEL are involved in transmission of the allosteric signal: A time-resolved infrared difference study. Protein Sci 2005, 14, 2267–2274. [Google Scholar]
- Cheng, M.Y.; Hartl, F.U.; Martin, J.; Pollock, R.A.; Kalousek, F.; Neupert, W.; Hallberg, E.M.; Hallberg, R.L.; Horwich, A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 1989, 337, 620–625. [Google Scholar]
- Ellis, R.J.; Hartl, F.U. Principles of protein folding in the cellular environment. Curr. Opin. Struct. Biol 1999, 9, 102–110. [Google Scholar]
- Feldman, D.E.; Frydman, J. Protein folding in vivo: The importance of molecular chaperones. Curr. Opin. Struct. Biol 2000, 10, 26–33. [Google Scholar]
- Sørensen, H.P.; Kristensen, J.E.; Sperling-Petersen, H.U.; Mortensen, K.K. Soluble expression of aggregating proteins by covalent coupling to the ribosome. Biochem. Biophys. Res. Commun 2004, 319, 715–719. [Google Scholar]
- Schimmele, B.; Gräfe, N.; Plückthun, A. Ribosome display of mammalian receptor domains. Protein Eng. Des. Sel 2005, 18, 285–294. [Google Scholar]
- Wittrup, K.D. Protein engineering by cell-surface display. Curr. Opin. Biotechnol 2001, 12, 395–399. [Google Scholar]
- Ngo, K.X.; Umakoshi, H.; Shimanouchi, T.; Sugaya, H.; Kuboi, R. Chitosanase displayed on liposome can increase its activity and stability. J. Bacteriol 2010, 146, 105–113. [Google Scholar]
- Kim, C.W.; Han, K.S.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Choi, S.I.; Seong, B.L. N-terminal domains of native multidomain proteins have the potential to assist de novo folding of their downstream domains in vivo by acting as solubility enhancers. Protein Sci 2007, 16, 635–643. [Google Scholar]
- Pelham, H.R. Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell 1986, 46, 959–961. [Google Scholar]
- Wegrzyn, R.D.; Deuerling, E. Molecular guardians for newborn proteins: Ribosome-associated chaperones and their role in protein folding. Cell. Mol. Life Sci 2005, 62, 2727–2738. [Google Scholar]
- Craig, E.A.; Eisenman, H.C.; Hundley, H.A. Ribosome-tethered molecular chaperones: The first line of defense against protein misfolding? Curr. Opin. Microbiol 2003, 6, 157–162. [Google Scholar]
- Peisker, K.; Chiabudini, M.; Rospert, S. The ribosome-bound Hsp70 homolog Ssb of Saccharomyces cerevisiae. Biochim. Biophys. Acta 2010, 1803, 662–672. [Google Scholar]
- Choi, S.I.; Lim, K.H.; Seong, B.L. Chaperoning roles of macromolecules interacting with proteins in vivo. Int. J. Mol. Sci 2011, 12, 1979–1990. [Google Scholar]
- Svetlov, M.S.; Kolb, V.A.; Spirin, A.S. Folding of the firefly luciferase polypeptide chain with immobilized C-terminus. Mol. Biol (Mosk) 2007, 41, 96–102. [Google Scholar]
- Choi, S.I.; Han, K.S.; Kim, C.W.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Kim, S.I.; Kang, T.H.; Shin, H.C.; Lim, K.H.; et al. Protein solubility and folding enhancement by interaction with RNA. PLoS One 2008, 3, e2677. [Google Scholar]
- Kaiser, C.M.; Goldman, D.H.; Chodera, J.D.; Tinoco, I., Jr; Bustamante, C. The ribosome modulates nascent protein folding. Science 2011, 334, 1723–1727. [Google Scholar]
- Das, B.; Chattopadhyay, S.; Bera, A.K.; DasGupta, C. In vitro protein folding by ribosomes from Escherichia coli, wheat germ and rat liver: The role of the 50S particle and its 23S rRNA. Eur. J. Biochem 1996, 235, 613–621. [Google Scholar]
- Chattopadhyay, S.; Das, B.; DasGupta, C. Reactivation of denatured proteins by 23S ribosomal RNA: Role of domain V. Proc. Natl. Acad. Sci. USA 1996, 93, 8284–8287. [Google Scholar]
- Samanta, D.; Mukhopadhyay, D.; Chowdhury, S.; Ghosh, J.; Pal, S.; Basu, A.; Bhattacharya, A.; Das, A.; Das, D.; DasGupta, C. Protein folding by domain V of Escherichia coli 23S rRNA: Specificity of RNA-protein interactions. J. Bacteriol 2008, 190, 3344–3352. [Google Scholar]
- Chen, W.; Georgiou, G. Cell-Surface display of heterologous proteins: From high-throughput screening to environmental applications. Biotechnol. Bioeng 2002, 79, 496–503. [Google Scholar]
- Cornelis, P. Expressing genes in different Escherichia coli compartments. Curr. Opin. Biotechnol 2000, 11, 450–454. [Google Scholar]
- Jose, J. Autodisplay: Efficient bacterial surface display of recombinant proteins. Appl. Microbiol. Biotechnol 2006, 69, 607–614. [Google Scholar]
- Chen, Y.P.; Hwang, I.E.; Lin, C.J.; Wang, H.J.; Tseng, C.P. Enhancing the stability of xylanase from Cellulomonas fimi by cell-surface display on Escherichia coli. J. Appl. Microbiol. 2012, 112, 455–463. [Google Scholar]
- Polizzi, K.M.; Bommarius, A.S.; Broering, J.M.; Chaparro-Riggers, J.F. Stability of biocatalysts. Curr. Opin. Chem. Biol 2007, 11, 220–225. [Google Scholar]
- O’Fágáin, C. Engineering protein stability. Methods Mol. Biol 2011, 681, 103–136. [Google Scholar]
- Hebert, D.N.; Zhang, J.X.; Chen, W.; Foellmer, B.; Helenius, A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell. Biol 1997, 139, 613–623. [Google Scholar]
- Garvey, M.; Griesser, S.S.; Griesser, H.J.; Thierry, B.; Nussio, M.R.; Shapter, J.G.; Ecroyd, H.; Giorgetti, S.; Bellotti, V.; Gerrard, J.A.; et al. Enhanced molecular chaperone activity of the small heat-shock protein αB-cystallin following covalent immobilization onto a solid-phase support. Biopolymers 2011, 95, 376–389. [Google Scholar]
- Netzer, W.J.; Hartl, F.U. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature 1997, 388, 343–349. [Google Scholar]
- Braun, P.; LaBaer, J. High throughput protein production for functional proteomics. Trends Biotechnol 2003, 21, 383–388. [Google Scholar]
- Esposito, D.; Chatterjee, D.K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol 2006, 17, 353–358. [Google Scholar]
- Van den Berg, B.; Ellis, R.J.; Dobson, C.M. Effects of macromolecular crowding on protein folding and aggregation. EMBO J 1999, 18, 6927–6933. [Google Scholar]
- Kramer, G.; Boehringer, D.; Ban, N.; Bukau, B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol 2009, 16, 589–597. [Google Scholar]
- Meinnel, T.; Giglione, C. Tools for analyzing and predicting N-terminal protein modifications. Proteomics 2008, 8, 626–649. [Google Scholar]
- Kauzmann, W. Factors in interpretation of protein denaturation. Adv. Protein Chem 1959, 14, 1–63. [Google Scholar]
- Baldwin, R.L. Energetics of protein folding. J. Mol. Biol 2007, 10, 283–301. [Google Scholar]
- Bolen, D.W.; Rose, G.D. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu. Rev. Biochem 2008, 77, 339–362. [Google Scholar]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta. Biochim. Pol 2004, 51, 349–371. [Google Scholar]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar]
- Fernández, A.; Crespo, A. Protein wrapping: A molecular marker for association, aggregation and drug design. Chem. Soc. Rev 2008, 37, 2373–2382. [Google Scholar]
- Misselwitz, B.; Staeck, O.; Rapoport, T.A. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell 1998, 2, 593–603. [Google Scholar]
- Aoki, K.; Taguchi, H.; Shindo, Y.; Yoshida, M.; Ogasahara, K.; Yutani, K.; Tanaka, N. Calorimetric observation of a GroEL-protein binding reaction with little contribution of hydrophobic interaction. J. Biol. Chem 1997, 272, 32158–32162. [Google Scholar]
- Martinez-Hackert, E.; Hendrickson, W.A. Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell 2009, 138, 923–934. [Google Scholar]
- Trombetta, E.S.; Helenius, A. Lectins as chaperones in glycoprotein folding. Curr. Opin. Struct. Biol 1998, 8, 587–592. [Google Scholar]
- Ellis, R.J. Protein folding: Importance of the Anfinsen cage. Curr. Biol 2003, 13, R881–R883. [Google Scholar]
- Apetri, A.C.; Horwich, A.L. Chaperonin chamber accelerates protein folding through passive action of preventing aggregation. Proc. Natl. Acad. Sci. USA 2008, 105, 17351–17355. [Google Scholar]
- Tang, Y.C.; Chang, H.C.; Roeben, A.; Wischnewski, D.; Wischnewski, N.; Kerner, M.J.; Hartl, F.U.; Hayer-Hartl, M. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell 2006, 125, 903–914. [Google Scholar]
- Lin, Z.; Rye, HS. GroEL-mediated protein folding: Making the impossible, possible. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 211–239. [Google Scholar]
- Todd, M.J.; Lorimer, G.H.; Thirumalai, D. Chaperonin-facilitated protein folding: Optimization of rate and yield by an iterative annealing mechanism. Proc. Natl. Acad. Sci. USA 1996, 93, 4030–4035. [Google Scholar]
- Kmiecik, S.; Kolinski, A. Simulation of chaperonin effect on protein folding: A shift from nucleation-condensation to framework mechanism. J. Am. Chem. Soc 2011, 133, 10283–10289. [Google Scholar]
- Shaw, B.F.; Moustakas, D.T.; Whitelegge, J.P.; Faull, K.F. Taking charge of proteins from neurodegeneration to industrial biotechnology. Adv. Protein Chem. Struct. Biol 2010, 79, 127–164. [Google Scholar]
- Schmittschmitt, J.P.; Scholtz, J.M. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci 2003, 12, 2374–2378. [Google Scholar]
- Shaw, K.L.; Grimsley, G.R.; Yakovlev, G.I.; Makarov, A.A.; Pace, C.N. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci 2001, 10, 1206–1215. [Google Scholar]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J 2004, 18, 1169–1175. [Google Scholar]
- Chiti, F.; Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 16419–16426. [Google Scholar]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc 2007, 129, 10110–10112. [Google Scholar]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar]
- Marqusee, S.; Robbins, V.H.; Baldwin, R.L. Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. USA 1989, 86, 5286–5290. [Google Scholar]
- Chen, J.; Skehel, J.J.; Wiley, D.C. A polar octapeptide fused to the N-terminal fusion peptide solubilizes the influenza virus HA2 subunit ectodomain. Biochemistry 1998, 37, 13643–13649. [Google Scholar]
- Zhang, Y.B.; Howitt, J.; McCorkle, S.; Lawrence, P.; Springer, K.; Freimuth, P. Protein aggregation during overexpression limited by peptide extensions with large net negative charge. Protein Expr. Purif 2004, 36, 207–216. [Google Scholar]
- Kapust, R.B.; Waugh, D.S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci 1999, 8, 1668–1674. [Google Scholar]
- Speed, M.A.; Wang, D.I.; King, J. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nat. Biotechnol 1996, 14, 1283–1287. [Google Scholar]
- Rajan, R.S.; Illing, M.E.; Bence, N.F.; Kopito, R.R. Specificity in intracellular protein aggregation and inclusion body formation. Proc. Natl. Acad. Sci. USA 2001, 98, 13060–13065. [Google Scholar]
- Wright, C.F.; Teichmann, S.A.; Clarke, J.; Dobson, C.M. The importance of sequence diversity in the aggregation and evolution of proteins. Nature 2005, 438, 878–881. [Google Scholar]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des 1998, 3, R9–R23. [Google Scholar]
- Høiberg-Nielsen, R.; Fuglsang, C.C.; Arleth, L.; Westh, P. Interrelationships of glycosylation and aggregation kinetics for Peniophora lycii phytase. Biochemistry 2006, 45, 5057–5066. [Google Scholar]
- Rajan, R.S.; Li, T.; Aras, M.; Sloey, C.; Sutherland, W.; Arai, H.; Briddell, R.; Kinstler, O.; Lueras, A.M.; Zhang, Y.; et al. Modulation of protein aggregation by polyethylene glycol conjugation: GCSF as a case study. Protein Sci 2006, 15, 1063–1075. [Google Scholar]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J 1997, 16, 1501–1507. [Google Scholar]
- Ryu, K.; Kim, C.W.; Kim, B.H.; Han, K.S.; Kim, K.H.; Choi, S.I.; Seong, B.L. Assessment of substrate-stabilizing factors for DnaK on the folding of aggregation-prone proteins. Biochem. Biophys. Res. Commun 2008, 373, 74–79. [Google Scholar]
- Hiemenz, P.C.; Rajagopalan, R. In Principles of Colloid and Surface Chemistry, 3rd ed; CRC Press: London, UK, 1997; pp. 575–621. [Google Scholar]
- Wayne, N.; Bolon, D.N. Charge-rich regions modulate the anti-aggregation activity of Hsp90. J. Mol. Biol 2010, 401, 931–939. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choi, S.I.; Son, A.; Lim, K.-H.; Jeong, H.; Seong, B.L. Macromolecule-Assisted de novo Protein Folding. Int. J. Mol. Sci. 2012, 13, 10368-10386. https://doi.org/10.3390/ijms130810368
Choi SI, Son A, Lim K-H, Jeong H, Seong BL. Macromolecule-Assisted de novo Protein Folding. International Journal of Molecular Sciences. 2012; 13(8):10368-10386. https://doi.org/10.3390/ijms130810368
Chicago/Turabian StyleChoi, Seong Il, Ahyun Son, Keo-Heun Lim, Hotcherl Jeong, and Baik L. Seong. 2012. "Macromolecule-Assisted de novo Protein Folding" International Journal of Molecular Sciences 13, no. 8: 10368-10386. https://doi.org/10.3390/ijms130810368
APA StyleChoi, S. I., Son, A., Lim, K.-H., Jeong, H., & Seong, B. L. (2012). Macromolecule-Assisted de novo Protein Folding. International Journal of Molecular Sciences, 13(8), 10368-10386. https://doi.org/10.3390/ijms130810368