Development and Validation of a Stability-Indicating High Performance Liquid Chromatographic (HPLC) Method for the Determination of Related Substances of Micafungin Sodium in Drug Substances

Abstract
:
1. Introduction
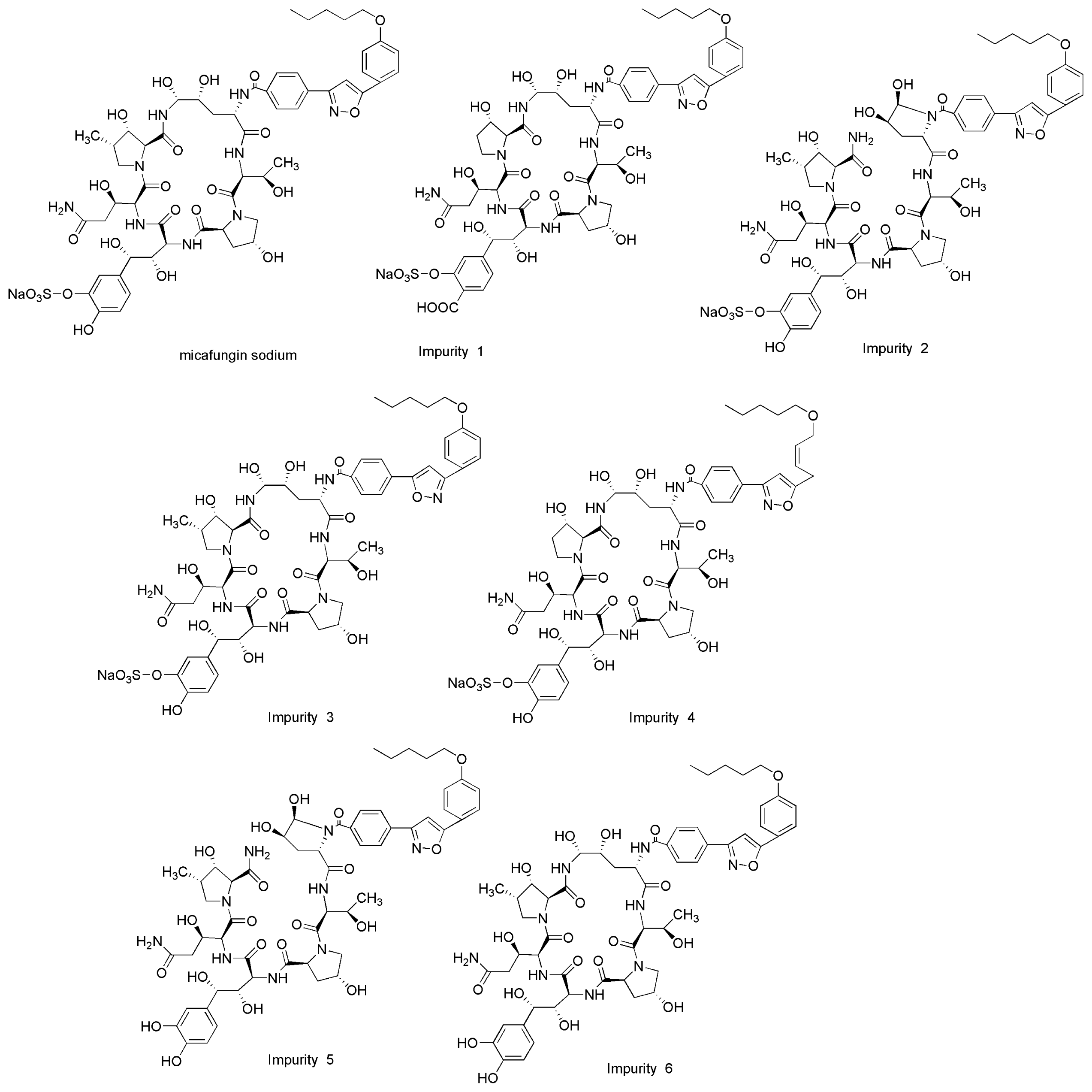
2. Results and Discussions
2.1. Method Development
2.2. Method Validation
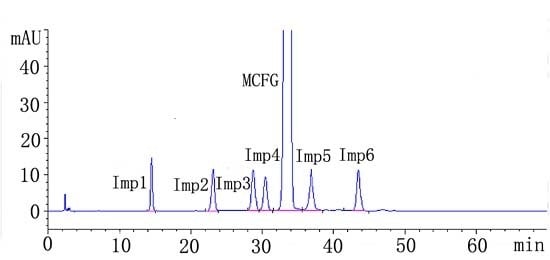
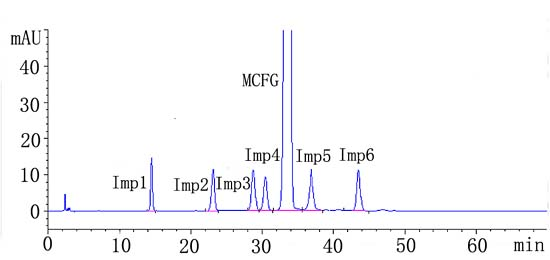
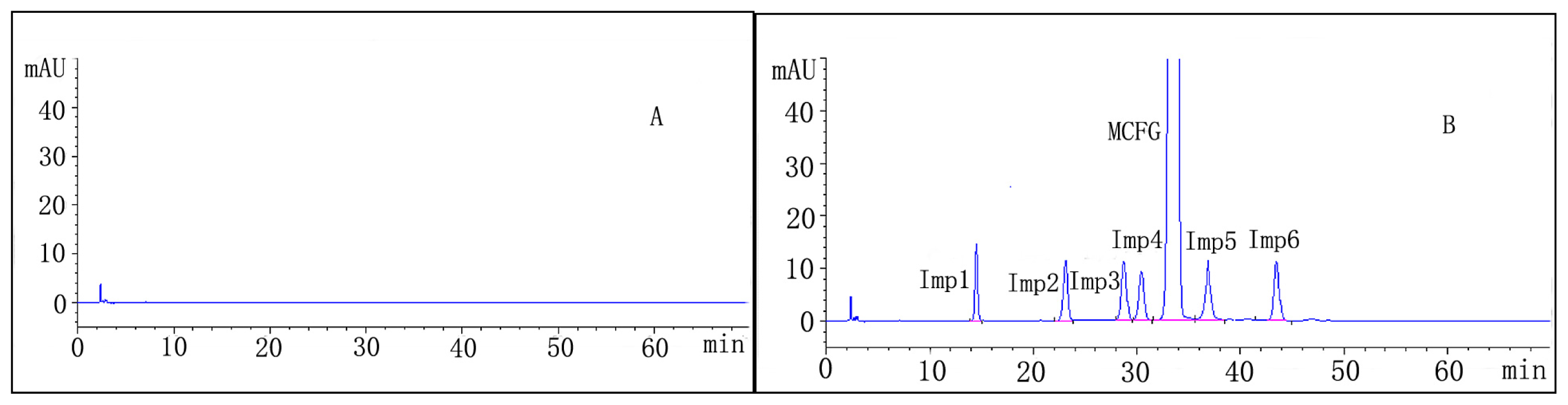
2.2.1. System Suitability
2.2.2. Specificity
2.2.3. Precision
2.2.4. Limit of Detection and Limit of Quantification
2.2.5. Linearity and Range
2.2.6. Accuracy
2.2.7. Robustness
2.2.8. Solution Stability and Mobile Phase Stability
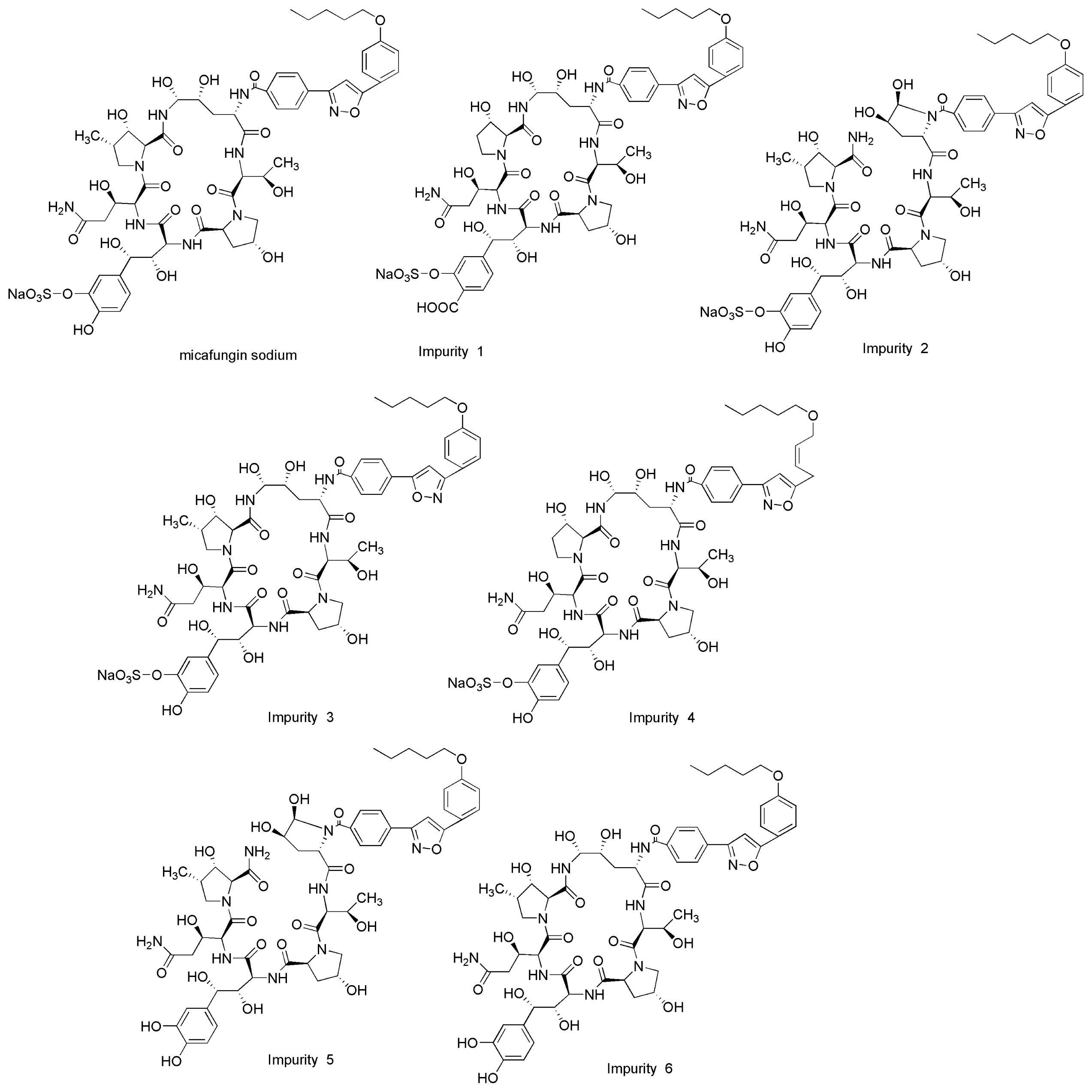
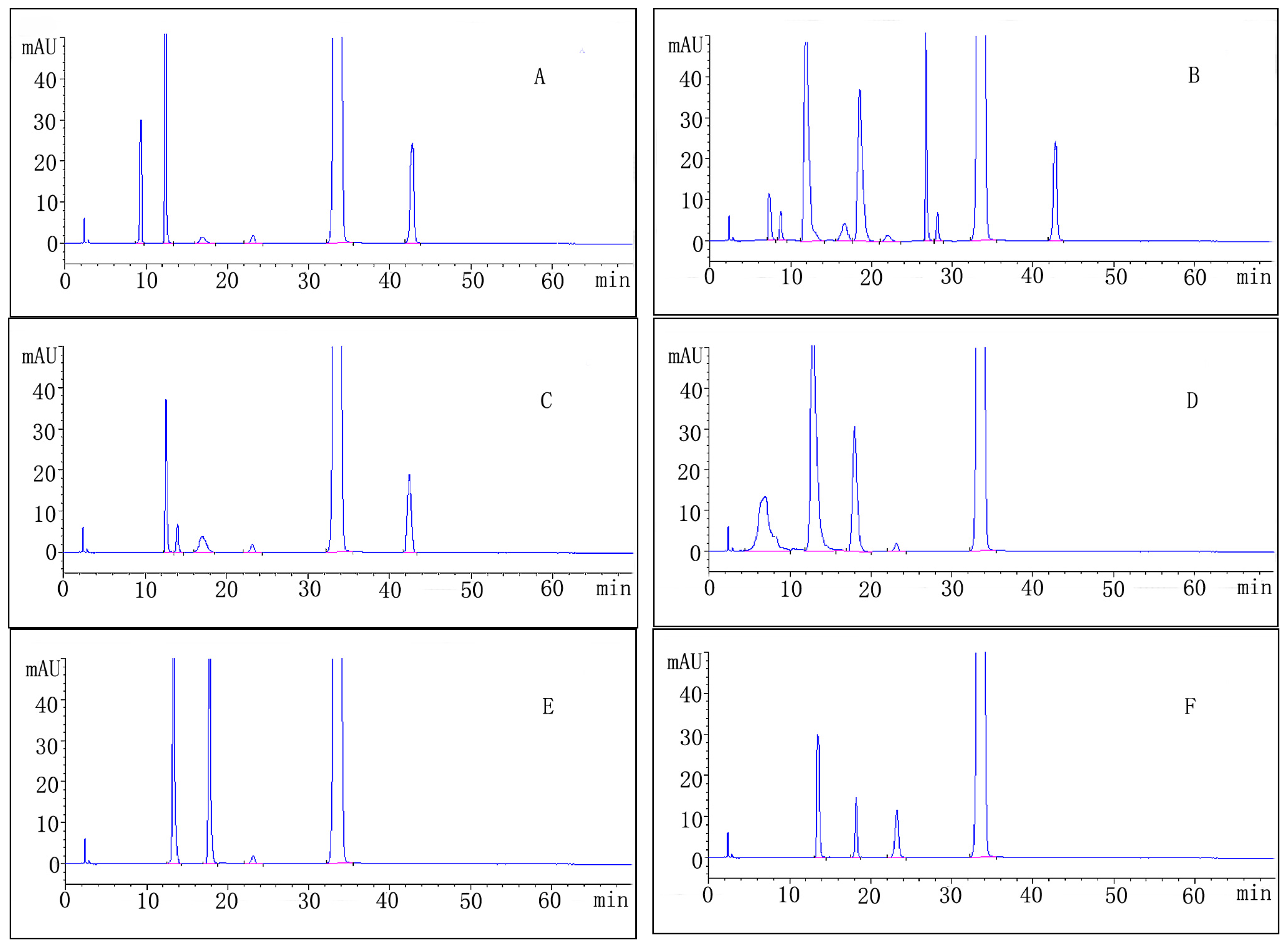
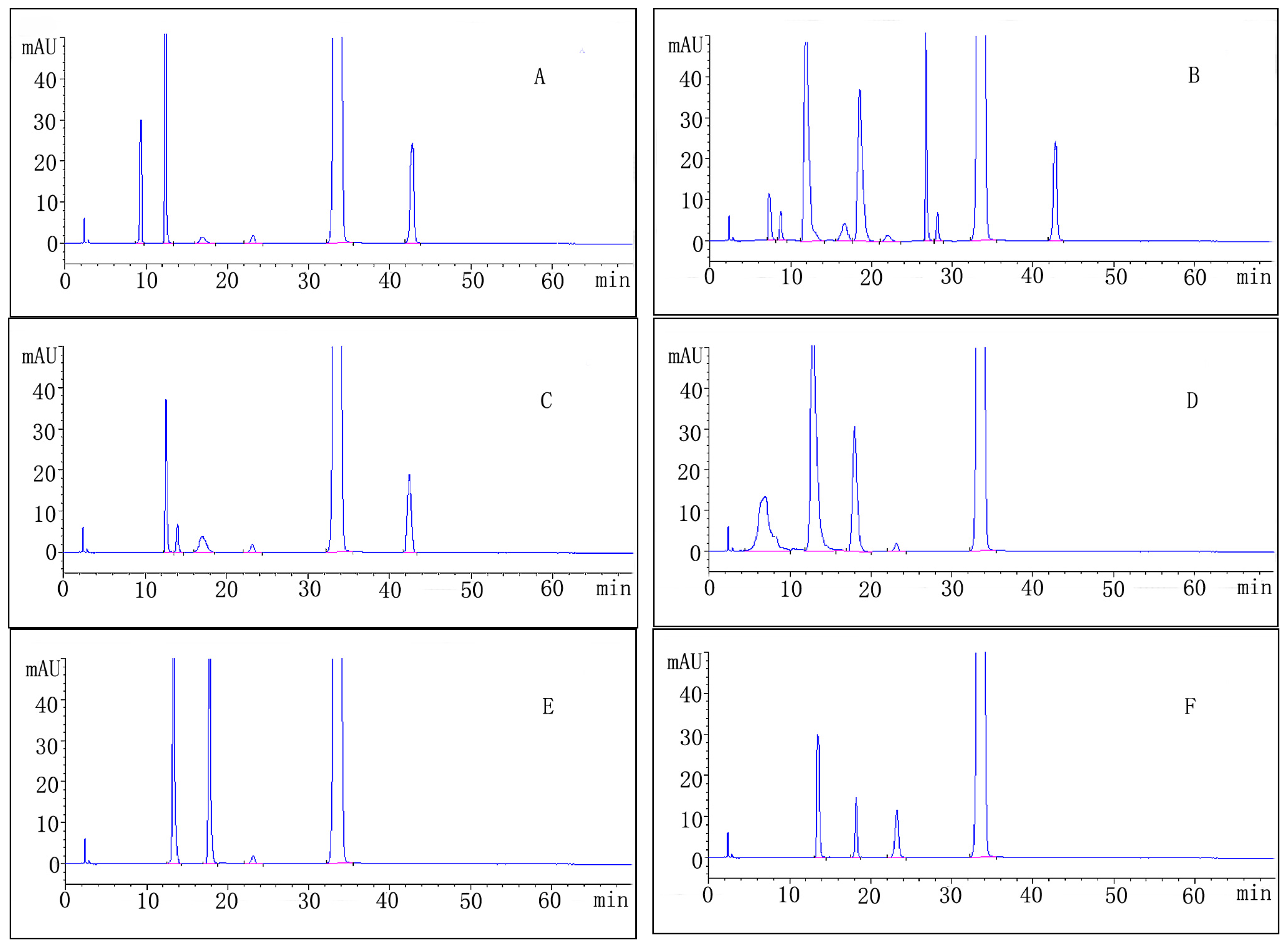
2.3. Forced Degradation Behavior
2.3.1. Acid Degradation
2.3.2. Base Degradation
2.3.3. Water Degradation
2.3.4. Oxidation Degradation
2.3.5. Photolytic Degradation
2.3.6. Thermal Degradation
2.4. Application of the Method: Analysis of Bulk Drug
3. Experimental
3.1. Instrumentation
3.2. Materials and Reagents
3.3. Chromatographic Conditions
3.4. Preparation of the Solutions
3.4.1. Diluent Solution
3.4.2. Working Solution
3.5. Forced Degradation Studies
3.5.1. Acid Degradation
3.5.2. Base Degradation
3.5.3. Hydrolytic Degradation
3.5.4. Oxidative Degradation
3.5.5. Thermal Degradation
3.5.6. Photolytic Degradation
4. Conclusions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | R | N | Symmetry factor | RRT | tR ± SD (min) | RSD (%) |
|---|---|---|---|---|---|---|
| 1 | / | 14010 | 1.08 | 0.44 | 14.50 ± 0.05 | 0.34 |
| 2 | 13.11 | 14700 | 1.12 | 0.69 | 23.12 ± 0.07 | 0.30 |
| 3 | 7.55 | 15588 | 1.22 | 0.85 | 28.63 ± 0.08 | 0.27 |
| 4 | 1.65 | 17556 | 1.24 | 0.91 | 30.34 ± 0.07 | 0.23 |
| a M | 3.18 | 18109 | 1.09 | 1.00 | 33.51 ± 0.07 | 0.21 |
| 5 | 3.22 | 18299 | 1.11 | 1.10 | 36.88 ± 0.07 | 0.19 |
| 6 | 4.87 | 17666 | 1.22 | 1.27 | 42.77 ± 0.08 | 0.17 |
| Analyst | % RSD impurities 1–6 | |||||
|---|---|---|---|---|---|---|
| Imp-1 | Imp-2 | Imp-3 | Imp-4 | Imp-5 | Imp-6 | |
| Analyst 1 (n = 6) | 0.22 | 0.17 | 0.27 | 0.18 | 0.16 | 0.18 |
| Analyst 2 (n = 6) | 0.33 | 0.26 | 0.34 | 0.26 | 0.24 | 0.25 |
| Analyst 1 and 2 (n =12) | 0.36 | 0.55 | 0.28 | 0.38 | 0.37 | 0.35 |
| Analyte | LOD (μg/mL) | LOQ (μg/mL) | Repeatability at LOQ (% RSD, n = 6) | Linearity range (μg/mL) | Calibration equation (y = area, x = μg/mL) | 95% confidence interval for intercept | Residual standard deviation |
|---|---|---|---|---|---|---|---|
| Imp-1 | 0.06 | 0.19 | 1.36 | 0.19–7.50 | y = 23.382x − 1.3075 | (−3.81,1.20) | 1.28 |
| Imp-2 | 0.11 | 0.26 | 1.55 | 0.26–7.50 | y = 25.474x + 0.136 | (−1.20,1.47) | 0.62 |
| Imp-3 | 0.12 | 0.29 | 1.28 | 0.29–7.50 | y = 25.664x − 2.9776 | (−3.81,2.60) | 1.61 |
| Imp-4 | 0.12 | 0.31 | 1.83 | 0.31–7.50 | y = 21.13x + 0.4211 | (−1.20,2.03) | 0.81 |
| Imp-5 | 0.08 | 0.25 | 1.15 | 0.25–7.50 | y = 20.019x − 0.5906 | (−2.67,1.49) | 1.05 |
| Imp-6 | 0.13 | 0.35 | 1.66 | 0.31–7.50 | y = 20.455x − 0.6923 | (−2.63,1.26) | 0.97 |
| a M | 0.09 | 0.21 | 1.27 | 0.21–7.50 | y = 21.049x + 0.4519 | (−1.07,1.97) | 0.79 |
| Analyte | Addd (μg/mL) | Measured mean ± SD (μg/mL) | Recovery (%) |
|---|---|---|---|
| 1 | 2.50 | 2.48 ± 0.012 | 99.2 |
| 5.00 | 4.92 ± 0.039 | 98.4 | |
| 7.50 | 7.63 ± 0.072 | 101.7 | |
| 2 | 2.50 | 2.51 ± 0.026 | 100.4 |
| 5.00 | 5.03 ± 0.037 | 100.6 | |
| 7.50 | 7.59 ± 0.108 | 101.2 | |
| 3 | 2.50 | 2.55 ± 0.028 | 102.0 |
| 5.00 | 4.95 ± 0.066 | 99.0 | |
| 7.50 | 7.39 ± 0.113 | 98.5 | |
| 4 | 2.50 | 2.52 ± 0.023 | 100.8 |
| 5.00 | 5.08 ± 0.056 | 101.6 | |
| 7.50 | 7.61 ± 0.087 | 101.5 | |
| 5 | 2.50 | 2.53 ± 0.019 | 101.2 |
| 5.00 | 4.91 ± 0.036 | 98.2 | |
| 7.50 | 7.62 ± 0.106 | 101.6 | |
| 6 | 2.50 | 2.48 ± 0.021 | 99.2 |
| 5.00 | 4.98 ± 0.046 | 99.6 | |
| 7.50 | 7.64 ± 0.107 | 101.9 | |
| Type of change | Variation | Retention time of principal (min) | Resolution | |||||
|---|---|---|---|---|---|---|---|---|
| 1/2 | 2/3 | 3/4 | 4/a M | a M/5 | 5/6 | |||
| flow rate (mL/min) | 0.9 | 37.17 | 13.67 | 7.50 | 1.64 | 3.21 | 3.21 | 4.88 |
| 1.0 | 33.51 | 13.32 | 7.45 | 1.65 | 3.15 | 3.14 | 4.82 | |
| 1.1 | 30.58 | 13.06 | 6.95 | 1.54 | 3.08 | 3.07 | 4.59 | |
| Temperature (°C) | 40 | 34.79 | 13.11 | 7.18 | 1.42 | 3.08 | 3.28 | 5.19 |
| 45 | 33.51 | 13.32 | 7.45 | 1.65 | 3.15 | 3.14 | 4.82 | |
| 50 | 32.74 | 13.61 | 7.8 | 1.79 | 3.14 | 3.11 | 4.48 | |
| Acetonitrile ratio (%) | 37 | 42.76 | 14.53 | 7.41 | 1.46 | 2.8 | 3.27 | 4.74 |
| 38 | 33.51 | 13.32 | 7.45 | 1.65 | 3.15 | 3.14 | 4.82 | |
| 39 | 28.53 | 12.96 | 7.35 | 1.28 | 2.85 | 3.01 | 4.58 | |
| pH | 2.7 | 36.05 | 13.84 | 7.56 | 1.55 | 3.19 | 3.15 | 4.61 |
| 2.9 | 33.51 | 13.32 | 7.45 | 1.65 | 3.15 | 3.14 | 4.82 | |
| 3.1 | 37.49 | 13.82 | 7.73 | 1.47 | 3.23 | 3.2 | 4.61 | |
| Stress condition/media/duration | Degradation (%) | Number of impurities | Retention time (tR) (min) | Peak purity |
|---|---|---|---|---|
| Acidic/0.1M HCl/RT/1 h | 11.1 | 5 | 9.43; 12.25; 17.32; 23.13; 42.44 | 0.99965 |
| Alkaline/0.1M NaOH/RT/1 h | 25.1 | 9 | 7.72; 8.82; 12.21; 17.12; 19.08; 23.13; 27.35; 28.11; 42.44 | 0.99972 |
| Neutral/H2O/70 °C/1 h | 5.5 | 5 | 12.18; 14.14; 17.18; 23.13; 42.44 | 0.99981 |
| Oxidative/3% H2O2/1 h | 17.6 | 4 | 6.53; 12.25; 18.32; 23.13 | 0.99989 |
| Photolytic/UV-lamp/72 h | 9.2 | 3 | 13.67; 18.29; 23.13 | 0.99977 |
| Thermal/105 °C/24 h | 5.2 | 3 | 13.67; 18.29; 23.13 | 0.99998 |
| Analyte | Impurities% | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | Any unknown Impurity (%) | Total impurities (%) | |
| 1228065 | <LOD | 0.19 | 0.068 | <LOD | 0.057 | <LOD | ND | 0.315 |
| 1228066 | <LOD | 0.19 | <LOD | <LOD | 0.068 | <LOD | ND | 0.258 |
| 1228067 | 0.11 | 0.16 | <LOD | <LOD | 0.059 | <LOD | ND | 0.219 |
Acknowledgments
Conflicts of Interest
References
- Tawara, S.; Ikeda, F.; Maki, K.; Morishita, Y.; Otomo, K.; Teratani, N.; Goto, T.; Tomishima, M.; Ohki, H.; Yamada, A.; et al. In vitro activities of a new lipopeptide antifungal agent, FK463, against a variety of clinically important fungi. Antimicrob. Agents Chemother 2000, 44, 57–62. [Google Scholar]
- Olatinwo, R.O.; Schilder, A.; Kravchenko, A.N. Incidence and causes of postharvest fruit rot in storedMichigan cranberries. Plant Dis 2004, 88, 1277–1282. [Google Scholar]
- Vehreschild, J.J.; Cornely, O.A. Micafungin sodium, the second of the echinocandin class of antifungals: Theory and practice. Future Microbiol 2006, 1, 161–170. [Google Scholar]
- Fritz, J.M.; Brielmaier, B.D.; Dubberke, E.R. Micafungin for the prophylaxis and treatment of Candida infections. Expert Rev. Anti-Infect. Ther 2008, 6, 153–162. [Google Scholar]
- Scott, L.J. Micafungin: A review of its use in the prophylaxis and treatment of invasive Candida infections. Drugs 2012, 72, 2141–2165. [Google Scholar]
- Hatano, K.; Morishita, Y.; Nakai, T.; Ikeda, F. Antifungal mechanism of FK463 against Candida albicans andAspergillus fumigatus. J. Antibiot 2002, 55, 219–222. [Google Scholar]
- Bormann, A.M.; Morrison, V.A. Review of the pharmacology and clinical studies of micafungin. Drug Des. Dev. Ther 2009, 3, 295–302. [Google Scholar]
- Carver, P.L. Micafungin. Ann. Pharmacother 2004, 38, 1707–1721. [Google Scholar]
- Zaas, A.K.; Steinbach, W.J. Micafungin: The US perspective. Expert Rev. Anti-Infect. Ther 2005, 3, 183–190. [Google Scholar]
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal 2002, 28, 1011–1040. [Google Scholar]
- Nageswara, R.R.; Nagaraju, V. An overview of the recent trends in development of HPLC methods for determination of impurities in drugs. J. Pharm. Biomed. Anal 2003, 33, 335–377. [Google Scholar]
- Noda, K.; Yokoyama, T.; Akiyama, Y.; Tadano, K. A liquid chromatographic method for quantifying unbound micafungin in human plasma and the relationship between the concentrations of unbound and total micafungin in plasma. Yakugaku Zasshi 2013, 133, 397–404. [Google Scholar]
- Nakagawa, S.; Kuwabara, N.; Kobayashi, H.; Shimoeda, S.; Ohta, S.; Yamato, S. Simple column-switching HPLC method for determining levels of the antifungal agent micafungin in human plasma and application to patient samples. Biomed. Chromatogr 2013, 27, 551–555. [Google Scholar]
- Undre, N.A.; Stevenson, P.; Freire, A.; Arrieta, A. Pharmacokinetics of micafungin in pediatric patients with invasive candidiasis and candidemia. Pediatr. Infect. Dis. J 2012, 31, 630–632. [Google Scholar]
- Martens-Lobenhoffer, J.; Rupprecht, V.; Bode-Boger, S.M. Determination of micafungin and anidulafungin in human plasma: UV- or mass spectrometric quantification? J. Chromatogr. B 2011, 879, 2051–2056. [Google Scholar]
- Uranishi, H.; Nakamura, M.; Nakamura, H.; Ikeda, Y.; Otsuka, M.; Kato, Z.; Tsuchiya, T. Direct-injection HPLC method of measuring micafungin in human plasma using a novel hydrophobic/hydrophilic hybrid ODS column. J. Chromatogr. B 2011, 879, 1029–1032. [Google Scholar]
- Tabata, K.; Katashima, M.; Kawamura, A.; Tanigawara, Y.; Sunagawa, K. Linear pharmacokinetics of micafungin and its active metabolites in Japanese pediatric patients with fungal infections. Biol. Pharm. Bull 2006, 29, 1706–1711. [Google Scholar]
- Yanni, S.B.; Augustijns, P.F.; Benjamin, D.J.; Brouwer, K.L.; Thakker, D.R.; Annaert, P.P. In vitro investigation of the hepatobiliary disposition mechanisms of the antifungal agent micafungin in humans and rats. Drug Metab. Dispos 2010, 38, 1848–1856. [Google Scholar]
- Q1A(R2) Stability Testing of New Drug Substances and Products. Available online: http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm128204.pdf (access on 17 October 2013).
- Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (access on 17 October 2013).
- Photostability Testing on New Drug Substances and Products Q1B. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1B/Step4/Q1B_Guideline.pdf (access on 17 October 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, S.; Meng, X.; Su, X.; Luo, Y.; Sun, Z. Development and Validation of a Stability-Indicating High Performance Liquid Chromatographic (HPLC) Method for the Determination of Related Substances of Micafungin Sodium in Drug Substances. Int. J. Mol. Sci. 2013, 14, 21202-21214. https://doi.org/10.3390/ijms141121202
Zhu S, Meng X, Su X, Luo Y, Sun Z. Development and Validation of a Stability-Indicating High Performance Liquid Chromatographic (HPLC) Method for the Determination of Related Substances of Micafungin Sodium in Drug Substances. International Journal of Molecular Sciences. 2013; 14(11):21202-21214. https://doi.org/10.3390/ijms141121202
Chicago/Turabian StyleZhu, Shengsheng, Xiang Meng, Xin Su, Yongwei Luo, and Zuyue Sun. 2013. "Development and Validation of a Stability-Indicating High Performance Liquid Chromatographic (HPLC) Method for the Determination of Related Substances of Micafungin Sodium in Drug Substances" International Journal of Molecular Sciences 14, no. 11: 21202-21214. https://doi.org/10.3390/ijms141121202