The NO/ONOO-Cycle as the Central Cause of Heart Failure

Abstract
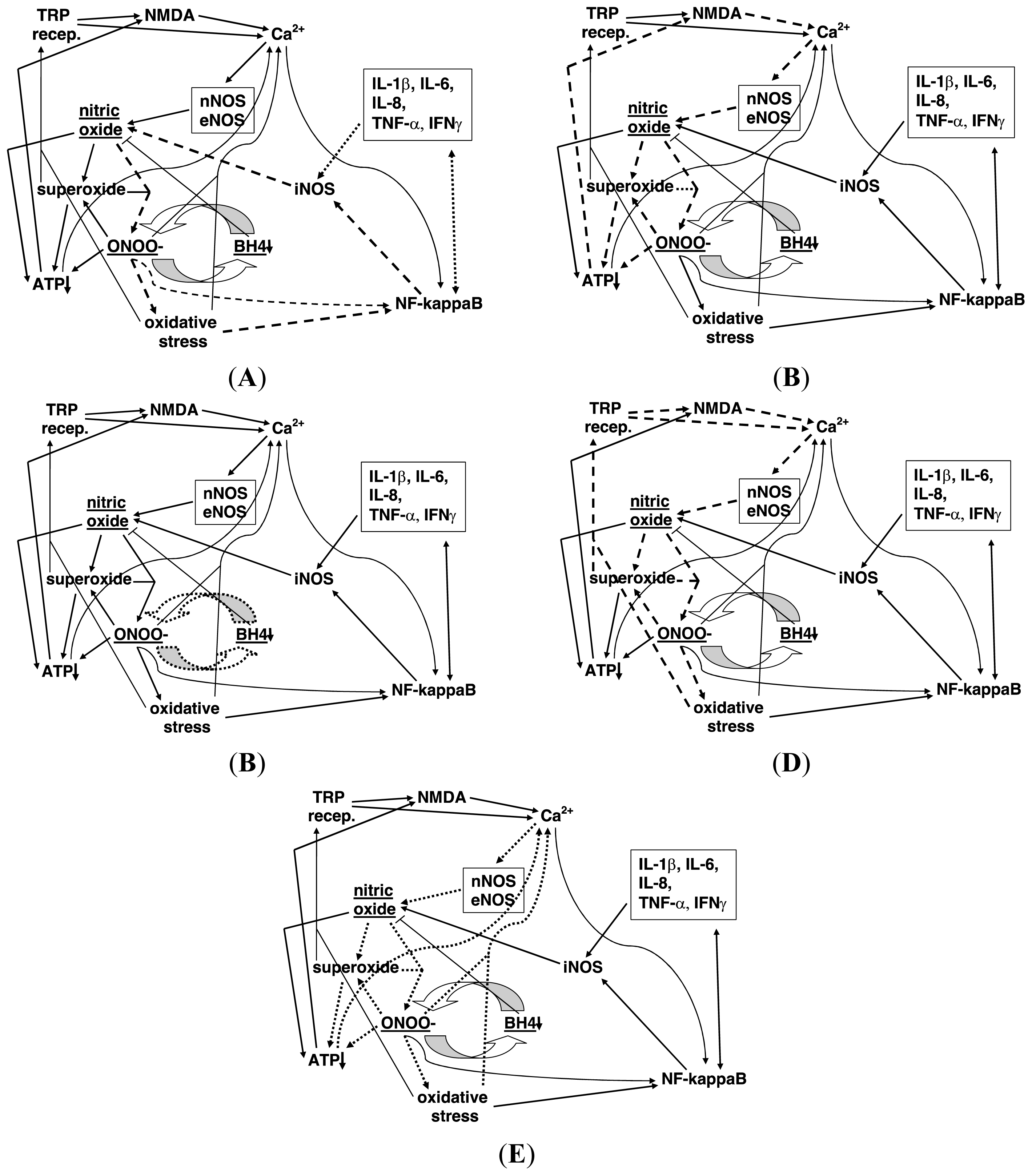
:1. Introduction
2. Proposed Properties of the NO/ONOO-Cycle
3. Thirty Four Specific NO/ONOO-Cycle Mechanisms
- Extremely rapid, diffusion limited reaction between nitric oxide (NO•) with superoxide (OO•−), forming peroxynitrite (ONOO-).
- ONOO-, a potent oxidant, can act to increase the activity of the transcription factor NF-κB.
- ONOO- breaks down both before and after reaction with carbon dioxide into the following free radicals, hydroxyl (HO•), carbonate (CO3•) and NO2 radical (NO2•), each of which are responsible for a number of consequences produced by ONOO-.
- ONOO- being a potent oxidant produces oxidative stress, an imbalance between oxidants and antioxidants.
- Oxidative stress also produces increases in NF-κB activity because its activity is stimulated by oxidants and inhibited by chain-breaking antioxidants.
- NF-κB produces increased transcription of the inducible nitric oxide synthase (iNOS), a gene whose transcription is known to be stimulated by NF-κB elevation and whose elevation also stimulates much of the inflammatory cascade.
- NF-κB also stimulates the transcription of several inflammatory cytokines, including IL-1β, IL-6, IL-8, TNF-α, and IFNγ.
- Each of the cytokines listed in 7 above, act directly and/or indirectly to stimulate the transcription of the iNOS gene, acting in some cytokines via the double headed arrow linking them to NF-κB and also, acting in some cytokines directly on iNOS induction.
- When iNOS is induced, it produces large amounts of NO.
- ONOO- inactivates the plasma membrane calcium-ATPase, leading to lowered calcium extrusion and increased levels of intracellular calcium.
- Other oxidants inactivate the plasma membrane calcium-ATPase, leading to increased levels of intracellular calcium; such inactivation of the calcium ATPase has substantial pathophysiological effects and may well contribute to the prolonged impairment of calcium extrusion seen under circumstances where the NO/ONOO-cycle may have a role.
- Lowered energy metabolism (decreased energy charge/ATP) also lowers calcium-ATPase activity, leading to increased levels of intracellular calcium, as predicted for such an ATPase.
- While modest elevation of mitochondrial calcium, leads to increased ATP synthesis, substantial elevation of intracellular calcium leads to substantial increases in intramitochondrial calcium, leading to increased superoxide generation in the mitochondrion; large increases in mitochondrial calcium will lead, in some circumstances, to apoptotic cell death.
- Intracellular calcium stimulates the nNOS and eNOS forms of nitric oxide synthase, both of which are calcium dependent enzymes.
- Increased nNOS and eNOS activity both produce increased NO synthesis.
- ONOO- oxidizes tetrahydrobiopterin (BH4), depleting BH4 levels.
- BH4 depletion produces partial uncoupling of the three NO synthases, such that these enzymes, when uncoupled, produce superoxide in place of NO. Because of the very rapid reaction of these two compounds to produce ONOO-, this partial uncoupling involving nearby NOS enzymes is expected to produce an increase in ONOO- production.
- Nicking of nuclear DNA by ONOO- and hydroxyl and other radicals can produce a massive stimulation of poly (ADP-ribose) polymerase (PARP) and consequent poly ADP ribosylation of chromosomal proteins, leading, in turn to a massive depletion of NAD/NADH pools, because NAD is the substrate for such poly ADP-ribosylation. NADH depletion lowers, in turn, ATP production in the mitochondrion.
- Other changes causing ATP depletion come from a cascade of events occurring within the mitochondrion. The cascade starts with NO, possibly produced by mitochondrial NO synthase (mtNOS which is thought to be largely a form of nNOS), with NO binding to cytochrome oxidase, competitively inhibiting the ability of molecular oxygen to bind. This inhibits the ability of cytochrome oxidase to serve as the terminal oxidase of the mitochondrial electron transport chain.
- The action of NO in 19 above, produces increased superoxide production by the electron transport chain.
- ONOO- in the mitochondrion also acts to produce increased superoxide from the electron transport chain.
- Peroxynitrite (ONOO-), superoxide and their products lead to lipid peroxidation of the cardiolipin in the inner membrane of the mitochondrion. Cardiolipin is highly susceptible to such peroxidation, because most of the fatty acids that make up its structure in mammals are polyunsaturated fatty acids, which are much more susceptible to peroxidation than are other fatty acids.
- Cardiolipin peroxidation leads to lowered activity of some of the enzymes in the electron transport chain, leading to further lowering of ATP synthesis.
- Cardiolipin peroxidation also leads to increased superoxide generation from the electron transport chain in the mitochondrion.
- ONOO- produces inactivation of the mitochondrial superoxide dismutase (Mn-SOD) as well as the copper-zinc superoxide dismutase, leading in turn to increased superoxide levels.
- ONOO-, superoxide and NO all inactivate or inhibit the aconitase enzyme, lowering citric acid cycle activity and subsequent ATP synthesis.
- Oxidative stress leads to oxidation of cysteine residues in the enzyme xanthine reductase, converting it into xanthine oxidase which produces superoxide as a product, thus increasing superoxide generation.
- Increased activity of the enzyme NADPH oxidase, which produces superoxide as a product, is an important part of the inflammatory cascade, and contributes, therefore, to the cascade by producing increased superoxide. (Note: Increased NADPH oxidase is produced through the action of angiotensin II in cardiovascular diseases, including HF).
- Activation of the NMDA receptors, produced as described in 31 and 32, below, allows calcium influx into the cell, raising intracellular calcium levels including mitochondrial calcium levels.
- Activity of transfer receptor potential (TRP) receptors also allows calcium influx into the cell, again raising intracellular calcium levels, presumably leading to increased nitric oxide production.
- The main physiological agonist of the NMDA receptors is glutamate whose extracellular concentration is lowered after release, by energy dependent transport. It follows that ATP depletion produces increased NMDA stimulation by lowering glutamate transport.
- The activity of the NMDA receptors is also greatly increased by ATP depletion within the cells containing these receptors. The mechanism here is that the ATP depletion produces partial depolarization of the plasma membrane, which produces, in turn, increased susceptibility of the NMDA receptors to stimulation.
- Several of the TRP group of receptors have been shown to be stimulated by increased superoxide and/or oxidative stress or their downstream consequences, these being the TRPV1, TRPA1, TRPC3, TRPC5, TRPM2 and TRPM7 receptors, being produced in part through the oxidation of cysteine residue side chains. Several TRP receptors are also activated by nitric oxide mediated nitrosylation.
- TRPV1, TRPA1 and probably several other TRP group receptors, receptor stimulation has each been repeatedly shown to lead to increased NMDA activity, with neurons containing these TRP family of receptors acting in part by releasing glutamate, the major physiological NMDA agonist.
4. Five Principles that Can Be Used to Test for NO/ONOO-Cycle Diseases
- Stressors that initiate the disease are able to act by raising cycle elements.
- The various elements of the cycle, with the possible exception of NO [9], should be elevated in the chronic phase of the disease.
- The correlates (symptoms and signs) of the disease should be produced by one or more elements of the cycle.
- The basic mechanism of the cycle is local and such that it is localized to different tissues in different individuals. The reason for this primarily local nature is that the three inorganic compounds involved, NO, superoxide and ONOO-, have limited half-lives in biological tissues. And the mechanisms of the cycle, those various arrows, act at the level of individual cells. This allows for great variations in tissue distribution from one patient to another, producing a huge spectrum of illness. The point here is not that there are no systemic changes—clearly antioxidant depletion, neuroendocrine and immune system changes, the actions of some inflammatory cytokines and BH4 depletion will be to some extent systemic. But rather this primarily local nature gives much inherent variation due to the varying tissue localization of the basic mechanism (see Chapter 4 in ref. [3]). A correlate of the primarily local nature of the cycle is that different NO/ONOO-cycle diseases will differ from one another in what tissue or tissues must be impacted by the cycle in order to be diagnosed as a specific cycle-caused disease.
- The cycle is the central cause of the disease, so that treatment of the disease should involve using agents that lower various parts of the cycle. In other words, we should treat the cause of the disease, not the symptoms. Other types of evidence showing causal roles for elements of the cycle, such as genetic evidence, also support this principle.
5. Role of Elevated NO/ONOO-Cycle Elements in HF
5.1. Peroxynitrite (ONOO-)
5.2. Oxidative Stress and Superoxide
5.3. NF-κB
5.4. Inflammatory Cytokines and Other Inflammatory Proteins
5.5. iNOS
5.6. NO
5.7. Mitochondrial Dysfunction (ATP Depletion)
5.8. BH4 Depletion
5.9. Cytosolic Calcium
5.10. NMDA
5.11. TRP Receptors
6. Initiation: via NO/ONOO-Cycle Elements?
7. Evidence Relating to Occurrence of the 34 Specific Mechanisms in HF
- #3 predicts that peroxynitrite and its CO2 adduct, break down to produce hydroxyl, carbonate and NO2 radicals. This is suggested in HF by the tyrosine nitration found in HF [11,13,14,17,19,212,226] which is thought to be produced not by peroxynitrite itself, but rather by NO2 and other radical products of peroxynitrite.
- #8 predicts that some cytokines raise NF-κB activity. The cytokine most extensively shown to do so in a general context is TNF-α and TNF-α stimulation of NF-κB has been shown to occur in HF [244,245], confirming part of this prediction. #8 also predicts that cytokines induce iNOS, shown in HF by [19,72].
- #10 predicts that the plasma membrane calcium-ATPase which pumps Ca2+ ions from the cytosol to the surrounding extracellular fluid is inactivated by tyrosine nitration. This has not been looked at in HF; however, the similar SERCA2A enzyme in the sarcoplasmic reticulum, which is similarly susceptible to inactivation by tyrosine nitration, has been shown to be nitrated and inactivated in HF [13,14]. In the myocardium, the SERCA2a inactivation is thought to be the more important of the two. The action of the NMDA receptors in producing apoptosis in HF [192,193] also strongly suggests a Ca2+ apoptotic role.
- #13 predicts that elevated Ca2+ levels in the mitochondrion can cause apoptosis. This is shown through the role of the calcium-dependent protease, calpain in causing apoptosis in HF [173].
- #16 predicts BH4 oxidation by peroxynitrite and #17 predicts NOS uncoupling, as a consequence of BH4 oxidation and depletion. Citations [142–149] have each shown BH4 depletion and consequent NOS uncoupling in HF, and a large number of other studies have also shown NOS uncoupling in HF. Studies [142,145,149] have each documented BH4 oxidation, with [149] showing that the oxidation is produced by peroxynitrite. It follows that both mechanisms #16 and 17 are well-documented in HF.
- #18, the activation of poly(ADP-ribose) polymerase (PARP) by peroxynitrite leading to extensive poly ADP-ribosylation of chromosomal proteins and NAD/NADH depletion has been shown on several studies of HF [246–249]. These studies also show a causal role of PARP in HF, based on studies of specific PARP inhibitors and also of a PARP gene knockout in the mouse [246–249].
- #19 predicts that NO binding to cytochrome oxidase in the mitochondrion will produce increased reduction of electron transport intermediates. Such increased reduction has been reported in [37] for coenzyme Q in HF.
- #13, 20–22 and 24 all predict elevated superoxide production from the mitochondrial electron transport chain and elevated mitochondrial superoxide levels. Evidence for such elevated superoxide generation from the mitochondrial electron transport chain in HF has been reviewed by Tsutsui et al. [54] and reported elsewhere [37].
8. High Level Endothelin-1 and RhoA as Causal Factors of HF: Both Act as NO/ONOO-Cycle Elements
9. Discussion and Conclusions


{kind=link}
| Receptor | Finding | Citation |
|---|---|---|
| Calpain | Right ventricular overload in the pig, produces both a lowering and aggregation of talin and right ventricular HF. The calpain inhibitor MDL-28170 normalized each of these and may also normalize aggregation of α-actinin and vinculin. | [167] |
| Calpain | Review: the role of calpains in myocardial remodeling and HF. Calpains may contribute to myocardial hypertrophy and inflammation, through activation of NF-κB. “They play an important role in the fibrosis process, partly by activating transforming growth factor β. They are also implicated in cell death as they cause the breakdown of sarcolemma and sarcomeres.” In addition, “calpains are indeed actively involved in common causes of HF, including hypertension, diabetes, atherosclerosis, ischemia-reperfusion injury, atrial fibrillation, congestive failure and mechanical unloading.” | [168] |
| Calpain | Study infers that “calpain mediates dystrophin loss and myofibril degradation in doxorubicin-treated rats.” | [169] |
| Calpain | Title: Calpain inhibition attenuates right ventricular contractile dysfunction after acute pressure overload. | [170] |
| Calpain | Calpain inhibitors lower the development of cardiac ventricular hypertrophy, an independent risk factor for HF. | [171] |
| Calpain | Overexpression of calpastatin, a naturally occurring inhibitor of calpain, attenuates myocardial dysfunction in response to endotoxin exposure. | [172] |
| Calpain | In cardiomyocytes, calpain 1 activates caspase 3 and poly-(ADP-ribose) polymerase (PARP), as well as apoptosis-inducing factor. | [173] |
| Calpain | Atrial fibrillation is a specific consequence of calpain activity in cardiac muscle | [174] |
| Calpain | “These results indicate that biochemical markers of cardiomyocyte cell death, sarcomeric disarray, gelsolin cleavage, and TUNEL-positive nuclei, are mediated, in part by calpain and that calpeptin may serve as a potential therapeutic agent…”. | [175] |
| Calpain | Calpain I produces Ca2+-dependent partial proteolysis of calcineurin, forming Ca2+/calmodulin-independent calcineurin. | [176] |
| CaMKII | One target of action of CaMKII in some types of HF, is phosphorylation and consequent loss of activity of Na(V)1.5 sodium channels in cardiac myocytes. | [177] |
| CaMKII | CaMKII phosphorylates the titin springs. Such “deranged” CaMKII-dependent phosphorylation occurs in HF and “contributes to altered diastolic stress.” | [178] |
| CaMKII | A mathematical modeling study suggests that lowering CaMKII phosphorylation along with lowering Ca2+ leak may be useful in HF therapy. | [179] |
| CaMKII | Review: CaMKII seems to be involved in both HF and arrythmias and may, therefore be a promising target for therapy. | [133] |
| CaMKII | CaMKII-dependent phosphorylation increases inner mitochondrial Ca2+ uniporter activity, producing lowered ΔPsim possibly opening the mitochondrial transition pore. CaMKII action may, therefore, have an important role in HF, including lowering mitochondrial function and increasing apoptotic cell death. | [180] |
| Calcineurin | A study of cardiac hypertrophy in isolated, adult animal hearts. The authors conclude “Although a direct cause-and-effect relationship between NFAT-luciferase activity and pathological hypertrophy was not proven here, our results support the hypothesis that separable signaling pathways regulate pathological versus physiological hypertrophic growth of the myocardium, with calcineurin-NFAT potentially serving a regulatory role that is more specialized for maladaptive hypertrophy and heart failure.” | [181] |
| Calcineurin | In a study of adaptive response to mouse aortic constriction, “Major calcineurin activation, associated with GSK3b inactivation, appeared to engage maladaptive hypertrophy and progression to HF.” | [182] |
| Calcineurin | Study of cardiac fibroblast proliferation and fibrosis, in response to electrical field exposure. Showed that field exposure acts to raise cytosolic Ca2+ via L-type calcium channel activation, leading to calcineurin and NFAT activation, producing fibroblast proliferation and fibrosis. | [183] |
| Calcineurin | Transgenic mouse carrying a tetracycline-inducible calcineurin gene; gene activation produced robust cardiac growth resembling pathological hypertrophy, followed by systolic dysfunction, fetal gene activation, fibrosis and HF. Each of these was reversed when the gene was inactivated, except fibrosis, which was partially reversed. | [184] |
| Calcineurin | Angiotensin II and norepinephrine, both of which can produce HF, were shown to activate the calcineurin, NFAT pathway in cardiomyocytes. | [185] |
| Initiating stressor | Raised NO/ONOO-cycle elements | Citation |
|---|---|---|
| Hypertension/pressure overload | Mitochondrial and general oxidative stress, peroxynitrite, superoxide, NF-κB, BH4 depletion | [44,61,62,142–144] |
| Mouse mitochondrial superoxide dismutase knockout | Superoxide, oxidative stress | [40,41] |
| Doxorubicin | Peroxynitrite, superoxide, oxidative stress, Ca2+ (particularly in the mitochondrion), NF-κB, iNOS, cytokines TNF-α | [11,38,39,66,209] |
| Homocysteine elevation | NMDA activity, NO, peroxynitrite, Ca2+, probable BH4 depletion | [188–190] |
| Transplantation—severe ischemia-reperfusion | Superoxide elevation, oxidative stress, mitochondrial dysfunction, peroxynitrite | [16] |
| Endothelin-1 (ET-1) | Superoxide, iNOS, oxidative stress, Ca2+ | [42,78,210] |
| Ovariectomy | BH4 depletion and oxidation; superoxide | [145] |
| Cardiomyocyte-specific NF-κB elevation (transgenic) | NF-κB, cytokine elevation | [63] |
| Transgenic calcineurin elevation | Ca2+, mitochondrial dysfunction, superoxide | [211] |
| Post-viral, autoimmune? | iNOS induction, peroxynitrite, inflammatory cytokines, NO | [212,213] |
| Duchenne muscular dystrophy | Ca2+, NO, iNOS induction, mitochondrial dysfunction, oxidative stress and elevated levels of several TRPC channels, superoxide | [214–216] |
| Endotoxin exposure; sepsis | iNOS, NF-κB, cytokines, superoxide, oxidative stress, Ca2+, mitochondrial dysfunction, NO, peroxynitrite | [28,79–82,217–222] |
| Cardiac-specific transgenic iNOS overexpression | iNOS, NO, oxidative stress, mitochondrial dysfunction | [48,83,84] |
| Tachypacing | iNOS, BH4 depletion, superoxide, peroxynitrite, Ca2+, oxidative stress, mitochondrial dysfunction | [146,223–225] |
| Myocardial infarction | Ca2+, oxidative stress, mitochondrial dysfunction, iNOS, peroxynitrite, NF-κB | [70,209,226] |
| Hypothyroid [227,228] | Oxidative stress, mitochondrial dysfunction, cytokines | [229,230] |
| Hyperthyroid [227,228] | Oxidative stress, mitochondrial dysfunction | [43,231–234] |
| Chagas disease | Ca2+, mitochondrial dysfunction, NO, cytokines, iNOS, oxidative stress, superoxide | [235–240] |
| Cytokines (IL-1β, IFNγ & TNF-α) | Cytokines, iNOS, NO, superoxide, peroxynitrite | [19] |
| Citation | Cycle element(s) | HF correlate changes produced by cycle element |
|---|---|---|
| [11] | Peroxynitrite and iNOS (both) | MMP activation, lipid peroxidation |
| [13,14] | Peroxynitrite, oxidative stress | Tyrosine nitration, oxidation, sulfonylation and consequent inactivation of SERCA2a; lowered rate of relaxation |
| [15] | peroxynitrite | Creatine kinase tyrosine nitration and inactivation; lowered energy storage and utilization in the myocardium |
| [18] | peroxynitrite | Cardiomyocyte action potential changes; slowed Ca2+ cycling |
| [20] | peroxynitrite | Decreased response to isoproterenol; lessened ability of isoproterenol to increase Ca2+ transients or shortening; increased Tyr284 nitration on protein phosphatase 2a; produces effect by decreasing Ser16 phosphorylation on phospholamban |
| [28] | peroxynitrite | Produces overall increase in protein-bound 3-NT, oxidative stress, NF-κB elevation, TNF-α elevation |
| [41] | Mitochondrial superoxide | Mitochondrial energy metabolism dysfunction |
| [44] | Hydrogen peroxide derived from mitochondrial superoxide | Changes in the mitochondrial proteome associated with HF |
| [45] | Oxidative stress, probably peroxynitrite | Ventricular remodeling; cavity dilatation and dysfunction |
| [49] | Mitochondrial oxidative stress | Oxidative changes in enzymes involved in mitochondrial ATP synthesis; energy metabolism dysfunction |
| [50,54] | Mitochondrial oxidative stress | Myocyte hypertrophy, apoptosis, interstitial fibrosis and MMP activation, producing maladaptive cardiac remodeling and failure; oxidative mtDNA damage and lowered mtDNA copy number |
| [51–53] | Mitochondrial oxidative stress | Cardiolipin peroxidation |
| [60] | Oxidative stress | Lowered myocardial Akt signaling, increased connective tissue growth factor |
| [106,107] | Oxidative stress | Oxidation of heme iron in soluble guanylate cyclase, lowered cGMP synthesis |
| [161,162] | Oxidative stress | Protein oxidation of RyR2, causes Ca2+ leakiness |
| [61] | NF-κB | Fibrosis, cardiomyocyte hypertrophy; MMP-2 activation; decreased fractional shortening |
| [62] | NF-κB | Fibrosis and associated increased collagen and fibronectin synthesis; increased connective tissue growth factor |
| [63] | NF-κB | Myocarditis, inflammatory dilated cardiomyopathy, muscle fiber atrophy; dilated ventricles and atria, strong systolic dysfunction and some diastolic dysfunction |
| [64,65] | NF-κB | Cardiac hypertrophy |
| [67–69] | NF-κB | Il-1β, TNF-α, IL-6 elevation |
| [70] | NF-κB | Systolic dysfunction, lowered chamber remodeling, cytokine expression, fibrosis and apoptosis |
| [72] | Cytokines, NO | Lowered contractility |
| [73,74] | Cytokine (TNF-α) | Cardiomyopathy [75] Cytokine signaling Cardiomyocyte mortality, contractile dysfunction, ventricular arrhythmia |
| [19] | Il-1β, TNF-α, IL-6 | iNOS, NO, superoxide, peroxynitrite, lowered cardiac function |
| [69] | IL-6 | Fetal gene expression, cardiomyocyte growth |
| [80] | iNOS | Cardiac contractile dysfunction |
| [81] | iNOS, NO | TNF-α elevation, oxidative stress, energy metabolism dysfunction |
| [48,83] | iNOS, NO | Cardiac hypertrophy, ventricular dilatation, interstitial fibrosis, reactivation of the fetal gene expression; reduced contractility, ejection fraction, and cardiac energetics; up-regulation of peroxiredoxins (a possible protective response) |
| [84] | iNOS | Mild inflammatory cell infiltrate, cardiac fibrosis, hypertrophy, dilatation; bradyarrhythmia |
| [85] | iNOS, NO | Lowered isoproterenol responsiveness [87] iNOS Cardiac contractile dysfunction |
| [126] | Mitochondrial dysfunction (caused by mtDNA mutation) | Dilated cardiomyopathy |
| [130] | Mitochondrial dysfunction | Cardiac hypertrophy, remodeling |
| [133] | Mitochondrial Ca2+ via CaMKII | Mitochondrial transition pore opening; myocyte apoptosis |
| [134] | Mitochondrial dysfunction | Lowered cardiomyocyte shortening; aberrant Ca2+ cycling |
| [135] | Mitochondrial dysfunction | Systolic dysfunction; hypertrophy |
| [140] | Mitochondrial dysfunction | Lowered ejection fraction |
| [142] | BH4 depletion | NOS partial uncoupling, dephosphorylated phospholamban, diastolic dysfunction, impaired relaxation |
| [143] | BH4 depletion (acting via eNOS partial uncoupling) | Fibrosis, myocyte hypertrophy, fetal gene expression, oxidative stress, peroxynitrite, MMP-2/9 activation |
| [144] | BH4 depletion | Hypertrophy, fibrosis, NO synthase uncoupling, oxidative stress |
| [146,149] | BH4 depletion, iNOS | Both have roles in producing atrial fibrillation and probably cardiomyopathy; NO synthase uncoupling |
| [167–176] (see Table 1) | Ca2+ stimulating calpain(s) | Calpain(s) partial proteolysis is thought to: degrade dystrophin, myofibrils, gelsolin, sarcolemma proteins; activate TGF-β, caspase-3, apoptosis inducing factor, NF-κB; aggregate talin, α-actinin and vinculin. These, in turn are thought to contribute to: fibrosis and remodeling, apoptosis, necrosis, hypertrophy, right ventricular dysfunction, atrial fibrillation |
| [133, 177–180] (see Table 1) | Ca2+ stimulating CaMKII | Phosphorylation of titin springs (contributes to diastolic stress, of Na(V)1.5 (changes action potential, stimulates arrythmia), of mitochondrial Ca2+ uniporter (lowers ΔPsi, may stimulate opening of mitochondrial transition pore and apoptosis) |
| [181–185] (see Table 1) | Ca2+ stimulating calcineurin | NFAT pathway, leading to maladaptive hypertrophy; systolic dysfunction, fetal gene expression, fibroblast growth and fibrosis |
| [186] | NMDA | Negative inotropic effects |
| [187] | NMDA | Sudden cardiomyopathic death |
| [188–193] | NMDA | MMP-9 elevation; decreased cell shortening, maximal contraction and relaxation rate, decay of Ca2+ transient; raised levels of NO, cytosolic Ca2+, calpain activity; cardiac arrhythmia and sudden cardiac death; oxidative stress, mitochondrial dysfunction, NO, cytokines, apoptosis |
| [198] | TRPC3/TRPC6 | Ventricular tachyarrhythmia |
| [199] | TRPC6 | Cardiac hypertrophy, calcineurin/NFAT signaling, beta-myosin overexpression, pathologic remodeling |
| [204] | TRPC3/TRPC6/TRPC4 | Pathologic cardiac hypertrophy, calcineurin/NFAT signaling |
| [205] | TRPC1 | Maladaptive cardiac hypertrophy |
| [206] | TRPM4 | Arrhythmia |
Conflicts of Interest
References
- Pall, M.L. Pulmonary hypertension is a probable NO/ONOO-cycle disease: A review. ISRN Hypertens 2013, 2013, 742418:1–742418:27. [Google Scholar]
- Pall, M.L. Multiple Chemical Sensitivity: Toxicological Questions and Mechanisms. In General and Applied Toxicology, 3rd ed; Ballantyne, B., Marrs, T.C., Syversen, T., Eds.; John Wiley & Sons: London, UK, 2009; pp. 2303–2352. [Google Scholar]
- Pall, M.L. Explaining “Unexplained Illnesses”: Disease Paradigm for Chronic Fatigue Syndrome, Multiple Chemical Sensitivity, Fibromyalgia, Post-Traumatic Stress Disorder, Gulf War Syndrome and Others; Harrington Park Press: New York, NY, USA, 2007. [Google Scholar]
- Pall, M.L.; Bedient, S.A. The NO/ONOO-cycle as the etiological mechanism of tinnitus. Int. Tinnitus J 2007, 13, 99–104. [Google Scholar]
- Pall, M.L. Teufelskreis NO/ONOO−-Zyklus, oxidaver Stress, mitochondriale, inflammatorische und neurologische Dysfunktion. Umw. Med. Ges 2010, 23, 281–293. [Google Scholar]
- Pall, M.L. How can we cure NO/ONOO-cycle diseases? Approaches to curing chronic fatigue syndrome/myalgic encephalomyelitis, fibromyalgia, multiple chemical sensitivity, Gulf War syndrome and possibly many others. Townsend Lett 2010, 75–84. [Google Scholar]
- Pall, M.L. Elevated, sustained peroxynitrite levels as the cause of chronic fatigue syndrome. Med. Hypotheses 2000, 54, 115–125. [Google Scholar]
- Pall, M.L. NMDA sensitization and stimulation by peroxynitrite, nitric oxide and organic solvents as the mechanism of chemical sensitivity in multiple chemical sensitivity. FASEB J 2002, 16, 1407–1417. [Google Scholar]
- Pall, M.L. Nitric oxide synthase partial uncoupling as a key switching mechanism for the NO/ONOO-cycle. Med. Hypotheses 2007, 69, 821–825. [Google Scholar]
- Nazýrođlu, M. Molecular role of catalase on oxidative stress-induced Ca2+ signaling and TRP cation channel activation in nervous system. J. Recept. Signal. Transduct. Res 2012, 32, 134–141. [Google Scholar]
- Pacher, P.; Liaudet, L.; Bai, P.; Mabley, J.G.; Kaminski, P.M.; Virág, L.; Deb, A.; Szabó, E.; Ungvári, Z.; Wolin, M.S.; et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation 2003, 107, 896–904. [Google Scholar]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; van der Vliet, A.; Maeda, H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J. Biol. Chem 2001, 276, 29596–29602. [Google Scholar]
- Lokuta, A.J.; Maertz, N.A.; Meethal, S.V.; Potter, K.T.; Kamp, T.J.; Valdivia, H.H.; Haworth, R.A. Increased nitration of sarcoplamic reticulum Ca2+-ATPase in human heart failure. Circulation 2005, 111, 988–995. [Google Scholar]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res 2010, 107, 228–232. [Google Scholar]
- Mihm, M.J.; Coyle, C.M.; Schanbacher, B.L.; Weinstein, D.M.; Bauer, J.A. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc. Res 2001, 49, 798–807. [Google Scholar]
- Lauzier, B.; Sicard, P.; Bouchot, O.; Delemasure, S.; Moreau, D.; Vergely, C.; Rochette, L. A peroxynitrite decomposition catalyst: FeTPPS confers cardioprotection during reperfusion after cardioplegic arrest in a working isolated rat heart model. Fundam. Clin. Pharmacol 2007, 21, 173–180. [Google Scholar]
- Eleuteri, E.; Magno, F.; Gnemmi, I.; Carbone, M.; Colombo, M.; la Rocca, G.; Anzalone, R.; Tarro Genta, F.; Zummo, G.; di Stephano, A.; et al. Role of oxidative and nitrosative stress biomarkers in chronic heart failure. Front. Biosci 2009, 14, 2230–2237. [Google Scholar]
- Bonilla, I.M.; Sridhar, A.; Nishijima, Y.; Györke, S.; Cardounel, A.J.; Carnes, C.A. Differential effects of the peroxynitrite donor, SIN-1, on atrial and ventricular myocyte electrophysiology. J. Cardiovasc. Pharmacol 2013, 61, 401–407. [Google Scholar]
- Ferdinandy, P.; Danial, H.; Ambrus, I.; Rothery, R.A.; Schulz, R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ. Res 2000, 87, 241–247. [Google Scholar]
- Kohr, M.J.; Wang, H.; Wheeler, D.G.; Velayutham, M.; Zweier, J.L.; Ziolo, M.T. Targeting of phospholamban by peroxynitrite decreases beta-adrenergic stimulation in cardiomyocytes. Cardiovasc. Res 2008, 77, 353–361. [Google Scholar]
- Ohama, T.; Brautigan, D.L. Endotoxin conditioning induces VCP/p97-mediated and inducible nitric-oxide synthase-dependent Tyr284 nitration in protein phosphatase 2A. J. Biol. Chem 2010, 285, 8711–8718. [Google Scholar]
- Katori, T.; Donzelli, S.; Tocchetti, C.G.; Miranda, K.M.; Cormaci, G.; Thomas, D.D.; Ketner, E.A.; Lee, M.J.; Mancardi, D.; Wink, D.A.; et al. Peroxynitrite and myocardial contractility: In vivo versus in vitro effects. Free Radic. Biol. Med 2006, 41, 1606–1618. [Google Scholar]
- Kohr, M.J.; Traynham, C.J.; Roof, S.R.; Davis, J.P.; Ziolo, M.T. cAMP-independent activation of protein kinase A by the peroxynitrite generator SIN-1 elicits positive inotropic effects in cardiomyocytes. J. Mol. Cell. Cardiol 2010, 48, 645–648. [Google Scholar]
- Paolocci, N.; Ekelund, U.E.; Isoda, T.; Ozaki, M.; Vandegaer, K.; Georgakopoulos, D.; Harrison, R.W.; Kass, D.A.; Hare, J.M. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: Potential role for nitrosylation. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H1982–H1988. [Google Scholar]
- Madej, E.; Folkes, L.K.; Wardman, P.; Czapski, G.; Goldstein, S. Thiyl radicals react with nitric oxide to form S-nitrosothiols with rate constants near the diffusion-controlled limit. Free Radic. Biol. Med 2008, 44, 2013–2018. [Google Scholar]
- Lancel, S.; Zhang, J.; Evangelista, A.; Trucillo, M.P.; Tong, X.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ. Res 2009, 104, 720–723. [Google Scholar]
- Daiber, A.; Oelze, M.; Coldewey, M.; Kaiser, K.; Huth, C.; Schildknecht, S.; Bachschmid, M.; Nazirisadeh, Y.; Ullrich, V.; Mülsch, A.; et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem. Biophys. Res. Commun 2005, 338, 1865–1874. [Google Scholar]
- Lancel, S.; Tissier, S.; Mordon, S.; Marechal, X.; Depontieu, F.; Scherpereel, A.; Chopin, C.; Neviere, R. Peroxynitrite decomposition catalysts prevent myocardial dysfunction and inflammation in endotoxemic rats. J. Am. Coll. Cardiol 2004, 43, 2348–2358. [Google Scholar]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J 1991, 65, 245–248. [Google Scholar]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ. Res 2000, 86, 152–157. [Google Scholar]
- Sobotka, P.A.; Brottman, M.D.; Weitz, Z.; Birnbaum, A.J.; Skosey, J.L.; Zarling, E.J. Elevated breath pentane in heart failure reduced by free radical scavenger. Free Radic. Biol. Med 1993, 14, 643–647. [Google Scholar]
- Wolfram, R.; Oguogho, A.; Palumbo, B.; Sinzinger, H. Enhanced oxidative stress in coronary heart disease and chronic heart failure as indicated by an increased 8-epi-PGF(2alpha). Eur. J. Heart Fail 2005, 7, 167–172. [Google Scholar]
- Freeman, L.M.; Rush, J.E.; Milbury, P.E.; Blumberg, J.B. Antioxidant status and biomarkers of oxidative stress in dogs with congestive heart failure. J. Vet. Intern. Med 2005, 19, 537–541. [Google Scholar]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res 1999, 85, 357–363. [Google Scholar]
- Mak, S.; Newton, G.E. The oxidative stress hypothesis of congestive heart failure: Radical thoughts. Chest 2001, 120, 2035–2046. [Google Scholar]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res 2007, 75, 770–781. [Google Scholar]
- Konorev, E.A.; Kennedy, M.C.; Kalyanaraman, B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: The role of reactive oxygen and nitrogen intermediates. Arch. Biochem. Biophys 1999, 368, 421–428. [Google Scholar]
- Faulk, A.; Weissig, V.; Elbayoumi, T. Mitochondria-specific nano-emulsified therapy for myocardial protection against Doxorubicin-induced cardiotoxicity. Methods Mol. Biol 2013, 991, 99–112. [Google Scholar]
- Melov, S.; Schneider, J.A.; Day, B.J.; Hinerfeld, D.; Coskun, P.; Mirra, S.S.; Crapo, J.D.; Wallace, D.C. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat. Genet 1998, 18, 159–163. [Google Scholar]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Tachibana, T.; Ishikawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem 2006, 281, 33789–33801. [Google Scholar]
- Kubin, A.M.; Skoumal, R.; Tavi, P.; Konyi, A.; Perjes, A.; Leskinen, H.; Ruskoaho, H.; Szokodi, I. Role of reactive oxygen species in regulation of cardiac contractility. J. Mol. Cell. Cardiol 2011, 50, 884–893. [Google Scholar]
- Ghosh, G.; De, K.; Maity, S.; Bandyopadhyay, D.; Bhattacharya, S.; Reiter, R.J.; Bandyopadhyay, A. Melatonin protects against oxidative damage and restores expression of GLUT4 gene in the hyperthyroid rat heart. J. Pineal Res 2007, 42, 71–82. [Google Scholar]
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc. Res 2012, 93, 79–88. [Google Scholar]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006, 113, 1779–1786. [Google Scholar]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar]
- Dubuisson, M.; Vander Stricht, D.; Clippe, A.; Etienne, F.; Nauser, T.; Kissner, R.; Koppenol, W.H.; Rees, J.F.; Knoops, B. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett 2004, 571, 161–165. [Google Scholar]
- Reinartz, M.; Ding, Z.; Flögel, U.; Gödecke, A.; Schrader, J. Nitrosative stress leads to protein glutathiolation, increased S-nitrosation, and up-regulation of peroxiredoxins in the heart. J. Biol. Chem 2008, 283, 17440–17449. [Google Scholar]
- Wang, S.B.; Murray, C.I.; Chung, H.S.; van Eyk, J.E. Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med 2013, 23, 14–18. [Google Scholar]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ. J 2008, 72, A31–A37. [Google Scholar]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol 2009, 53, 290–301. [Google Scholar]
- Sparagna, G.C.; Chicco, A.J.; Murphy, R.C.; Bristow, M.R.; Johnson, C.A.; Rees, M.L.; Maxey, M.L.; McCune, S.A.; Moore, R.L. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res 2007, 48, 1559–1570. [Google Scholar]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650. [Google Scholar]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res 2009, 81, 449–456. [Google Scholar]
- Siveski-Iliskovic, N.; Hill, M.; Chow, D.A.; Singal, P.K. Probucol protects against adriamycin cardiomyopathy without interfering with its antitumor effect. Circulation 1995, 91, 10–15. [Google Scholar]
- Rautiainen, S.; Levitan, E.B.; Mittleman, M.A.; Wolk, A. Total antioxidant capacity of diet and risk of heart failure: A population-based prospective cohort of women. Am. J. Med 2013, 126, 494–500. [Google Scholar]
- Korantzopoulos, P.; Galaris, D.; Papaioannides, D.; Siogas, K. The possible role of oxidative stress in heart failure and the potential of antioxidant intervention. Med. Sci. Monit 2003, 9, RA120–RA125. [Google Scholar]
- Castro, P.; Pérez, O.; Greig, D.; Díaz-Araya, G.; Moraga, F.; Chiong, M.; Troncoso, R.; Padilla, I.; Vukasovic, J.L.; Corbalán, R.; et al. Effects of carvedilol on functional capacity, left ventricular function, catecholamines and oxidative stress in patients with chronic heart failure. Rev. Esp. Cardiol 2004, 57, 1053–1058. (in Spanish). [Google Scholar]
- Witman, M.A.; McDaniel, J.; Fjeldstad, A.S.; Ives, S.J.; Zhao, J.; Nativi, J.N.; Stehlik, J.; Wray, D.W.; Richardson, R.S. A differing role of oxidative stress in the regulation of central and peripheral hemodynamics during exercise in heart failure. Am. J. Physiol. Heart Circ. Physiol 2012, 303, H1237–H1244. [Google Scholar]
- Wang, G.; Li, W.; Lu, X.; Bao, P.; Zhao, X. Luteolin ameliorates cardiac failure in type I diabetic cardiomyopathy. J. Diabetes Complicat 2012, 26, 259–265. [Google Scholar]
- Tanaka, T.; Ogawa, M.; Suzuki, J.; Sekinishi, A.; Itai, A.; Hirata, Y.; Nagai, R.; Isobe, M. Inhibition of IκB phosphorylation prevents load-induced cardiac dysfunction in mice. Am. J. Physiol. Heart Circ. Physiol 2012, 303, H1435–H1445. [Google Scholar]
- Cai, Y.; Yu, S.S.; Chen, T.T.; Gao, S.; Geng, B.; Yu, Y.; Ye, J.T.; Liu, P.Q. EGCG inhibits CTGF expression via blocking NF-κB activation in cardiac fibroblast. Phytomedicine 2013, 20, 106–113. [Google Scholar]
- Maier, H.J.; Schips, T.G.; Wietelmann, A.; Krüger, M.; Brunner, C.; Sauter, M.; Klingel, K.; Böttger, T.; Braun, T.; Wirth, T. Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 11794–11799. [Google Scholar]
- Purcell, N.H.; Tang, G.; Yu, C.; Mercurio, F.; DiDonato, J.A.; Lin, A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 6668–6673. [Google Scholar]
- Li, Y.; Ha, T.; Gao, X.; Kelley, J.; Williams, D.L.; Browder, I.W.; Kao, R.L.; Li, C. NF-kappaB activation is required for the development of cardiac hypertrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1712–H1720. [Google Scholar]
- Xu, Z.; Lin, S.; Wu, W.; Tan, H.; Wang, Z.; Cheng, C.; Lu, L.; Zhang, X. Ghrelin prevents doxorubicin-induced cardiotoxicity through TNF-alpha/NF-kappaB pathways and mitochondrial protective mechanisms. Toxicology 2008, 247, 133–138. [Google Scholar]
- Xing, L.; Jiang, M.; Dong, L.; Gao, J.; Hou, Y.; Bai, G.; Luo, G. Cardioprotective effects of the YiQiFuMai injection and isolated compounds on attenuating chronic heart failure via NF-κB inactivation and cytokine suppression. J. Ethnopharmacol 2013, 148, 239–245. [Google Scholar]
- Ock, S.; Ahn, J.; Lee, S.H.; Park, H.; Son, J.W.; Oh, J.G.; Yang, D.K.; Lee, W.S.; Kim, H.S.; Rho, J.; et al. Receptor activator of nuclear factor-κB ligand is a novel inducer of myocardial inflammation. Cardiovasc. Res 2012, 94, 105–114. [Google Scholar]
- Del Vescovo, C.D.; Cotecchia, S.; Diviani, D. A-kinase-anchoring protein-Lbc anchors IκB kinase β to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol. Cell. Biol 2013, 33, 14–27. [Google Scholar]
- Hamid, T.; Guo, S.Z.; Kingery, J.R.; Xiang, X.; Dawn, B.; Prabhu, S.D. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc. Res 2011, 89, 129–138. [Google Scholar]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 2013, 1832, 1049–1060. [Google Scholar]
- Finkel, M.S.; Oddis, C.V.; Jacob, T.D.; Watkins, S.C.; Hattler, B.G.; Simmons, R.L. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992, 257, 387–389. [Google Scholar]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Demetris, A.J.; Feldman, A.M. Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. J. Card. Fail 1997, 3, 117–124. [Google Scholar]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res 1997, 81, 627–635. [Google Scholar]
- Yajima, T.; Murofushi, Y.; Zhou, H.; Park, S.; Housman, J.; Zhong, Z.H.; Nakamura, M.; Machida, M.; Hwang, K.K.; Gu, Y.; et al. Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation 2011, 124, 2690–2701. [Google Scholar]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem 1997, 272, 14899–14907. [Google Scholar]
- Schulz, R.; Nava, E.; Moncada, S. Induction and potential biological relevance of a Ca2+-independent nitric oxide synthase in the myocardium. Br. J. Pharmacol 1992, 105, 575–580. [Google Scholar]
- Kalk, P.; Westermann, D.; Herzfeld, S.; Relle, K.; Pfab, T.; Bauer, C.; Tschöpe, C.; Stasch, J.P.; Hocher, B. Additional lack of iNOS attenuates diastolic dysfunction in aged ET-1 transgenic mice. Can. J. Physiol. Pharmacol 2008, 86, 353–357. [Google Scholar]
- Madonna, R.; Jiang, J.; Geng, Y.J. Attenuated expression of gelsolin in association with induction of aquaporin-1 and nitric oxide synthase in dysfunctional hearts of aging mice exposed to endotoxin. Int. J. Immunopathol. Pharmacol 2012, 25, 911–922. [Google Scholar]
- Tatsumi, T.; Akashi, K.; Keira, N.; Matoba, S.; Mano, A.; Shiraishi, J.; Yamanaka, S.; Kobara, M.; Hibino, N.; Hosokawa, S.; et al. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J. Mol. Cell. Cardiol 2004, 37, 775–784. [Google Scholar]
- Motawi, T.K.; Darwish, H.A.; Abd, El; Tawab, A.M. The relative efficacy of aminoguanidine and pentoxifylline in modulating endotoxin-induced cardiac stress. Cell Biochem. Funct 2011, 29, 694–702. [Google Scholar]
- Panaro, M.A.; Pricci, M.; Meziani, F.; Ragot, T.; Andriantsitohaina, R.; Mitolo, V.; Tesse, A. Cyclooxygenase-2-derived prostacyclin protective role on endotoxin-induced mouse cardiomyocyte mortality. Cardiovasc. Toxicol 2011, 11, 347–356. [Google Scholar]
- Gödecke, A.; Molojavyi, A.; Heger, J.; Flögel, U.; Ding, Z.; Jacoby, C.; Schrader, J. Myoglobin protects the heart from inducible nitric-oxide synthase (iNOS)-mediated nitrosative stress. J. Biol. Chem 2003, 278, 21761–21766. [Google Scholar]
- Mungrue, I.N.; Gros, R.; You, X.; Pirani, A.; Azad, A.; Csont, T.; Schulz, R.; Butany, J.; Stewart, D.J.; Husain, M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Investig 2002, 109, 735–743. [Google Scholar]
- Drexler, H.; Kästner, S.; Strobel, A.; Studer, R.; Brodde, O.E.; Hasenfuss, G. Expression, activity and functional significance of inducible nitric oxide synthase in the failing human heart. J. Am. Coll. Cardiol 1998, 32, 955–963. [Google Scholar]
- Haywood, G.A.; Tsao, P.S.; von der Leyen, H.E.; Mann, M.J.; Keeling, P.J.; Trindade, P.T.; Lewis, N.P.; Byrne, C.D.; Rickenbacher, P.R.; Bishopric, N.H.; et al. Expression of inducible nitric oxide synthase in human heart failure. Circulation 1996, 93, 1087–1094. [Google Scholar]
- Dias, F.A.L.; Urboniene, D.; Wolska, B.M. Ablation of iNOS delays cardiac contractile dysfunction in chronic hypertension. Front. Biosci 2010, 2, 312–324. [Google Scholar]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.L.; Heymes, C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar]
- Winlaw, D.S.; Smythe, G.A.; Keogh, A.M.; Schyvens, C.G.; Spratt, P.M.; Macdonald, P.S. Increased nitric oxide production in heart failure. Lancet 1994, 344, 373–374. [Google Scholar]
- Sugamori, T.; Ishibashi, Y.; Shimada, T.; Takahashi, N.; Sakane, T.; Ohata, S.; Kunizawa, Y.; Inoue, S.; Nakamura, K.; Ohta, Y.; et al. Increased nitric oxide in proportion to the severity of heart failure in patients with dilated cardiomyopathy: Close correlation of tumor necrosis factor-alpha with systemic and local production of nitric oxide. Circ. J 2002, 66, 627–632. [Google Scholar]
- Usui, M.; Matsuoka, H.; Miyazaki, H.; Ueda, S.; Okuda, S.; Imaizumi, T. Increased endogenous nitric oxide synthase inhibitor in patients with congestive heart failure. Life Sci 1998, 62, 2425–2430. [Google Scholar]
- De Laforcade, A.M.; Freeman, L.M.; Rush, J.E. Serum nitrate and nitrite in dogs with spontaneous cardiac disease. J. Vet. Intern. Med 2003, 17, 315–318. [Google Scholar]
- Yu, C.M.; Fung, P.C.; Chan, G.; Lai, K.W.; Wang, Q.; Lau, C.P. Plasma nitric oxide level in heart failure secondary to left ventricular diastolic dysfunction. Am. J. Cardiol 2001, 88, 867–870. [Google Scholar]
- Bernstein, R.D.; Zhang, X.; Zhao, G.; Forfia, P.; Tuzman, J.; Ochoa, F.; Ochoa, M.; Vogel, T.; Hintze, T.H. Mechanisms of nitrate accumulation in plasma during pacing-induced heart failure in conscious dogs. Nitric Oxide 1997, 1, 386–396. [Google Scholar]
- Kaye, D.M.; Chin-Dusting, J.; Esler, M.D.; Jennings, G.L. The failing human heart does not release nitrogen oxides. Life Sci 1998, 62, 883–887. [Google Scholar]
- Archer, S. Measurement of nitric oxide in biological models. FASEB J 1993, 7, 349–360. [Google Scholar]
- Murphy, M.E.; Noack, E. Nitric oxide assay using hemoglobin method. Methods Enzymol 1994, 233, 240–250. [Google Scholar]
- Rodnenkov, O.V.; Luneva, O.G.; Ulyanova, N.A.; Maksimov, G.V.; Rubin, A.B.; Orlov, S.N.; Chazov, E.I. Erythrocyte membrane fluidity and haemoglobin haemoporphyrin conformation: Features revealed in patients with heart failure. Pathophysiology 2005, 11, 209–213. [Google Scholar]
- Sumino, H.; Sato, K.; Sakamaki, T.; Masuda, H.; Nakamura, T.; Kanda, T.; Nagai, R. Decreased basal production of nitric oxide in patients with heart disease. Chest 1998, 113, 317–322. [Google Scholar]
- Katz, S.D.; Khan, T.; Zeballos, G.A.; Mathew, L.; Potharlanka, P.; Knecht, M.; Whelan, J. Decreased activity of the l-arginine-nitric oxide metabolic pathway in patients with congestive heart failure. Circulation 1999, 99, 2113–2117. [Google Scholar]
- Otani, H. The role of nitric oxide in myocardial repair and remodeling. Antioxid. Redox Signal 2009, 11, 1913–1928. [Google Scholar]
- McDonald, L.J.; Murad, F. Nitric oxide and cyclic GMP signaling. Proc. Soc. Exp. Biol. Med 1996, 211, 1–6. [Google Scholar]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev 2010, 62, 525–563. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Pryor, W.A.; Squadrito, G.L. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. Am. J. Physiol 1995, 268, L699–L722. [Google Scholar]
- Costell, M.H.; Ancellin, N.; Bernard, R.E.; Zhao, S.; Upson, J.J.; Morgan, L.A.; Maniscalco, K.; Olzinski, A.R.; Ballard, V.L.; Herry, K.; et al. Comparison of soluble guanylate cyclase stimulators and activators in models of cardiovascular disease associated with oxidative stress. Front. Pharmacol 2012, 3, 128. [Google Scholar]
- Gheorghiade, M.; Marti, C.N.; Sabbah, H.N.; Roessig, L.; Greene, S.J.; Böhm, M.; Burnett, J.C.; Campia, U.; Cleland, J.G.; Collins, S.P.; et al. Academic Research Team in Heart Failure (ART-HF). Soluble guanylate cyclase: A potential therapeutic target for heart failure. Heart Fail. Rev 2013, 18, 123–134. [Google Scholar]
- Irvine, J.C.; Ganthavee, V.; Love, J.E.; Alexander, A.E.; Horowitz, J.D.; Stasch, J.P.; Kemp-Harper, B.K.; Ritchie, R.H. The soluble guanylyl cyclase activator bay 58–2667 selectively limits cardiomyocyte hypertrophy. PLoS One 2012, 7, e44481. [Google Scholar]
- Lin, E.Q.; Irvine, J.C.; Cao, A.H.; Alexander, A.E.; Love, J.E.; Patel, R.; McMullen, J.R.; Kaye, D.M.; Kemp-Harper, B.K.; Ritchie, R.H. Nitroxyl (HNO) stimulates soluble guanylyl cyclase to suppress cardiomyocyte hypertrophy and superoxide generation. PLoS One 2012, 7, e34892. [Google Scholar]
- Blanton, R.M.; Takimoto, E.; Lane, A.M.; Aronovitz, M.; Piotrowski, R.; Karas, R.H.; Kass, D.A.; Mendelsohn, M.E. Protein kinase G Iα inhibits pressure overload-induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J. Am. Heart Assoc. 2012, 1, e003731. [Google Scholar]
- Westermann, D.; Becher, P.M.; Lindner, D.; Savvatis, K.; Xia, Y.; Fröhlich, M.; Hoffmann, S.; Schultheiss, H.P.; Tschöpe, C. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res. Cardiol. 2012, 107, 308. [Google Scholar]
- Crassous, P.A.; Couloubaly, S.; Huang, C.; Zhou, Z.; Baskaran, P.; Kim, D.D.; Papapetropoulos, A.; Fioramonti, X.; Durán, W.N.; Beuve, A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am. J. Physiol. Heart Circ. Physiol 2012, 303, H597–H604. [Google Scholar]
- Schmidt, K.; Neubauer, A.; Kolesnik, B.; Stasch, J.P.; Werner, E.R.; Gorren, A.C.; Mayer, B. Tetrahydrobiopterin protects soluble guanylate cyclase against oxidative inactivation. Mol. Pharmacol 2012, 82, 420–427. [Google Scholar]
- Aggarwal, S.; Gross, C.M.; Kumar, S.; Datar, S.; Oishi, P.; Kalkan, G.; Schreiber, C.; Fratz, S.; Fineman, J.R.; Black, S.M. Attenuated vasodilatation in lambs with endogenous and exogenous activation of cGMP signaling: Role of protein kinase G nitration. J. Cell. Physiol 2011, 226, 3104–3113. [Google Scholar]
- Negash, S.; Gao, Y.; Zhou, W.; Liu, J.; Chinta, S.; Raj, J.U. Regulation of cGMP-dependent protein kinase-mediated vasodilation by hypoxia-induced reactive species in ovine fetal pulmonary veins. Am. J. Physiol. Lung Cell. Mol. Physiol 2007, 293, L1012–L1020. [Google Scholar]
- Cruz, J.A.; Bauer, E.M.; Rodriguez, A.I.; Gangopadhyay, A.; Zeineh, N.S.; Wang, Y.; Shiva, S.; Champion, H.C.; Bauer, P.M. Chronic hypoxia induces right heart failure in caveolin-1−/− mice. Am. J. Physiol. Heart Circ. Physiol 2012, 302, H2518–H2527. [Google Scholar]
- Zhao, Y.Y.; Malik, A.B. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc. Med 2009, 19, 238–242. [Google Scholar]
- Liu, Y.; Dillon, A.R.; Tillson, M.; Makarewich, C.; Nguyen, V.; Dell’italia, L.; Sabri, A.K.; Rizzo, V.; Tsai, E.J. Volume overload induces differential spatiotemporal regulation of myocardial soluble guanylyl cyclase in eccentric hypertrophy and heart failure. J. Mol. Cell. Cardiol 2013, 60C, 72–83. [Google Scholar]
- Lu, Z.; Xu, X.; Hu, X.; Lee, S.; Traverse, J.H.; Zhu, G.; Fassett, J.; Tao, Y.; Zhang, P.; dos Remedios, C.; et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 2010, 121, 1474–1483. [Google Scholar]
- Takahashi, M.; Takeda, S.; Kurokawa, S.; Kubo, T.; Fukuda, N.; Izumi, T. Cyclic GMP production by ANP, BNP, and NO during worsening and improvement of chronic heart failure. Jpn. Heart J 2003, 44, 713–724. [Google Scholar]
- Woodard, G.E.; Rosado, J.A. Natriuretic peptides in vascular physiology and pathology. Int. Rev. Cell. Mol. Biol 2008, 268, 59–93. [Google Scholar]
- Dickey, D.M.; Flora, D.R.; Bryan, P.M.; Xu, X.; Chen, Y.; Potter, L.R. Differential regulation of membrane guanylyl cyclases in congestive heart failure: Natriuretic peptide receptor (NPR)-B, not NPR-A, is the predominant natriuretic peptide receptor in the failing heart. Endocrinology 2007, 148, 3518–3522. [Google Scholar]
- Arstall, M.A.; Sawyer, D.B.; Fukazawa, R.; Kelly, R.A. Cytokine-mediated apoptosis in cardiac myocytes: The role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ. Res 1999, 85, 829–840. [Google Scholar]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol 2013, 61, 599–610. [Google Scholar]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta 2011, 1813, 1360–1372. [Google Scholar]
- Casademont, J.; Miró, O. Electron transport chain defects in heart failure. Heart Fail. Rev 2002, 7, 131–139. [Google Scholar]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol 2013, 55, 31–41. [Google Scholar]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res 2010, 88, 40–50. [Google Scholar]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol 2004, 555, 1–13. [Google Scholar]
- Verdejo, H.E.; del Campo, A.; Troncoso, R.; Gutierrez, T.; Toro, B.; Quiroga, C.; Pedrozo, Z.; Munoz, J.P.; Garcia, L.; Castro, P.F.; et al. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr. Hypertens. Rep 2012, 14, 532–539. [Google Scholar]
- Scolletta, S.; Biagioli, B. Energetic myocardial metabolism and oxidative stress: Let’s make them our friends in the fight against heart failure. Biomed. Pharmacother 2010, 64, 203–207. [Google Scholar]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 2011, 1813, 1333–1350. [Google Scholar]
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res 2011, 109, 1327–1331. [Google Scholar]
- Hal, M.E.; Smith, G.; Hall, J.E.; Stec, D.E. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H253–H260. [Google Scholar]
- Shin, J.; Lee, S.H.; Kwon, M.C.; Yang, D.K.; Seo, H.R.; Kim, J.; Kim, Y.Y.; Im, S.K.; Abel, E.D.; Kim, K.T.; et al. Cardiomyocyte specific deletion of Crif1 causes mitochondrial cardiomyopathy in mice. PLoS One 2013, 8, e53577. [Google Scholar]
- Ferrari, R.; Merli, E.; Cicchitelli, G.; Mele, D.; Fucili, A.; Ceconi, C. Therapeutic effects of l-carnitine and propionyl-l-carnitine on cardiovascular diseases: A review. Ann. N. Y. Acad. Sci 2004, 1033, 79–91. [Google Scholar]
- Mingorance, C.; Rodríguez-Rodríguez, R.; Justo, M.L.; Alvarez de Sotomayor, M.; Herrera, M.D. Pharmacological effects and clinical applications of propionyl-l-carnitine. Nutr. Rev 2011, 69, 279–290. [Google Scholar]
- Malaguarnera, M. Carnitine derivatives: Clinical usefulness. Curr. Opin. Gastroenterol 2012, 28, 166–176. [Google Scholar]
- Fotino, A.D.; Thompson-Paul, A.M.; Bazzano, L.A. Effect of coenzyme Q10 supplementation on heart failure: A meta-analysis. Am. J. Clin. Nutr 2013, 97, 268–275. [Google Scholar]
- Stocker, R.; Macdonald, P. The benefit of coenzyme Q10 supplements in the management of chronic heart failure: A long tale of promise in the continued absence of clear evidence. Am. J. Clin. Nutr 2013, 97, 233–234. [Google Scholar]
- Silberman, G.A.; Fan, T.H.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010, 121, 519–528. [Google Scholar]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig 2005, 115, 1221–1231. [Google Scholar]
- Moens, A.L.; Takimoto, E.; Tocchetti, C.G.; Chakir, K.; Bedja, D.; Cormaci, G.; Ketner, E.A.; Majmudar, M.; Gabrielson, K.; Halushka, M.K.; et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: Efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 2008, 117, 2626–2636. [Google Scholar]
- Jessup, J.A.; Zhang, L.; Chen, A.F.; Presley, T.D.; Kim-Shapiro, D.B.; Chappell, M.C.; Wang, H.; Groban, L. Neuronal nitric oxide synthase inhibition improves diastolic function and reduces oxidative stress in ovariectomized mRen2.Lewis rats. Menopause 2011, 18, 698–708. [Google Scholar]
- Nishijima, Y.; Sridhar, A.; Bonilla, I.; Velayutham, M.; Khan, M.; Terentyeva, R.; Li, C.; Kuppusamy, P.; Elton, T.S.; Terentyev, D.; et al. Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc. Res 2011, 91, 71–79. [Google Scholar]
- Crijns, H.J.; Schotten, U.; Moens, A.L. Is NOS uncoupling the missing link between atrial fibrillation and chronic non-ischaemic cardiomyopathy? Cardiovasc. Res 2011, 91, 556. [Google Scholar]
- Nishijima, Y.; Sridhar, A.; Zweier, J.L.; Cardounel, A.J.; Carnes, C.A. Is NOS uncoupling the missing link between atrial fibrillation and chronic non-ischaemic cardiomyopathy? Author reply. Cardiovasc. Res 2011, 91, 557–558. [Google Scholar]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling of cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep 2012, 9, 200–210. [Google Scholar]
- Tang, W.H.; Shrestha, K.; Tong, W.; Wang, Z.; Troughton, R.W.; Borowski, A.G.; Klein, A.L.; Hazen, S.L. Nitric oxide bioavailability and adiponectin production in chronic systolic heart failure: Relation to severity of cardiac dysfunction. Transl. Res 2013, 162, 26–33. [Google Scholar]
- Xi, W.; Satoh, H.; Kase, H.; Suzuki, K.; Hattori, Y. Stimulated HSP90 binding to eNOS and activation of the PI3-Akt pathway contribute to globular adiponectin-induced NO production: Vasorelaxation in response to globular adiponectin. Biochem. Biophys. Res. Commun 2005, 332, 200–205. [Google Scholar]
- Lin, L.Y.; Lin, C.Y.; Su, T.C.; Liau, C.S. Angiotensin II-induced apoptosis in human endothelial cells is inhibited by adiponectin through restoration of the association between endothelial nitric oxide synthase and heat shock protein 90. FEBS Lett 2004, 574, 106–110. [Google Scholar]
- Pall, M.L. Do sauna therapy and exercise act by raising the availability of tetrahydrobiopterin? Med. Hypotheses 2009, 73, 610–613. [Google Scholar]
- Wang, X.; Hattori, Y.; Satoh, H.; Iwata, C.; Banba, N.; Monden, T.; Uchida, K.; Kamikawa, Y.; Kasai, K. Tetrahydrobiopterin prevents endothelial dysfunction and restores adiponectin levels in rats. Eur. J. Pharmacol 2007, 555, 48–53. [Google Scholar]
- Sharma, S.; Sun, X.; Kumar, S.; Rafikov, R.; Aramburo, A.; Kalkan, G.; Tian, J.; Rehmani, I.; Kallarackal, S.; Fineman, J.R.; et al. Preserving mitochondrial function prevents the proteasomal degradation of GTP cyclohydrolase I. Free Radic. Biol. Med 2012, 53, 216–229. [Google Scholar]
- Morgan, J.P.; Erny, R.E.; Allen, P.D.; Grossman, W.; Gwathmey, J.K. Abnormal intracellular calcium handling, a major cause of systolic and diastolic dysfunction in ventricular myocardium from patients with heart failure. Circulation 1990, 81, III21–III32. [Google Scholar]
- Reuter, H.; Schwinger, R.H. Calcium handling in human heart failure—Abnormalities and target for therapy. Wien. Med. Wochenschr 2012, 162, 297–301. [Google Scholar]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig 2013, 123, 46–52. [Google Scholar]
- Knyushko, T.V.; Sharov, V.S.; Williams, T.D.; Schöneich, C.; Bigelow, D.J. 3-Nitrotyrosine modification of SERCA2a in the aging heart: A distinct signature of the cellular redox environment. Biochemistry 2005, 44, 13071–13081. [Google Scholar]
- Squier, T.C.; Bigelow, D.J. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci 2000, 5, D504–D526. [Google Scholar]
- Kawakami, M.; Okabe, E. Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol 1998, 53, 497–503. [Google Scholar]
- Salama, G.; Menshikova, E.V.; Abramson, J.J. Molecular interaction between nitric oxide and ryanodine receptors of skeletal and cardiac sarcoplasmic reticulum. Antioxid. Redox Signal 2000, 2, 5–16. [Google Scholar]
- Kanski, J.; Hong, S.J.; Schöneich, C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J. Biol. Chem 2005, 280, 24261–24266. [Google Scholar]
- McCauley, M.D.; Wehrens, X.H. Targeting ryanodine receptors for anti-arrhythmic therapy. Acta Pharmacol. Sin 2011, 32, 749–757. [Google Scholar]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—Targets for heart failure therapy. Nat. Rev. Cardiol 2012, 9, 717–733. [Google Scholar]
- Currie, S.; Elliott, E.B.; Smith, G.L.; Loughrey, C.M. Two candidates at the heart of dysfunction: The ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention—An in vivo perspective. Pharmacol. Ther 2011, 131, 204–220. [Google Scholar]
- Ahmad, H.A.; Lu, L.; Ye, S.; Schwartz, G.G.; Greyson, C.R. Calpain inhibition preserves talin and attenuates right heart failure in acute pulmonary hypertension. Am. J. Respir. Cell Mol. Biol 2012, 47, 379–386. [Google Scholar]
- Letavernier, E.; Zafrani, L.; Perez, J.; Letavernier, B.; Haymann, J.P.; Baud, L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res 2012, 96, 38–45. [Google Scholar]
- Campos, E.C.; O’Connell, J.L.; Malvestio, L.M.; Romano, M.M.; Ramos, S.G.; Celes, M.R.; Prado, C.M.; Simões, M.V.; Rossi, M.A. Calpain-mediated dystrophin disruption may be a potential structural culprit behind chronic doxorubicin-induced cardiomyopathy. Eur. J. Pharmacol 2011, 670, 541–553. [Google Scholar]
- Greyson, C.R.; Schwartz, G.G.; Lu, L.; Ye, S.; Helmke, S.; Xu, Y.; Ahmad, H. Calpain inhibition attenuates right ventricular contractile dysfunction after acute pressure overload. J. Mol. Cell. Cardiol 2008, 44, 59–68. [Google Scholar]
- Patterson, C.; Portbury, A.L.; Schisler, J.C.; Willis, M.S. Tear me down: Role of calpain in the development of cardiac ventricular hypertrophy. Circ. Res 2011, 109, 453–462. [Google Scholar]
- Li, X.; Li, Y.; Shan, L.; Shen, E.; Chen, R.; Peng, T. Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc. Res 2009, 83, 72–79. [Google Scholar]
- Smith, M.A.; Schnellmann, R.G. Calpains, mitochondria, and apoptosis. Cardiovasc. Res 2012, 96, 32–37. [Google Scholar]
- Bukowska, A.; Lendeckel, U.; Bode-Böger, S.M.; Goette, A. Physiologic and pathophysiologic role of calpain: Implications for the occurrence of atrial fibrillation. Cardiovasc. Ther 2012, 30, e115–e127. [Google Scholar]
- Mani, S.K.; Shiraishi, H.; Balasubramanian, S.; Yamane, K.; Chellaiah, M.; Cooper, G.; Banik, N.; Zile, M.R.; Kuppuswamy, D. In vivo administration of calpeptin attenuates calpain activation and cardiomyocyte loss in pressure-overloaded feline myocardium. Am. J. Physiol. Heart Circ. Physiol 2008, 295, H314–H326. [Google Scholar]
- Wang, J.C.; Zhao, Y.; Li, X.D.; Zhou, N.N.; Sun, H.; Sun, Y.Y. Proteolysis by endogenous calpain I leads to the activation of calcineurin in human heart. Clin. Lab 2012, 58, 1145–1152. [Google Scholar]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem 2012, 287, 19856–19869. [Google Scholar]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; Dos Remedios, C.G.; Maier, L.S.; Krüger, M.; Backs, J.; Linke, W.A. Crucial role for Ca2+/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ. Res 2013, 112, 664–674. [Google Scholar]
- Zang, Y.; Dai, L.; Zhan, H.; Dou, J.; Xia, L.; Zhang, H. Theoretical investigation of the mechanism of heart failure using a canine ventricular cell model: Especially the role of up-regulated CaMKII and SR Ca2+ leak. J. Mol. Cell. Cardiol 2013, 56, 34–43. [Google Scholar]
- Fischer, T.H.; Neef, S.; Maier, L.S. The Ca-calmodulin dependent kinase II: A promising target for future antiarrhythmic therapies? J. Mol. Cell. Cardiol 2013, 58, 182–187. [Google Scholar]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res 2004, 94, 110–118. [Google Scholar]
- Prévilon, M.; Pezet, M.; Dachez, C.; Mercadier, J.J.; Rouet-Benzineb, P. Sequential alterations in Akt, GSK3β, and calcineurin signalling in the mouse left ventricle after thoracic aortic constriction. Can. J. Physiol. Pharmacol 2010, 88, 1093–1101. [Google Scholar]
- Chen, Q.Q.; Zhang, W.; Chen, X.F.; Bao, Y.J.; Wang, J.; Zhu, W.Z. Electrical field stimulation induces cardiac fibroblast proliferation through the calcineurin-NFAT pathway. Can. J. Physiol. Pharmacol 2012, 90, 1611–1622. [Google Scholar]
- Berry, J.M.; Le, V.; Rotter, D.; Battiprolu, P.K.; Grinsfelder, B.; Tannous, P.; Burchfield, J.S.; Czubryt, M.; Backs, J.; Olson, E.N.; et al. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ. Res 2011, 109, 407–417. [Google Scholar]
- Lunde, I.G.; Kvaløy, H.; Austbø, B.; Christensen, G.; Carlson, C.R. Angiotensin II and norepinephrine activate specific calcineurin-dependent NFAT transcription factor isoforms in cardiomyocytes. J. Appl. Physiol 2011, 111, 1278–1289. [Google Scholar]
- Huang, C.F.; Su, M.J. Positive inotropic action of NMDA receptor antagonist (+)-MK801 in rat heart. J. Biomed. Sci 1999, 6, 387–398. [Google Scholar]
- Matsuoka, N.; Kodama, H.; Arakawa, H.; Yamaguchi, I. N-Methyl-d-aspartate receptor blockade by dizocilpine prevents stress-induced sudden death in cardiomyopathic hamsters. Brain Res 2002, 944, 200–204. [Google Scholar]
- Moshal, K.S.; Tipparaju, S.M.; Vacek, T.P.; Kumar, M.; Singh, M.; Frank, I.E.; Patibandla, P.K.; Tyagi, N.; Rai, J.; Metreveli, N.; et al. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am. J. Physiol. Heart Circ. Physiol 2008, 295, H890–H897. [Google Scholar]
- Moshal, K.S.; Kumar, M.; Tyagi, N.; Mishra, P.K.; Metreveli, N.; Rodriguez, W.E.; Tyagi, S.C. Restoration of contractility in hyperhomocysteinemia by cardiac-specific deletion of NMDA-R1. Am. J. Physiol. Heart Circ. Physiol 2009, 296, H887–H892. [Google Scholar]
- Moshal, K.S.; Metreveli, N.; Frank, I.; Tyagi, S.C. Mitochondrial MMP activation, dysfunction and arrhythmogenesis in hyperhomocysteinemia. Curr. Vasc. Pharmacol 2008, 6, 84–92. [Google Scholar]
- Maldonado, C.; Soni, C.V.; Todnem, N.D.; Pushpakumar, S.; Rosenberger, D.; Givvimani, S.; Villafane, J.; Tyagi, S.C. Hyperhomocysteinemia and sudden cardiac death: Potential arrhythmogenic mechanisms. Curr. Vasc. Pharmacol 2010, 8, 64–74. [Google Scholar]
- Gao, X.; Xu, X.; Pang, J.; Zhang, C.; Ding, J.M.; Peng, X.; Liu, Y.; Cao, J.M. NMDA receptor activation induces mitochondrial dysfunction, oxidative stress and apoptosis in cultured neonatal rat cardiomyocytes. Physiol. Res 2007, 56, 559–569. [Google Scholar]
- Tejero-Taldo, M.I.; Kramer, J.H.; Mak, I.T.; Komarov, A.M.; Weglicki, W.B. The nerve-heart connection in the pro-oxidant response to Mg-deficiency. Heart Fail. Rev 2006, 11, 35–44. [Google Scholar]
- Kreuder, C.; Miller, M.A.; Lowenstine, L.J.; Conrad, P.A.; Carpenter, T.E.; Jessup, D.A.; Mazet, J.A. Evaluation of cardiac lesions and risk factors associated with myocarditis and dilated cardiomyopathy in southern sea otters (Enhydra lutris nereis). Am. J. Vet. Res 2005, 66, 289–299. [Google Scholar]
- Rowell, J.; Koitabashi, N.; Kass, D.A. TRP-ing up heart and vessels: Canonical transient receptor potential channels and cardiovascular disease. J. Cardiovasc. Transl. Res 2010, 3, 516–524. [Google Scholar]
- Watanabe, H.; Murakami, M.; Ohba, T.; Takahashi, Y.; Ito, H. TRP channel and cardiovascular disease. Pharmacol. Ther 2008, 118, 337–351. [Google Scholar]
- Stiber, J.A.; Tang, Y.; Li, T.; Rosenberg, P.B. Cytoskeletal regulation of TRPC channels in the cardiorenal system. Curr. Hypertens. Rep 2012, 14, 492–497. [Google Scholar]
- Hirose, M.; Takeishi, Y.; Niizeki, T.; Nakada, T.; Shimojo, H.; Kashihara, T.; Horiuchi-Hirose, M.; Kubota, I.; Mende, U.; Yamada, M. Diacylglycerol kinase ζ inhibits ventricular tachyarrhythmias in a mouse model of heart failure. Circ. J 2011, 75, 2333–2342. [Google Scholar]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig 2006, 116, 3114–3126. [Google Scholar]
- Poteser, M.; Schleifer, H.; Lichtenegger, M.; Schernthaner, M.; Stockner, T.; Kappe, C.O.; Glasnov, T.N.; Romanin, C.; Groschner, K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10556–10561. [Google Scholar]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem 2006, 281, 33487–33496. [Google Scholar]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 2006, 25, 5305–5316. [Google Scholar]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J 2006, 20, 1660–1670. [Google Scholar]
- Wu, X.; Eder, P.; Chang, B.; Molkentin, J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 7000–7005. [Google Scholar]
- Seth, M.; Zhang, Z.S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res 2009, 105, 1023–1030. [Google Scholar]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br. J. Pharmacol 2012, 165, 2354–2364. [Google Scholar]
- Hazari, M.S.; Haykal-Coates, N.; Winsett, D.W.; Krantz, Q.T.; King, C.; Costa, D.L.; Farraj, A.K. TRPA1 and sympathetic activation contribute to increased risk of triggered cardiac arrhythmias in hypertensive rats exposed to diesel exhaust. Environ. Health Perspect 2011, 119, 951–957. [Google Scholar]
- Carll, A.P.; Hazari, M.S.; Perez, C.M.; Krantz, Q.T.; King, C.J.; Winsett, D.W.; Costa, D.L.; Farraj, A.K. Whole and particle-free diesel exhausts differentially affect cardiac electrophysiology, blood pressure, and autonomic balance in heart failure-prone rats. Toxicol. Sci 2012, 128, 490–499. [Google Scholar]
- Shaheen, M.; Cheema, Y.; Shahbaz, A.U.; Bhattacharya, S.K.; Weber, K.T. Intracellular calcium overloading and oxidative stress in cardiomyocyte necrosis via a mitochondriocentric signal-transducer-effector pathway. Exp. Clin. Cardiol 2011, 16, 109–115. [Google Scholar]
- Kawanabe, Y.; Nauli, S.M. Involvement of extracellular Ca2+ influx through voltage-independent Ca2+ channels in endothelin-1 function. Cell. Signal 2005, 17, 911–916. [Google Scholar]
- Sayen, M.R.; Gustaffsson, A.B.; Sussman, M.A.; Molkentin, J.D.; Gottlieb, R.A. Calcineurin transgenic mice have mitochondrial dysfunction and elevated superoxide production. Am. J. Physiol. Cell Physiol 2003, 284, C562–C570. [Google Scholar]
- Bachmaier, K.; Neu, N.; Pummerer, C.; Duncan, G.S.; Mak, T.W.; Matsuyama, T.; Penninger, J.M. iNOS expression and nitrotyrosine formation in the myocardium in response to inflammation is controlled by the interferon regulatory factor 1. Circulation 1997, 96, 585–591. [Google Scholar]
- Glück, B.; Merkle, I.; Dornberger, G.; Stelzner, A. Expression of inducible nitric oxide synthase in experimental viral myocarditis. Herz 2000, 25, 255–260. [Google Scholar]
- Mosqueira, M.; Zeiger, U.; Förderer, M.; Brinkmeier, H.; Fink, R.H. Cardiac and respiratory dysfunction in duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev 2013, 33, 1174–1213. [Google Scholar]
- Judge, D.P.; Kass, D.A.; Thompson, W.R.; Wagner, K.R. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am. J. Cardiovasc. Drugs 2011, 11, 287–294. [Google Scholar]
- Burelle, Y.; Khairallah, M.; Ascah, A.; Allen, B.G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Alterations in mitochondrial function as a harbinger of cardiomyopathy: Lessons from the dystrophic heart. J. Mol. Cell. Cardiol 2010, 48, 310–321. [Google Scholar]
- An, J.; Du, J.; Wei, N.; Guan, T.; Camara, A.K.; Shi, Y. Differential sensitivity to LPS-induced myocardial dysfunction in the isolated brown Norway and Dahl S rat hearts: Roles of mitochondrial function, NF-κB activation, and TNF-α production. Shock 2012, 37, 325–332. [Google Scholar]
- Stamm, C.; Cowan, D.B.; Friehs, I.; Noria, S.; del Nido, P.J.; McGowan, F.X., Jr. Rapid endotoxin-induced alterations in myocardial calcium handling: Obligatory role of cardiac TNF-alpha. Anesthesiology 2001, 95, 1396–1405. [Google Scholar]
- Matsuno, K.; Iwata, K.; Matsumoto, M.; Katsuyama, M.; Cui, W.; Murata, A.; Nakamura, H.; Ibi, M.; Ikami, K.; Zhang, J.; et al. NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte apoptosis. Free Radic. Biol. Med 2012, 53, 1718–1728. [Google Scholar]
- Lorigados, C.B.; Soriano, F.G.; Szabo, C. Pathomechanisms of myocardial dysfunction in sepsis. Endocr. Metab. Immune Disord. Drug Targets 2010, 10, 274–284. [Google Scholar]
- Flesch, M.; Kilter, H.; Cremers, B.; Laufs, U.; Südkamp, M.; Ortmann, M.; Müller, F.U.; Böhm, M. Effects of endotoxin on human myocardial contractility involvement of nitric oxide and peroxynitrite. J. Am. Coll. Cardiol 1999, 33, 1062–1070. [Google Scholar]
- Alvarez, S.; Boveris, A. Mitochondrial nitric oxide metabolism in rat muscle during endotoxemia. Free Radic. Biol. Med 2004, 37, 1472–1478. [Google Scholar]
- Shiroshita-Takeshita, A.; Brundel, B.J.; Burstein, B.; Leung, T.K.; Mitamura, H.; Ogawa, S.; Nattel, S. Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc. Res 2007, 74, 75–84. [Google Scholar]
- Pepe, M.; Mamdani, M.; Zentilin, L.; Csiszar, A.; Qanud, K.; Zacchigna, S.; Ungvari, Z.; Puligadda, U.; Moimas, S.; Xu, X.; et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ. Res 2010, 106, 1893–1903. [Google Scholar]
- Sachse, F.B.; Torres, N.S.; Savio-Galimberti, E.; Aiba, T.; Kass, D.A.; Tomaselli, G.F.; Bridge, J.H. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ. Res 2012, 110, 588–597. [Google Scholar]
- Liu, Y.H.; Carretero, O.A.; Cingolani, O.H.; Liao, T.D.; Sun, Y.; Xu, J.; Li, L.Y.; Pagano, P.J.; Yang, J.J.; Yang, X.P. Role of inducible nitric oxide synthase in cardiac function and remodeling in mice with heart failure due to myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H2616–H2623. [Google Scholar]
- Biondi, B. Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur. J. Endocrinol 2012, 167, 609–618. [Google Scholar]
- Schmidt-Ott, U.M.; Ascheim, D.D. Thyroid hormone and heart failure. Curr. Heart Fail. Rep 2006, 3, 114–119. [Google Scholar]
- Athéa, Y.; Garnier, A.; Fortin, D.; Bahi, L.; Veksler, V.; Ventura-Clapier, R. Mitochondrial and energetic cardiac phenotype in hypothyroid rat. Relevance to heart failure. Pflugers Arch 2007, 455, 431–442. [Google Scholar]
- Papanas, N.; Papatheodorou, K.; Papazoglou, D.; Gioka, T.; Antonoglou, C.; Kotsiou, S.; Maltezos, E. Post-thyroidectomy thyroxine replacement dose in patients with or without compensated heart failure: The role of cytokines. Cytokine 2008, 41, 121–126. [Google Scholar]
- Asayama, K.; Dobashi, K.; Hayashibe, H.; Megata, Y.; Kato, K. Lipid peroxidation and free radical scavengers in thyroid dysfunction in the rat: A possible mechanism of injury to heart and skeletal muscle in hyperthyroidism. Endocrinology 1987, 121, 2112–2118. [Google Scholar]
- Callas, G.; Hayes, J.R. Alterations in the fine structure of cardiac muscle mitochondria induced by hyperthyroidism. Anat. Rec 1974, 178, 539–549. [Google Scholar]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol 2012, 32, 4493–4504. [Google Scholar]
- Boehm, E.A.; Jones, B.E.; Radda, G.K.; Veech, R.L.; Clarke, K. Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am. J. Physiol. Heart Circ. Physiol 2001, 280, H977–H983. [Google Scholar]
- Roman-Campos, D.; Sales-Júnior, P.; Duarte, H.L.; Gomes, E.R.; Guatimosim, S.; Ropert, C.; Gazzinelli, R.T.; Cruz, J.S. Cardiomyocyte dysfunction during the chronic phase of Chagas disease. Mem. Inst. Oswaldo Cruz 2013, 108, 243–245. [Google Scholar]
- Báez, A.L.; Lo Presti, M.S.; Fretes, R.; Díaz, C.; Pons, P.; Bazán, P.C.; Strauss, M.; Rivarola, H.W.; Paglini-Oliva, P. Chronic indeterminate phase of Chagas’ disease: Mitochondrial involvement in infection with two strains. Parasitology 2013, 140, 414–421. [Google Scholar]
- Roman-Campos, D.; Sales-Junior, P.; Duarte, H.L.; Gomes, E.R.; Lara, A.; Campos, P.; Rocha, N.N.; Resende, R.R.; Ferreira, A.; Guatimosim, S.; et al. Novel insights into the development of chagasic cardiomyopathy: Role of PI3Kinase/NO axis. Int. J. Cardiol 2013, 167, 3011–3020. [Google Scholar]
- Rocha Rodrigues, D.B.; dos Reis, M.A.; Romano, A.; de Lima Pereira, S.A.; de Paula Autunes Teixeira, V.; Tostes, S., Jr.; Rodrigues, V., Jr. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas’ disease patients with heart failure. Clin. Dev. Immunol 2012, 2012, 361730:1–361730:7. [Google Scholar]
- Teixeira, P.C.; Santos, R.H.; Fiorelli, A.I.; Bilate, A.M.; Benvenuti, L.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Selective decrease of components of the creatine kinase system and ATP synthase complex in chronic Chagas disease cardiomyopathy. PLoS Negl. Trop. Dis 2011, 5, e1205. [Google Scholar]
- Teixeira, P.C.; Iwai, L.K.; Kuramoto, A.C.; Honorato, R.; Fiorelli, A.; Stolf, N.; Kalil, J.; Cunha-Neto, E. Proteomic inventory of myocardial proteins from patients with chronic Chagas’ cardiomyopathy. Braz. J. Med. Biol. Res 2006, 39, 1549–1562. [Google Scholar]
- Hosseinzadeh, L.; Behravan, J.; Mosaffa, F.; Bahrami, G.; Bahrami, A.; Karimi, G. Curcumin potentiates doxorubicin-induced apoptosis in H9c2 cardiac muscle cells through generation of reactive oxygen species. Food Chem. Toxicol 2011, 49, 1102–1109. [Google Scholar]
- Liu, C.; Cao, F.; Tang, Q.Z.; Yan, L.; Dong, Y.G.; Zhu, L.H.; Wang, L.; Bian, Z.Y.; Li, H. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J. Nutr. Biochem 2010, 21, 1238–1250. [Google Scholar]
- Gao, G.; Dudley, S.C., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid. Redox Signal 2009, 11, 2265–2277. [Google Scholar]
- Ueland, T.; Yndestad, A.; Dahl, C.P.; Gullestad, L.; Aukrust, P. TNF revisited: Osteoprotegerin and TNF-related molecules in heart failure. Curr. Heart Fail. Rep 2012, 9, 92–100. [Google Scholar]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Laguna, J.C.; Vázquez-Carrera, M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res 2010, 87, 449–458. [Google Scholar]
- Bartha, E.; Solti, I.; Szabo, A.; Olah, G.; Magyar, K.; Szabados, E.; Kalai, T.; Hideg, K.; Toth, K.; Gero, D.; et al. Regulation of kinase cascade activation and heat shock protein expression by poly(ADP-ribose) polymerase inhibition in doxorubicin-induced heart failure. J. Cardiovasc. Pharmacol 2011, 58, 380–391. [Google Scholar]
- Bartha, E.; Kiss, G.N.; Kalman, E.; Kulcsár, G.; Kálai, T.; Hideg, K.; Habon, T.; Sumegi, B.; Toth, K.; Halmosi, R. Effect of L-2286, a poly(ADP-ribose)polymerase inhibitor and enalapril on myocardial remodeling and heart failure. J. Cardiovasc. Pharmacol 2008, 52, 253–261. [Google Scholar]
- Geng, B.; Cai, Y.; Gao, S.; Lu, J.; Zhang, L.; Zou, J.; Liu, M.; Yu, S.; Ye, J.; Liu, P. PARP-2 knockdown protects cardiomyocytes from hypertrophy via activation of SIRT1. Biochem. Biophys. Res. Commun 2013, 430, 944–950. [Google Scholar]
- Xiao, C.Y.; Chen, M.; Zsengellér, Z.; Li, H.; Kiss, L.; Kollai, M.; Szabó, C. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J. Pharmacol. Exp. Ther 2005, 312, 891–898. [Google Scholar]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar]
- Turakhia, S.; Venkatakrishnan, C.D.; Dunsmore, K.; Wong, H.; Kuppusamy, P.; Zweier, J.L.; Ilangovan, G. Doxorubicin-induced cardiotoxicity: Direct correlation of cardiac fibroblast and H9c2 cell survival and aconitase activity with heat shock protein 27. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H3111–H3121. [Google Scholar]
- Tziomalos, K.; Hare, J.M. Role of xanthine oxidoreductase in cardiac nitroso-redox imbalance. Front. Biosci 2009, 14, 237–262. [Google Scholar]
- Yamamoto, E.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Dong, Y.F.; Matsuba, S.; Ogawa, H.; Kim-Mitsuyama, S. Role of xanthine oxidoreductase in the reversal of diastolic heart failure by candesartan in the salt-sensitive hypertensive rat. Hypertension 2007, 50, 657–662. [Google Scholar]
- Rehsia, N.S.; Dhalla, N.S. Potential of endothelin-1 and vasopressin antagonists for the treatment of congestive heart failure. Heart Fail. Rev 2010, 15, 85–101. [Google Scholar]
- Drawnel, F.M.; Archer, C.R.; Roderick, H.L. The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br. J. Pharmacol 2013, 168, 296–317. [Google Scholar]
- Zarain-Herzberg, A.; Estrada-Avilés, R.; Fragoso-Medina, J. Regulation of sarco(endo)plasmic reticulum Ca2+-ATPase and calsequestrin gene expression in the heart. Can. J. Physiol. Pharmacol 2012, 90, 1017–1028. [Google Scholar]
- Murray, D.B.; Gardner, J.D.; Brower, G.L.; Janicki, J.S. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am. J. Physiol. Heart Circ. Physiol 2004, 287, H2295–H2299. [Google Scholar]
- Murray, D.B.; Gardner, J.D.; Brower, G.L.; Janicki, J.S. Effects of nonselective endothelin-1 receptor antagonism on cardiac mast cell-mediated ventricular remodeling in rats. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1251–H1257. [Google Scholar]
- Takefuji, M.; Wirth, A.; Lukasova, M.; Takefuji, S.; Boettger, T.; Braun, T.; Althoff, T.; Offermanns, S.; Wettschureck, N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation 2012, 126, 1972–1982. [Google Scholar]
- Haack, K.K.; Gao, L.; Schiller, A.M.; Curry, P.L.; Pellegrino, P.R.; Zucker, I.H. Central Rho kinase inhibition restores baroreflex sensitivity and angiotensin II type 1 receptor protein imbalance in conscious rabbits with chronic heart failure. Hypertension 2013, 61, 723–729. [Google Scholar]
- Dong, M.; Liao, J.K.; Fang, F.; Lee, A.P.; Yan, B.P.; Liu, M.; Yu, C.M. Increased Rho kinase activity in congestive heart failure. Eur. J. Heart Fail 2012, 14, 965–973. [Google Scholar]
- Surma, M.; Wei, L.; Shi, J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol 2011, 7, 657–671. [Google Scholar]
- Satoh, K.; Fukumoto, Y.; Shimokawa, H. Rho-kinase: Important new therapeutic target in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H287–H296. [Google Scholar]
- Shi, J.; Zhang, L.; Wei, L. Rho-kinase in development and heart failure: Insights from genetic models. Pediatr. Cardiol 2011, 32, 297–304. [Google Scholar]
- Wang, N.; Guan, P.; Zhang, J.P.; Li, Y.Q.; Chang, Y.Z.; Shi, Z.H.; Wang, F.Y.; Chu, L. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways. J. Cell. Biochem 2011, 112, 1920–1929. [Google Scholar]
- Wang, N.; Guan, P.; Zhang, J.P.; Chang, Y.Z.; Gu, L.J.; Hao, F.K.; Shi, Z.H.; Wang, F.Y.; Chu, L. Preventive effects of fasudil on adriamycin-induced cardiomyopathy: Possible involvement of inhibition of RhoA/ROCK pathway. Food Chem. Toxicol 2011, 49, 2975–2982. [Google Scholar]
- Zhao, X.S.; Pan, W.; Bekeredjian, R.; Shohet, R.V. Endogenous endothelin-1 is required for cardiomyocyte survival in vivo. Circulation 2006, 114, 830–837. [Google Scholar]
- Schorlemmer, A.; Matter, M.L.; Shohet, R.V. Cardioprotective signaling by endothelin. Trends Cardiovasc. Med 2008, 18, 233–239. [Google Scholar]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing in neurons. Mol. Neurobiol 2010, 41, 242–247. [Google Scholar]
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol 2003, 27, 325–355. [Google Scholar]
- Cole, R.T.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Gheorghiade, M.; Quyyumi, A.; Yancy, C.; Butler, J. Hydralazine and isosorbide dinitrate in heart failure: Historical perspective, mechanisms, and future directions. Circulation 2011, 123, 2414–2422. [Google Scholar]
- Arif, S.A.; Mergenhagen, K.A.; del Carpio, R.O.; Ho, C. Treatment of systolic heart failure in the elderly: An evidence-based review. Ann. Pharmacother 2010, 44, 1604–1614. [Google Scholar]
- Taylor, A.L.; Sabolinski, M.L.; Tam, S.W.; Ziesche, S.; Ghali, J.K.; Archambault, W.T.; Worcel, M.; Cohn, J.N. A-HeFT Investigators. Effect of fixed-dose combined isosorbide dinitrate/hydralazine in elderly patients in the African-American heart failure trial. J. Card. Fail 2012, 18, 600–606. [Google Scholar]
- Csiszar, A.; Labinskyy, N.; Podlutsky, A.; Kaminski, P.M.; Wolin, M.S.; Zhang, C.; Mukhopadhyay, P.; Pacher, P.; Hu, F.; de Cabo, R.; et al. Vasoprotective effects of resveratrol and SIRT1: Attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H2721–H2735. [Google Scholar]
- Csiszar, A.; Labinskyy, N.; Jimenez, R.; Pinto, J.T.; Ballabh, P.; Losonczy, G.; Pearson, K.J.; de Cabo, R.; Ungvari, Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: Role of circulating factors and SIRT1. Mech. Ageing Dev 2009, 130, 518–527. [Google Scholar]
- Huang, W.; Shang, W.L.; Wang, H.D.; Wu, W.W.; Hou, S.X. Sirt1 overexpression protects murine osteoblasts against TNF-α-induced injury in vitro by suppressing the NF-κB signaling pathway. Acta Pharmacol. Sin 2012, 33, 668–674. [Google Scholar]
- Xia, N.; Daiber, A.; Habermeier, A.; Closs, E.I.; Thum, T.; Spanier, G.; Lu, Q.; Oelze, M.; Torzewski, M.; Lackner, K.J.; et al. Resveratrol reverses endothelial nitric-oxide synthase uncoupling in apolipoprotein E knockout mice. J. Pharmacol. Exp. Ther 2010, 335, 149–154. [Google Scholar]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H1876–H1881. [Google Scholar]
- Biala, A.; Tauriainen, E.; Siltanen, A.; Shi, J.; Merasto, S.; Louhelainen, M.; Martonen, E.; Finckenberg, P.; Muller, D.N.; Mervaala, E. Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press 2010, 19, 196–205. [Google Scholar]
- Tsai, R.Y.; Chou, K.Y.; Shen, C.H.; Chien, C.C.; Tsai, W.Y.; Huang, Y.N.; Tao, P.L.; Lin, Y.S.; Wong, C.S. Resveratrol regulates N-methyl-d-aspartate receptor expression and suppresses neuroinflammation in morphine-tolerant rats. Anesth. Analg 2012, 115, 944–952. [Google Scholar]
- Quincozes-Santos, A.; Gottfried, C. Resveratrol modulates astroglial functions: Neuroprotective hypothesis. Ann. N. Y. Acad. Sci 2011, 1215, 72–78. [Google Scholar]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys 2010, 501, 79–90. [Google Scholar]
- Nadtochiy, S.M.; Yao, H.; McBurney, M.W.; Gu, W.; Guarente, L.; Rahman, I.; Brookes, P.S. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1506–H1512. [Google Scholar]
- Sundaresan, N.R.; Pillai, V.B.; Gupta, M.P. Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J. Mol. Cell. Cardiol 2011, 51, 614–618. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pall, M.L. The NO/ONOO-Cycle as the Central Cause of Heart Failure. Int. J. Mol. Sci. 2013, 14, 22274-22330. https://doi.org/10.3390/ijms141122274
Pall ML. The NO/ONOO-Cycle as the Central Cause of Heart Failure. International Journal of Molecular Sciences. 2013; 14(11):22274-22330. https://doi.org/10.3390/ijms141122274
Chicago/Turabian StylePall, Martin L. 2013. "The NO/ONOO-Cycle as the Central Cause of Heart Failure" International Journal of Molecular Sciences 14, no. 11: 22274-22330. https://doi.org/10.3390/ijms141122274