Oxidative Stress as an Underlying Contributor in the Development of Chronic Complications in Diabetes Mellitus


Abstract
:
1. Introduction
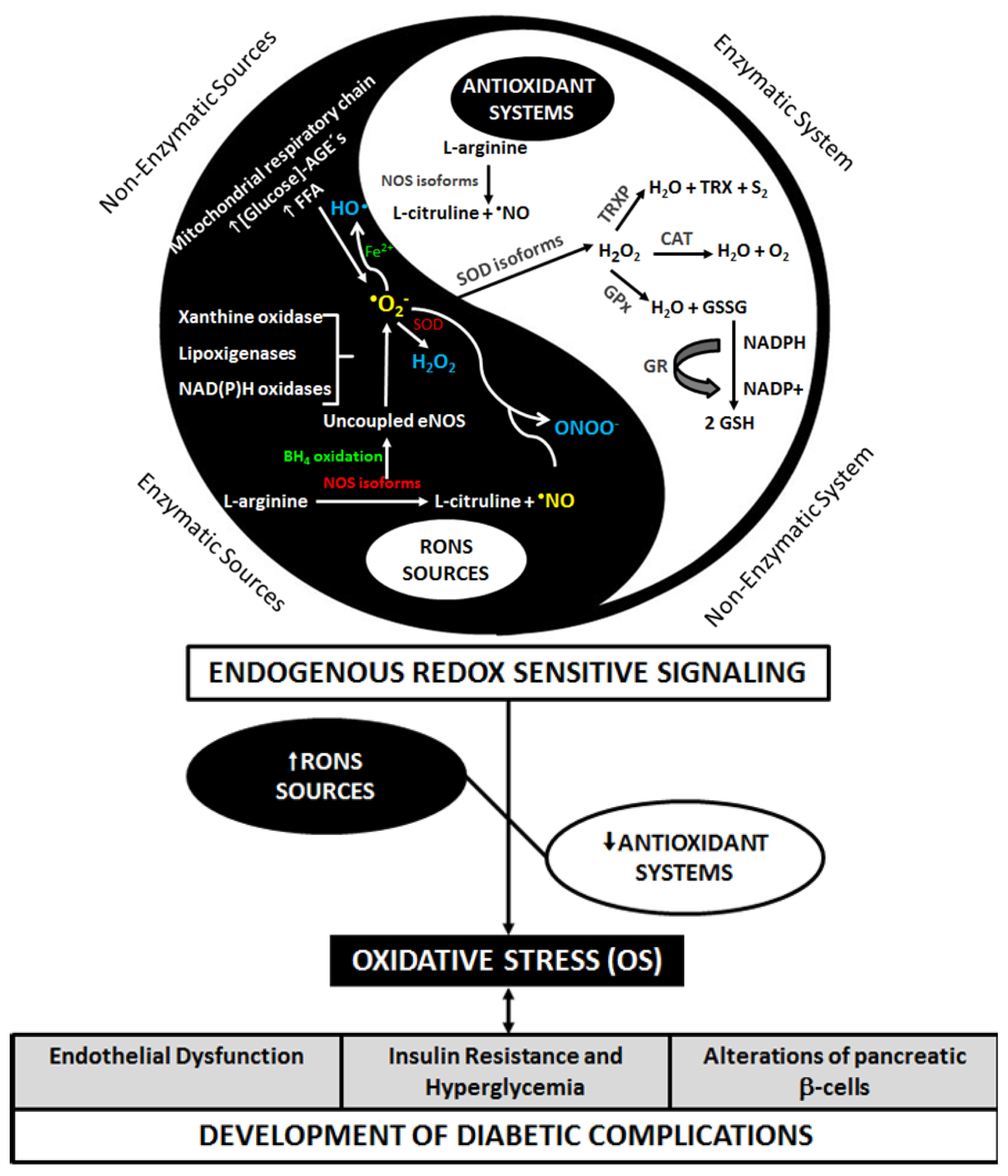
2. Diabetes and Oxidative Stress
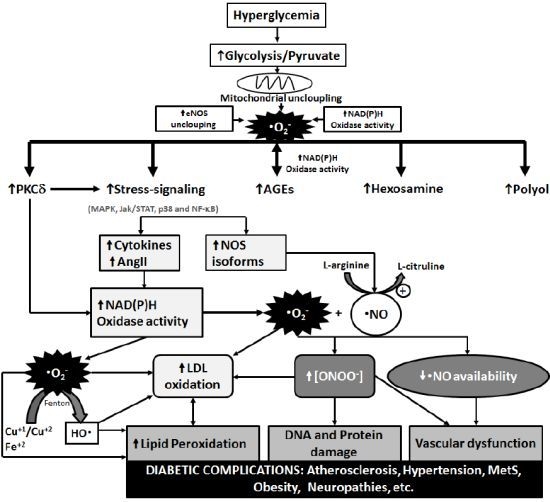
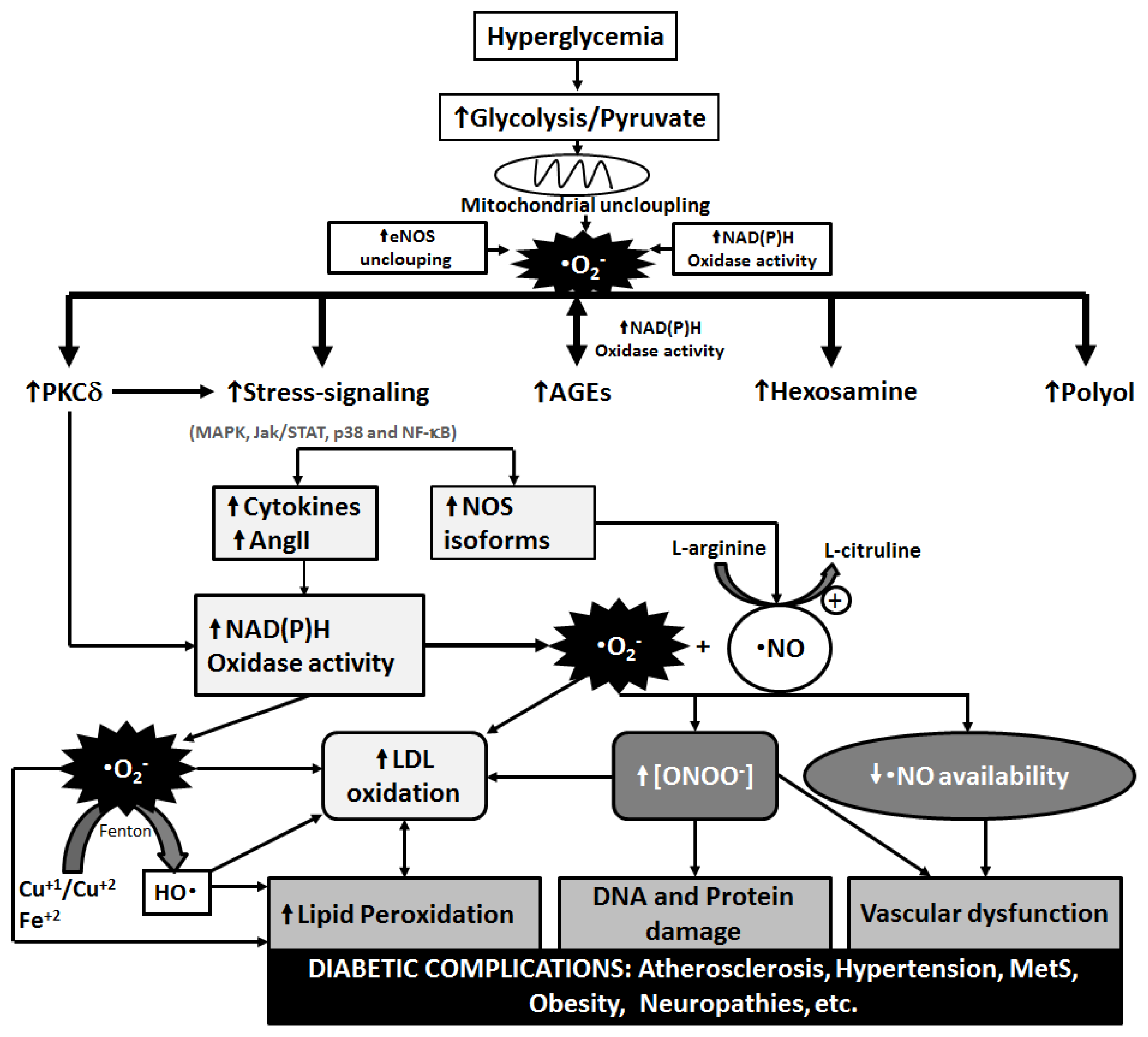
3. Metabolic Pathways of Diabetic Complications and the Role of OS
4. Studies Assessing the OS in T2DM Carriers
5. Main Biomarkers Evaluated in the Clinical Studies Presented
6. Conclusion
Acknowledgments
Conflict of Interest
References
- World Health Organization. Available online: http://www.who.int/diabetes/en accessed on 10 February 2012.
- International Diabetes Federation. Available online: http://www.diabetesatlas.org accessed on 12 April 2012.
- Sociedade Brasileira de Diabetes, Diretrizes da Sociedade Brasileira de Diabetes [in Portuguese]; Sociedade Brasileira de Diabetes: Rio de Janeiro, Brazil, 2006.
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2diabetes and in nondiabetic subjects with and without priormyocardial infarction. N. Engl. J. Med 1998, 339, 229–234. [Google Scholar]
- Schaan, B.D.; Harzheim, E.; Gus, I. Cardiac risk profile in diabetes mellitus and impaired fasting glucose [in Portuguese]. Rev. Saúde Pública 2004, 38, 529–536. [Google Scholar]
- Vehkavaara, S.; Seppälä-Lindroos, A.; Westerbacka, J.; Groop, P.; Yki-Järvinen, H. In vivo endothelial dysfunction characterizes patients with impaired fasting glucose. Diabetes Care 1999, 22, 2055–2060. [Google Scholar]
- Brownlee, M.; Cerami, A. The biochemistry of the complications of diabetes-mellitus. Ann. Rev. Biochem 1981, 50, 385–432. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazuz, M.; Telsen, J. Free radicals and antioxidants in normal physiological functions and human disease. IJBCB 2007, 39, 44–84. [Google Scholar]
- The Diabetes Control and Complications Trial (DCCT) Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. NEJM 1993, 329, 977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853.
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar]
- Miyazaki, Y.; Kawano, H.; Yoshida, T.; Miyamoto, S.; Hokomaki, J.; Nagayoshi, Y. Pancreatic B-cell function is altered by oxidative stress induced by acute hyperglycaemia. Diabet Med 2007, 24, 154–160. [Google Scholar]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.H.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice: A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest 1995, 96, 1395–1403. [Google Scholar]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar]
- Kaneto, H.; Kajimoto, Y.; Fujitani, Y.; Matsuoka, T.; Sakamoto, K.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Oxidative stress induces p21 expression in pancreatic islet cells: Possible implication in beta-cell dysfunction. Diabetologia 1999, 42, 1093–1097. [Google Scholar]
- Lee, A.Y.W.; Chung, S.S.M. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 1999, 13, 23–30. [Google Scholar]
- Ho, E.C.M.; Lam, K.S.L.; Chen, Y.S.; Yip, J.C.W.; Arvindakshan, M.; Yamagishi, S.-I.; Yagihashi, S.; Oates, P.J.; Ellery, C.A.; Chung, S.S.; et al. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 2006, 55, 1946–1953. [Google Scholar]
- Koya, D.; King, G.L. Protein kinase C activation and the development of diabetic complications. Diabetes 1998, 47, 859–866. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol 1997, 82, 291–295. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal 2006, 8, 1865–1879. [Google Scholar]
- Ceriello, A.; Testa, R. Antioxidant anti-inflammatory treatment in type 2 diabetes. Diabetes Care 2009, 32, S232–S236. [Google Scholar]
- Barbosa, J.H.P.; Oliveira, S.L.; Seara, L.T. The role of advanced glycation end-products (AGEs) in the development of vascular diabetic complications [in Portuguese]. Arq. Bras. Endocrinol. Metab 2008, 52, 940–950. [Google Scholar]
- Yamagishi, S.-i. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol 2011, 46, 217–224. [Google Scholar]
- Schaan, B.D. Role of protein kinase C in the development of vascular complications of diabetes mellitus [in Portuguese]. Arq. Bras. Endocrinol. Metab 2003, 47, 654–662. [Google Scholar]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest 2003, 112, 1049–1057. [Google Scholar]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev 2002, 23, 599–622. [Google Scholar]
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol 2005, 4, 5–16. [Google Scholar]
- Rabelo, L.A.; Souza, V.N.; Fonseca, L.J.S.; Sampaio, W.O. Redox imbalance: NADPH oxidase as therapeutic target in blood pressure control. Arq. Bras. Cardiol 2010, 94, 684–693. [Google Scholar]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal 2005, 7, 1040–1052. [Google Scholar]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol 2004, 24, 816–823. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar]
- Halliwell, B. Oxidants and human disease: Some new concepts. FASEBJ 1987, 1, 358–364. [Google Scholar]
- Barreiros, A.L.B.; David, J.M. Oxidative stress: Relations between the formation of reactive species and the organism’s defense. Quim. Nova 2005, 29, 113–123. [Google Scholar]
- Liu, Y.; Zhao, H.; Li, H.; Kalyanaraman, B.; Nicolosi, A.C.; Gutterman, D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res 2003, 93, 573–580. [Google Scholar]
- Ronsein, G.E.; Miyamoto, S.; Bechara, E.; di Mascio, P.; Martinez, G.R. Singlet oxygen-mediated protein oxidation: Damage mechanisms, detection techniques and biological implication. Quim. Nova 2006, 29, 563–568. [Google Scholar]
- Ziegler, D.; Sohr, C.; Nourooz-Zadeh, J. Oxidative stress and antioxidant defense in relation to severity of diabetic polineuropathy and cardiovascular autonomic neuropathy. Diabetes Care 2004, 27, 2178–2183. [Google Scholar]
- Monnier, V.M. Intervention against the Maillard reaction in vivo. Arch. Biochem. Biophys 2003, 419, 1–15. [Google Scholar]
- Giardino, I.; Edelstein, D.; Brownlee, M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity: A model for intracellular glycosylation in diabetes. J. Clin. Invest 1994, 94, 110–117. [Google Scholar]
- Lal, M.A.; Brismar, H.; Ekolof, A-C.; Aperia, A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002, 61, 2006–2014. [Google Scholar]
- Miyata, T.; Wada, Y.; Cal, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; de Strihou, C.V.Y. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int 1997, 51, 1170–1181. [Google Scholar]
- Kalousová, M.; Krha, J.; Zima, T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol. Res 2002, 51, 597–604. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar]
- He, Z.; Rask-Madsen, C.; King, G.L. Managing heart disease mechanisms of cardiovascular complications in diabetes and potential new pharmacological therapies. Eur. Heart J 2003, 5, B51–B57. [Google Scholar]
- Jay, D.; Hitomi, H.; Griendling, K.K. Oxidative stress and diabetic cardiovascular complications. Free Radic. Biol. Med 2006, 40, 183–192. [Google Scholar]
- Alikhani, M.; MacLellan, C.M.; Raptis, M.; Vora, S.; Trackman, P.C.; Graves, D.T. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am. J. Physiol. Cell Physiol 2007, 292, C850–C856. [Google Scholar]
- Wright, E., Jr; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract 2006, 60, 308–314. [Google Scholar]
- Rösen, P.; Nawroth, P.P.; King, G.; Moller, W.; Tritschler, H.J.; Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: A summary of a congress series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab. Res. Rev 2001, 17, 189–212. [Google Scholar]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol 2003, 14, S227–S232. [Google Scholar]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar]
- Ceriello, A.; Morocutti, A.; Mercuri, F.; Quagliaro, L.; Moro, M.; Damante, G. Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes 2000, 49, 2170–2177. [Google Scholar]
- Monnier, L.; Colette, C. Glycemic variability: Should we and can we prevent? Diabetes Care 2008, 31, S150–S154. [Google Scholar]
- Bhattacharya, B.; Ahmed, K.K.M.; Chakrabosty, S. Free radicals and cardiovascular disease: An update. Free Radic. Antioxid 2011, 1, 17–22. [Google Scholar]
- Vasconcelos, S.M.L.; Goulart, M.O.F.; Silva, M.A.M.; Manfredini, V.; Benfato, M.S.; Rabelo, L.A.; Fontes, G. Markers of redox imbalance in the blood of hypertensive patients of a community in northeastern brazil. Arq. Bras. Cardiol 2011, 97, 141–147. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem 2006, 52, 601–623. [Google Scholar]
- Piwowar, A.; Knapik-Kordecka, M.; Warwas, M. AOPP and its relations with selected markers of oxidative/antioxidative system in type 2 diabetes mellitus. Diabetes Res. Clin. Pr 2007, 77, 188–192. [Google Scholar]
- Girona, J.; Manzanares, J.M.; Marimóm, F.; Cabré, A.; Heras, M.; Guardiola, M. Oxidized to non-oxidized lipoprotein ratios are association with metabolic syndrome in diabetic patients. NMCD 2008, 18, 380–387. [Google Scholar]
- Kimura, F.; Hosegawa, G.; Obayashi, H.; Adachi, T.; Hara, H.; Ohta, M.L. Serum extracellular superoxide dismutase (EC-SOD) in patients with type 2 diabetes, relationship to the development of micro- and macrovascular complications. Diabetes Care 2003, 26, 1246–1250. [Google Scholar]
- Jaffar, N.; Ziegler, D.; Christopher, S.; John, B.D.; Jan, K.; John, H. The use of Pholasin® as a probe for the determination of plasma total antioxidant capacity. Clin. Biochem 2006, 39, 55–61. [Google Scholar]
- Kasznicki, J.; Kosmalski, M.; Sliwinska, A.; Mrowicka, M.; Stanczyk, M.; Majsterek, I.; Drzewoski, J. Evaluation of oxidative stress markers in pathogenesis of diabetic neuropathy. Mol. Biol. Rep 2012, 39, 8669–8678. [Google Scholar]
- Bravi, M.C.; Armiento, A.; Laurenti, O.; Cassone-Faldetta, M.; de Luca, O.; Moretti, A.; de Mattia, G. Insulin decreases intracellular oxidative stress in patients with type 2 diabetes mellitus. Metabolism 2006, 55, 691–695. [Google Scholar]
- Bandeira, S.M.; Guedes, G.; Fonseca, L.; Pires, A.; Gelain, D.P.; Moreira, J.C.F.; Rabelo, L.A.; Vasconcelos, S.M.L.; Goulart, M.O.F. Characterization of blood oxidative stress in type 2 diabetes mellitus patients: Increase in lipid peroxidation and SOD activity. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]
- Ganesh, S.; Dharmalingam, M.; Marcus, S.R. Oxidative stress in type 2 diabetes with iron deficiency in Asian Indians. J. Med. Biochem 2012, 30, 115–120. [Google Scholar]
- Moussa, S.A. Oxidative stress in diabetes mellitus. Romanian J. Biophys 2008, 18, 225–236. [Google Scholar]
- Lima, V.B.S.; Sampaio, F.H.; Bezerra, D.L.C.; Neto, J.M.M. Parameters of glycemic control and their relationship with zinc concentrations in blood and with superoxide dismutase enzyme activity in type 2 diabetes patients. Arq. Bras. Endocrinol. Metabol 2011, 55, 701–707. [Google Scholar]
- Soliman, G.Z.A. Blood lipid peroxidation (superoxide dismutase, malondialdehyde, glutathione) levels in Egyptian type 2 diabetic patients. Singapore Med. J 2008, 49, 129–136. [Google Scholar]
- Savu, O.; Ionescu-Tirgoviste, C.; Atanasiu, V.; Gaman, L.; Papacocea, R.; Stoian, I. Increase in total antioxidant capacity of plasma despite high levels of oxidative stress in uncomplicated type 2 diabetes mellitus. J. Int. Med. Res 2012, 40, 709–716. [Google Scholar]
- Al-Aubaidy, H.A.; Jenelek, H.F. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol 2011, 164, 899–904. [Google Scholar]
- Tabak, O.; Gelisgen, R.; Erman, H.; Erdenen, F.; Muderrisoglu, C.; Aral, H.; Uzun, H. Oxidative lipid, protein, and DNA damage as oxidative stress markers in vascular complications of diabetes mellitus. Clin. Invest. Med 2011, 34, E163–E171. [Google Scholar]
- Song, F.; Jia, W.; Yao, Y.; Hu, Y.; Lei, L.; Lin, J.; Sun, X.; Liu, L. Oxidative stress, antioxidants status and DNA damage in patients with impaired glucose regulation and newly diagnosed type 2 diabetes. Clin. Sci 2007, 112, 599–606. [Google Scholar]
- Moemen, L.A.; Shoier, A.T.; Ali, E.T. Oxidative stress and apoptosis in relation to the progression of diabetic retinopathy in diabetics. J. Appl. Sci. Res 2012, 8, 2713–2724. [Google Scholar]
- Čolak, E.; Dimitrijević-Srećković, V.; Djordjević, P.B.; Stanković, S.; Glišić, B.; Srećković, B.; Majkić-Singh, N. Biomarkers of enzymatic and non-enzimatic antioxidant defense in type 2 diabetes mellitus—comparative analysis. Biochemia Medica 2008, 18, 42–51. [Google Scholar]
- Memisogullari, R.; Bakan, E. Levels of ceruloplasmin, transferrin, and lipid peroxidation in the serum of patients with type 2 diabetes mellitus. J. Diabetes Complicat 2004, 18, 193–197. [Google Scholar]
- Pasaoglu, H.; Sancak, B.; Bukan, N. Lipid peroxidation and resistance to oxidation in patients with type 2 diabetes mellitus. Tohoku J. Exp. Med 2004, 203, 211–218. [Google Scholar]
- Genet, S.; Lema, Y.; Lutale, J. Oxidative stress correlates with complications among diabetic patients attending a diabetic clinic in Muhimbili National Hospital, Dar es Salaam, Tanzania. Ind. J. Clin. Biochem. 2012. [Google Scholar] [CrossRef]
- Desco, M.C.; Asensi, M.; Márquez, R.; Martínez-Valls, J.; Vento, M.; Pallardó, F.V.; Sastre, J.; Viña, J. Xanthine oxidase is involved in free radical production in type 1 diabetes: Protection by allopurinol. Diabetes 2002, 51, 1118–1124. [Google Scholar]
- Nojima, H.; Watanabe, H.; Yamane, K.; Kitahara, Y.; Sekikawa, K.; Yamamoto, H.; Yokoyama, A.; Inamizu, T.; Asahara, T.; Kohno, N. Hiroshima University Health Promotion Study Group. Effect of aerobic exercise training on oxidative stress in patients with type 2 diabetes mellitus. Metabolism 2008, 57, 170–176. [Google Scholar]
- Lima, E.S.; Abdalla, D.S.P. Lipid peroxidation: Mechanisms and evaluation in biological samples. RBCF 2001, 37, 293–303. [Google Scholar]
- Sánchez-Rodríguez, M.A.; Santiago-Osorio, E.; Vargas, L.A.; Mendoza-Núñez, V.M. Propuesta de un constructo para evaluar integralmente elestrés oxidativo [in Spanish]. Bioquimica 2004, 29, 81–90. [Google Scholar]
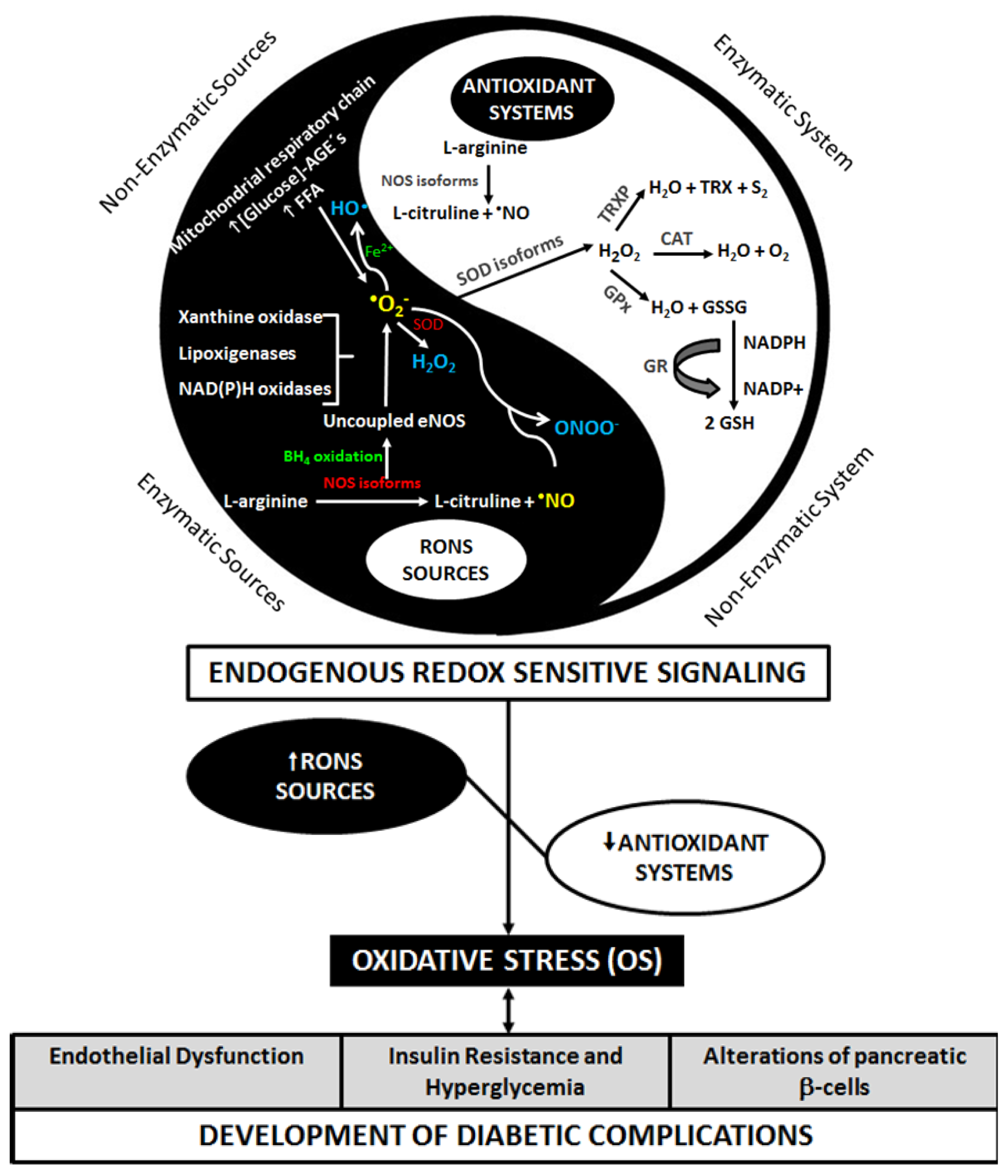
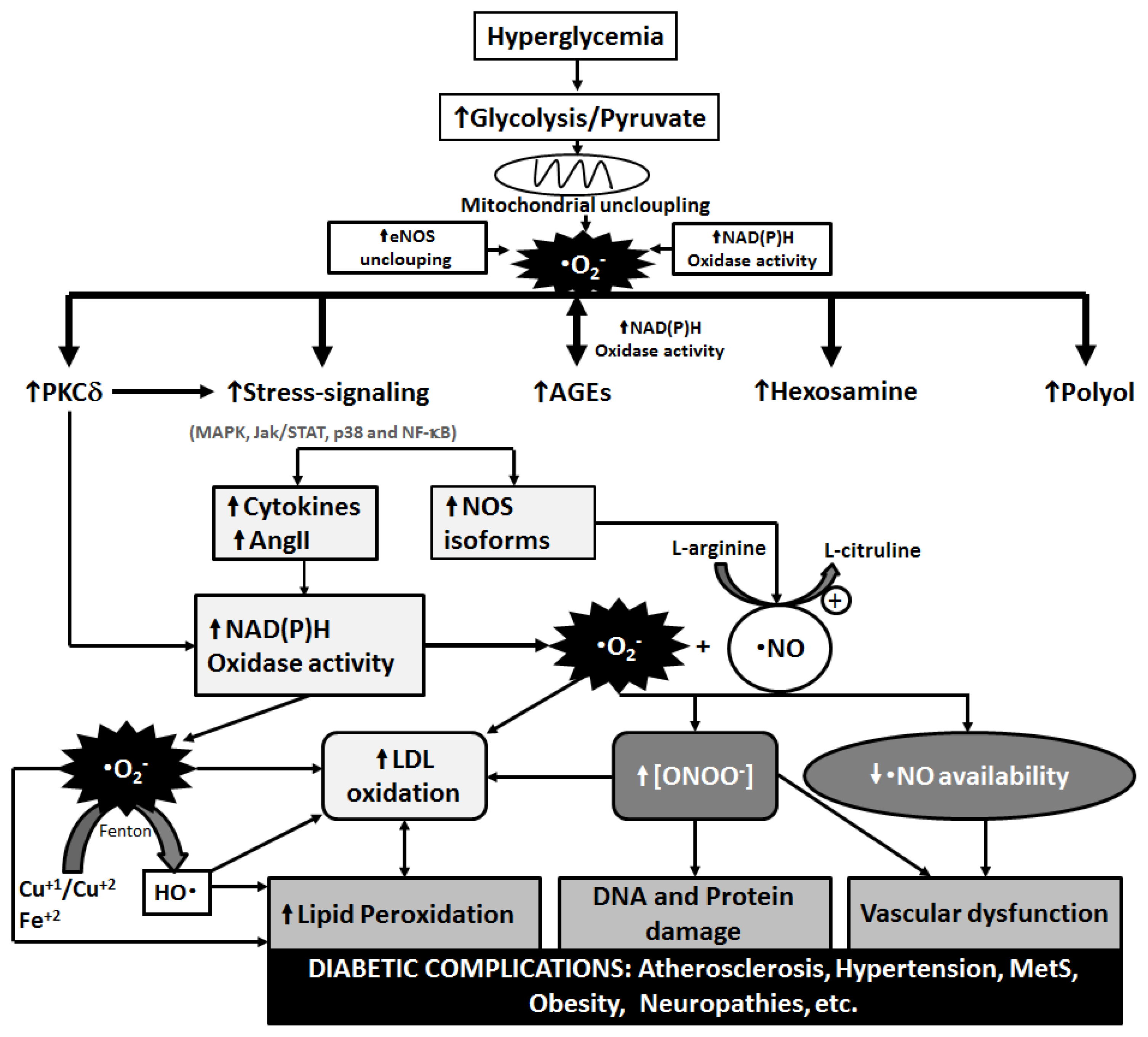

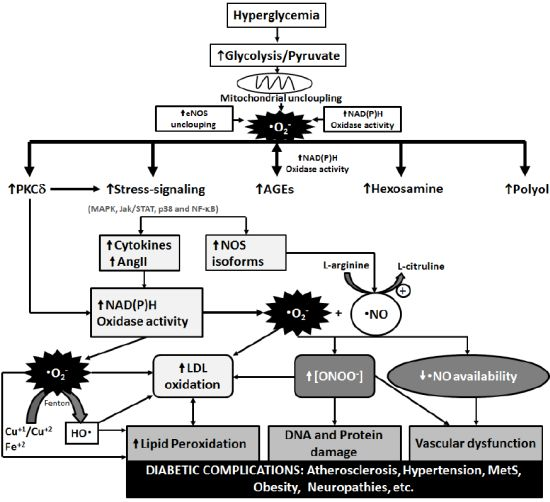{kind=link}
{kind=link}
{kind=link}
| Reactive Species | Reactivity | Source | Activity | Reference |
|---|---|---|---|---|
| •O2− | Low | ETC, phagocytic cells, autoxidation reactions (myoglobin, hemoglobin, catecholamines), enzymatic reactions (xanthine oxidase and NADPH oxidase), smoking | Vascular regulation; response to OS and maintenance of redox homeostasis | [34–36] |
| H2O2 | Low | Mitochondrial matrix, •O2−dismutation by SOD, xanthine dehydrogenase, smoking | May generate •OH radical in the presence of transition metals, inactivate enzymes through oxidation of essential -SH groups | [10,37,38] |
| •OH | High | Water radiolysis, reaction of •O2− with H2O2 (Haber-Weiss) and in the presence of transition metals (Fenton), water ozonation and ONOO− dissociation, smoking | May damage DNA, proteins, lipids and carbohydrates | [9,10,37] |
| O21 | High | Phagocytes, light induction, radiation, reactions catalyzed by peroxidases, smoking and others | Direct DNA damage, protein damage, initiates lipid peroxidation and produces alcoxyl and peroxyl radicals | [9,39] |
| •NO | High | NOS enzymes, smoking | Endothelium-derived relaxing factor. Reacts with other radical species, forming RNS that may modify DNA, proteins and lipids. Inhibits cytochrome P450 and some mitochondrial enzymes (ubiquinone oxidoreductase, oxidoreductase succinate and aconitase) | [34,40] |
| ONOO− | High | •O2− reaction with −NO, smoking | Lipid oxidation and nitration, DNA breaks | [9,10] |
| HClO | High | Reaction of H2O2 with Cl− ions (catalyzed by myeloperoxidase) | Acts as a cellular defense against bacteria; may generate chlorinated amines that are strong oxidants | [9] |
| Study sample | Biomarkers | Results with significant differences | Reference |
|---|---|---|---|
| T2DM = 94, C = 36 | TRAP, OPD (SH, CO, NH2), AOPP | ↓ TRAP and ↓SH; ↑ CO, ↑NH2, ↑ AOPP; ↑ AOPP in diabetic patients with macroangiopathy compared with those presenting microangiopathy; ↑ AOPP progressive with BMI | [59] |
| DA(+) = 73; DA(−) = 93 | Vit E OxLDL/LDL OxLDL/HDL OxLDL/Ab | DA(+) vs. DA(−): ↑ OxLDL/LDL, OxLDL/HDL and OxLDL/Ab; ↑ OxLDL/LDL, OxLDL/HDL in the presence of MetS | [60] |
| T2DM = 222; C = 75 | EC-SOD | ↑ EC-SOD; Positive correlation between EC-SOD levels and severity of micro and macrovascular complications | [61] |
| T2DM PN(−)/CAN(−) = 62; T2DM PN(+)/CAN(−) = 105; TDM PN(+)/CAN(+) = 22; C = 85 | 8isoPGF2α (antioxidant capacity – •O2−); (antioxidant capacity –ONOO−); Vit E/Lip; Vit C | PN(+)/CAN(−) vs. C: ↑ 8isoPGF 2α, •O2−; ↓ Vit E/Lip, Vit C and ONOO−; PN(+)/CAN(+) vs. C:↑ •O2−; ↓ Vit E/Lip and ONOO−; PN(+)/CAN(+) vs. PN(−)/CAN(−): ↑ •O2−; PN(+)/CAN(−) vs. PN(−)/CAN(−): ↑ •O2−; ↓ Vit E/Lip, Vit C | [40] |
| T1DM = 61; T2DM = 124; C = 70 | TAC | ↓ TAC in the presence of PN and/or CAN | [62] |
| T2DM(+)DSP N = 16; T2DM(−) DSPN = 16; C = 19 | CAT, SOD, TAS, •NO, DNA oxidative damage (Comet assay, endonuclease assay) | T2DM(+)DSPN vs. C: ↓ SOD, ↓ GPx, ↓ •NO, ↑ DNA damage; T2DM(−)DSPN vs. C: ↓ •NO, ↑ DNA damage; T2DM(+)DSPN vs. T2DM(−)DSPN: ↑ DNA damage | [63] |
| T2DM = 10 (Incubation of erythrocytes with insulin); T2DM = 14 (During clamp); C = 24 | GSH/GSSG; TBARS; During hyperinsulinemic euglycemic clamp; GSH/GSSG; Erythrocytes incubation | D vs. C: ↑ GSH/GSSG; ↑ GSH/GSSG—after 2 hours of insulin incubation; ↑ GSH/GSSG—crescent after 60 and 120 min (during clamp); TBARS—no alteration All vs. | [64] |
| T2DM = 55; T2DM(−)LPO = 29; 2DM(+)LPO = 25; Pre-DM = 9;C = 29 | SOD, CAT, GPx, uric acid, SH, CER, TRF, TBARS | T2DM vs. Pre-DM and C: ↑ SOD, ↑TBARS; T2DM and Pre-DM vs. C: ↑TRF; T2DM(+)LPO and Pre-DM vs. C: ↑TRF; T2DM(+)LPO vs. Pre-DM and C: ↑SOD; T2DM(+)LPO vs. T2DM(−)LPO, Pre-DM and C: ↑ FG, ↑ HbA1c | [65] |
| T2DM(+)Iron. def = 30; T2DM(−)Iron.def = 30; C = 30 | MDA, uric acid | T2DM(+)Iron.def vs. T2DM(−)Iron.def: ↑MDA, ↓ uric acid; T2DM(+)Iron.def vs. C: ↑ MDA, ↓ uric acid | [66] |
| T1DM= 95; T2DM= 30; C= 20 | GSH, GSH-red, GPx, SOD, MDA | T2DM vs. C: ↑ SOD, ↑ MDA; T1DM vs. C: ↑ SOD, ↑ MDA | [67] |
| T2DM= 36; C= 37 | SOD, serum and erythrocyte Zn | ↑ SOD, ↑ Zn | [68] |
| T2DM = 80; C = 80 | MDA, GSH, SOD | ↑ MDA, ↑ SOD, ↓ GSH | [69] |
| T2DM = 15; C = 18 | TAC, residual antioxidant activity, MDA, albumin, uric acid | ↑ TAC, ↑ residual antioxidant activity, ↑ MDA, ↑ acid uric | [70] |
| T2DM = 35; Pre-DM = 8; C = 119 | 8-OHdG | T2DM vs. Pre-DM and C: ↑ 8-OHdG; Pre-DM vs. C: ↑ 8-OHdG | [71] |
| T2DM = 69; T2DM(−)C = 20; T2DM(+)C = 49 C = 20 | HEL, AOPP, 8-OHdG, 15- F2t-IsoP, PON 1 | T2DM vs. C: ↑ AOPP, ↑ 8-OHdG, ↑ 15-F2t-IsoP, ↓ PON 1; T2DM(+)C vs. T2DM(−)C: ↑ AOPP, ↑ 8-OHdG, ↓ PON 1 | [72] |
| IGR = 16; T2DMrecent = 34; C = 27 | GSH, MDA, SOD, TAC, comet assay | T2DMrecent vs. IGR and C: ↑MDA, ↑ DNA damage, ↓ TAC; IGR vs. C: ↑ DNA damage; T2DMrecent and IGR vs. C: ↓ SOD | [73] |
| D = 35; NPDR = 29; PDR = 40; C = 32 | MDA; NOx; SOD; GPx | D vs. C: ↑ MDA, ↑ NOx, ↑ SOD; NPDR vs. C: ↑ MDA, ↑ NOx; NPDR vs. D: ↑MDA, ↑ NOx, ↓ SOD, NPDR vs. PDR: ↑ MDA, ↑ NOx, ↓ SOD; PDR vs. C: ↑ MDA, ↑ NOx, ↓ SOD, ↓ GPx; PDR vs. D: ↑ MDA, ↑ NOx, ↓ SOD, ↓ GPx | [74] |
| DC(+) = 69; DC(−) = 48; C = 42 | SOD, GPx, GR, TAS, Bilirubin, uric acid | DC(+) vs. C: ↓ SOD, GPx and GR, TAS, ↑ Bilirubin and uric acid; DC(+) vs. DC(−): ↓ SOD, GPx and GR, TAS | [75] |
| T2DM = 50; C = 21; T2DM(−)C = 29; T2DM(+)C = 21 | CER, TRF, MDA | T2DM vs. C: ↑ CER, ↑ MDA, ↓ TRF; T2DM(+)C vs. C: ↑ CER, ↑ MDA, ↓ TRF; T2DM(+)C vs. T2DM(−)C: ↑ CER, ↑ MDA | [76] |
| T2DMrecent = 20; T2DM = 20; C = 20 | Serum MDA, MDA after oxidation, erythrocyte MDA, total-SH, GSH, uric acid | T2DMrecent vs. C: ↑ Serum MDA, ↑ MDA after oxidation, ↑ erythrocyte MDA, ↓ GSH, ↓ total-SH; T2DM vs. C: ↑ Serum MDA, ↑ MDA after oxidation, ↑ erythrocyte MDA, ↓ GSH, ↓ total-SH; T2DM vs. T2DMrecent: ↑ MDA after oxidation, ↑ erythrocyte MDA | [77] |
| T2DM = 68; T1DM = 12; C = 30 | TBARS | DM vs. C: ↑TBARS | [78] |
| T1DM = 12; C = 5 | Plasma GSH, GSSG GSH/GSSG, and MDA | T1DM vs. C: ↑ GSSG, ↑ GSSH/GSH ↑ MDA | [79] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De M. Bandeira, S.; Da Fonseca, L.J.S.; Da S. Guedes, G.; Rabelo, L.A.; Goulart, M.O.F.; Vasconcelos, S.M.L. Oxidative Stress as an Underlying Contributor in the Development of Chronic Complications in Diabetes Mellitus. Int. J. Mol. Sci. 2013, 14, 3265-3284. https://doi.org/10.3390/ijms14023265
De M. Bandeira S, Da Fonseca LJS, Da S. Guedes G, Rabelo LA, Goulart MOF, Vasconcelos SML. Oxidative Stress as an Underlying Contributor in the Development of Chronic Complications in Diabetes Mellitus. International Journal of Molecular Sciences. 2013; 14(2):3265-3284. https://doi.org/10.3390/ijms14023265
Chicago/Turabian StyleDe M. Bandeira, Suziy, Lucas José S. Da Fonseca, Glaucevane Da S. Guedes, Luíza A. Rabelo, Marília O. F. Goulart, and Sandra Mary L. Vasconcelos. 2013. "Oxidative Stress as an Underlying Contributor in the Development of Chronic Complications in Diabetes Mellitus" International Journal of Molecular Sciences 14, no. 2: 3265-3284. https://doi.org/10.3390/ijms14023265