The dynamical potential of
H (
qi,
pi,
P) is the effective environment in which the
qi coordinate stays for each P in a certain molecule. This is achieved by calculating the maximal and minimal energies by varying
pi for each
qi under the condition that n
2 and n
3 are nonnegative. The dynamical potential composed of these maximal and minimal energies as a function of
qi is represented by a closed curve in which the quantal levels are enclosed. The dynamical potential also defines the
qi region for each level it encloses. Furthermore, the points in the dynamical potential corresponding to ∂
H ∂
qi= 0 are fixed points in the dynamical space [
5]. In this section, two main parts will be addressed as follows: (1) the dynamical potential influences of the H–O stretching model on the H–O–B bending model and the O–Br stretching model and (2) for instance, the phase space trajectories, the action integrals, and quantal environments of all energy levels in the dynamical potentials when
P is equal to 27 will be studied.
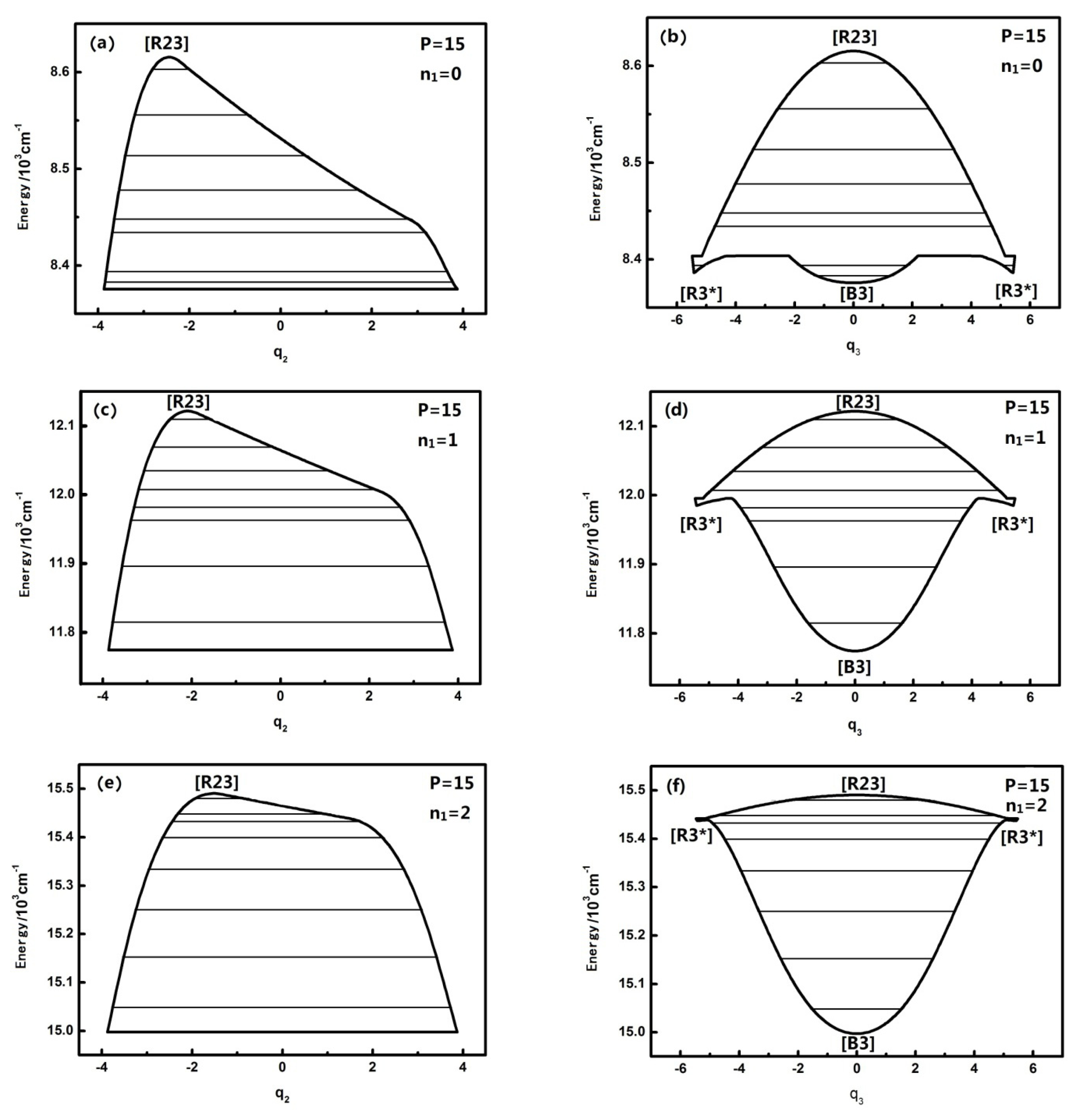
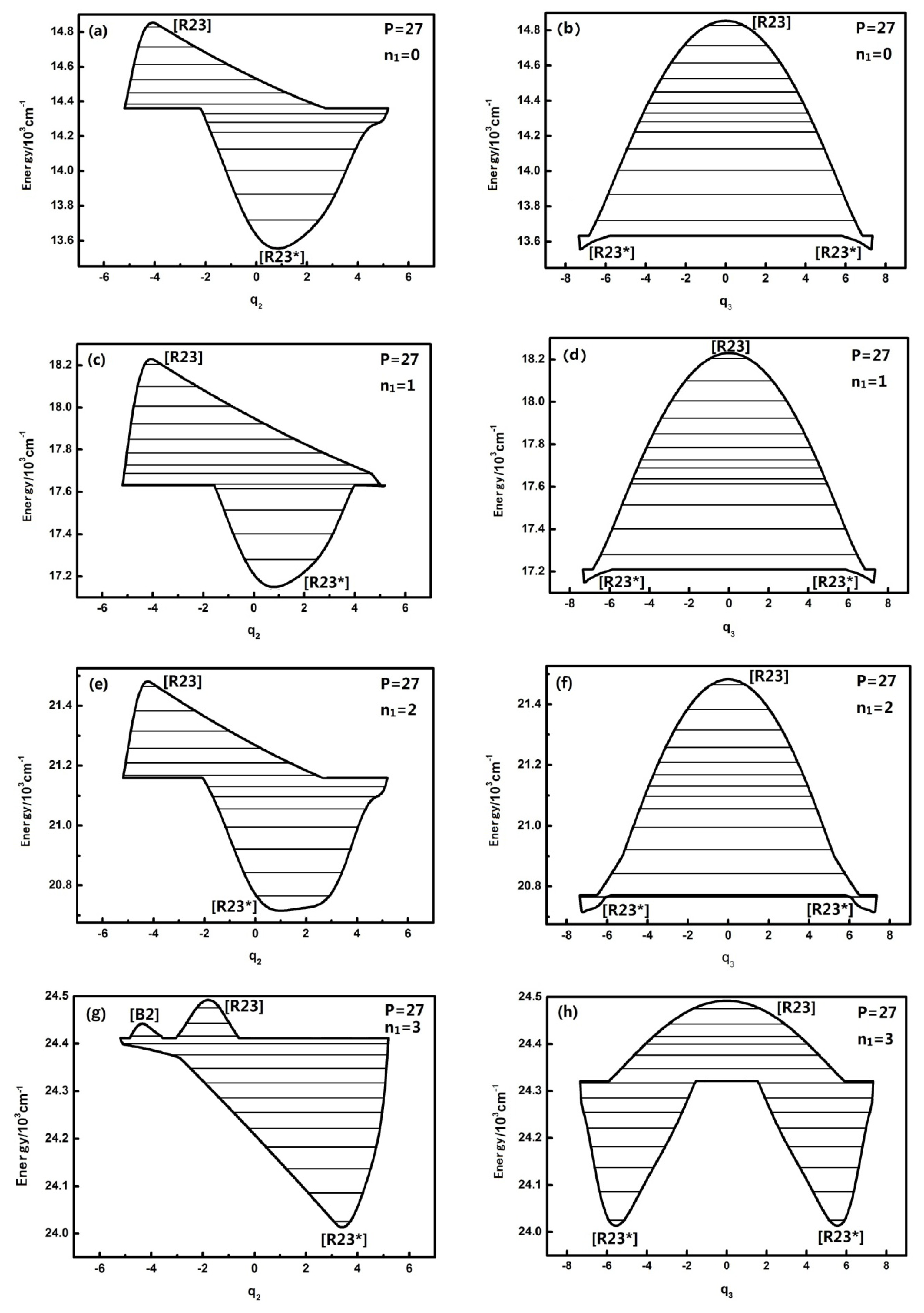
3.1. The Study of Dynamical Potentials and Their Characteristics of Different Quantum Numbers n1 Corresponding to Typical Polyad Numbers in HOBr
The dynamical potentials of HOBr (in coordinates
q2,
q3,) with
n1 = 0,1,2,3 of different P numbers is considered, and their levels can be calculated with the aforementioned Hamiltonian (ground state from the potential energy at the bottom of 2764.0 cm
−1). It is found that the dynamical potentials are different for small and large
P numbers. The dynamical potential is shown as follows (the path marked by the fixed point is in accordance with the literature [
13]) when
P is equal to 15:
Figure 1 shows that when
n1 is equal to 0,1,2,3, the dynamical potentials in the
q2 coordinates are all simple inverse Morse potentials, which shows that the n
1 mode has little effect on the dynamical potentials of the
q2 coordinates. In the theory of the dynamical potential [
9] it is noted that, for an inverse Morse potential, the stability of the lowest energy level is the worst in the inverted Morse potential corresponding to a certain
P, which indicates that H–O stretching has no influence on the stability of the energy level corresponding to the H–O–Br bending vibration mode under the same
P number. In the sense of the geometrical shape of the dynamical potential, it is found that the top of dynamical potentials only gradually flattens with the increase of n
1, which shows that the n
1 mode has almost no significant effect on the coupling of
n2 and
n3. In contrast, the dynamical potentials of HOBr in the
q3 coordinate consist of an inverted Morse potential and a positive Morse potential, and with the increase of
n1, the top of the inverted Morse potential gradually flattens and the positive Morse potential wells become deeper, which shows that the dynamical potential is turning into an almost pure positive Morse potential, especially when
n1 is equal to three. As a result, the stability of the high energy levels becomes worse while the original fixed-point still remains the same. All the above phenomena show that, although the n
1 mode is not coupled with the
n2 and
n3, it still has some effect on the resonant coupling of the other two modes, and this impact is reflected in the stability of the stretching mode (when
n1 is equal to three, this impact is particularly evident). Or said differently, the H–O stretching mode has an important influence on the dynamics of the other degrees of freedom. According to qualitative analysis, this is because the intramolecular vibrational relaxation (IVR), caused by the resonance between
n2 and
n3, which enhances the stability of the energy levels [
9], is weakened in the high-energy region. In the previous literature, the uncoupling modes in molecules have hardly ever been considered in the study of molecular dynamical behavior, and in this work, it is shown that the previous cognition maybe not comprehensive [
2,
3].
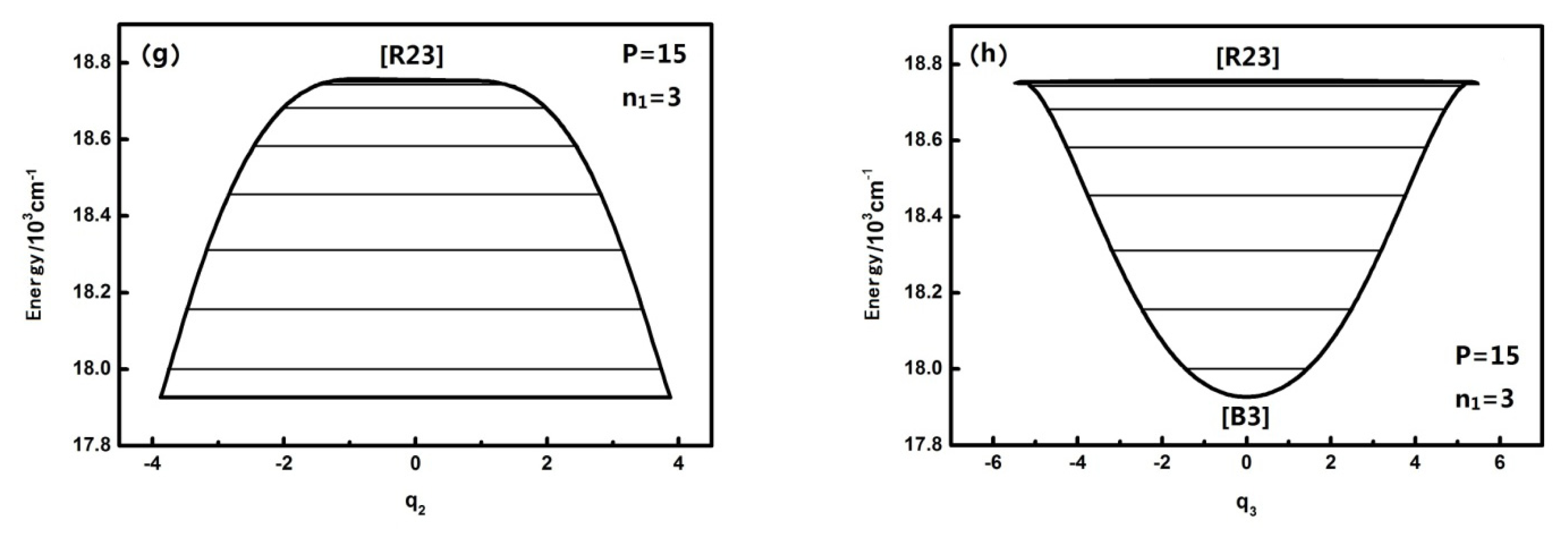
For larger P numbers, the change of the dynamical potentials is much more complicated. The dynamical potentials are shown as follows when P is, for instance, equal to 27:
When n1 is equal to three, the geometrical shape of the dynamical potentials of both q2 and q3 becomes increasingly complex and the number of the potential wells also increases. There are two inverted Morse in the q2 dynamical potentials, yet there are two positive Morse in the q3 dynamical potentials. This shows that with the increase of the n1 mode, the dynamical potential system consists of n2 and n3, which makes it more complex. This conclusion confirms once again that the n1 mode has some effect on the dynamical behavior of the other mutually coupled resonance modes and that this effect is more apparent compared with the case of P = 15, which shows that the uncoupling mode becomes much more important to the molecular dynamics behaviors with Polyad number.
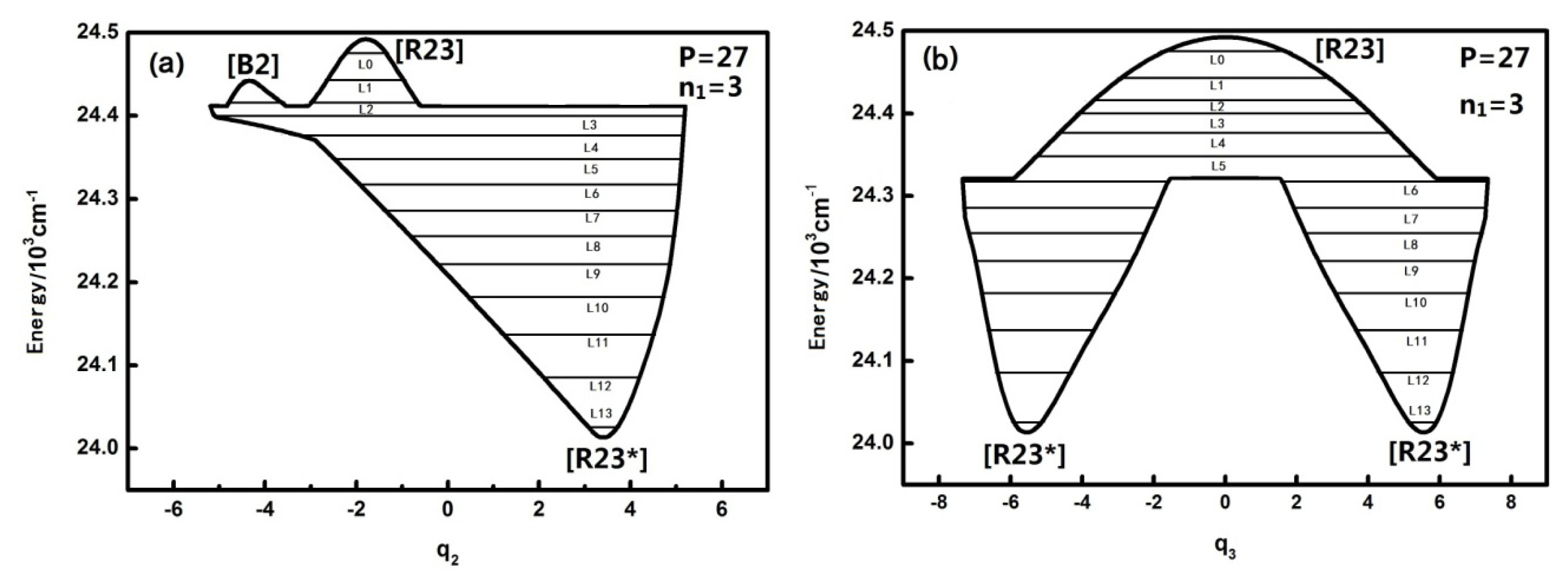
Figure 2 also shows that the
q2 dynamical potential with
n1 = 3 is more complex than the one with
n1 = 0,1,2 and that the [
B2] fixed point appears. This fixed point leads to the vibration modes corresponding to the highest three energy levels in the dynamical potential becoming localized vibrational when
n1 is equal to three (in the two inverted Morse potentials). In addition, the fixed points of the dynamical potentials in
q3 remain the same, but when
n1 is equal to three, the vibration mode corresponding to the low energy level becomes localized vibrational (in the two positive Morse potentials). Compared with the dynamical potentials when P is equal to 15, it is obvious that the effect of n
1 on
n2 (or
n3’s resonant coupling) is closely related to the value of the P number. The general conclusion is that n
1 has little effect on the geometrical shape of the dynamical potential of
q2,
q3 when the P number is small, however, this effect becomes larger when the
P number is larger.
In the above-mentioned study, it was found that although the H–O stretching mode was not coupled with the H–O–Br bending mode and the O–Br stretching mode, it still affected the resonant coupling of the two modes, thereby affecting the dynamics of HOBr. It is elucidated with qualitative analysis that the geometrical shape of the dynamical potentials and the corresponding fixed points are sensitive to the change of the H–O stretching mode in the sense of geometry. However, this phenomenon and the related explanation should be verified in further studies of other molecular systems.
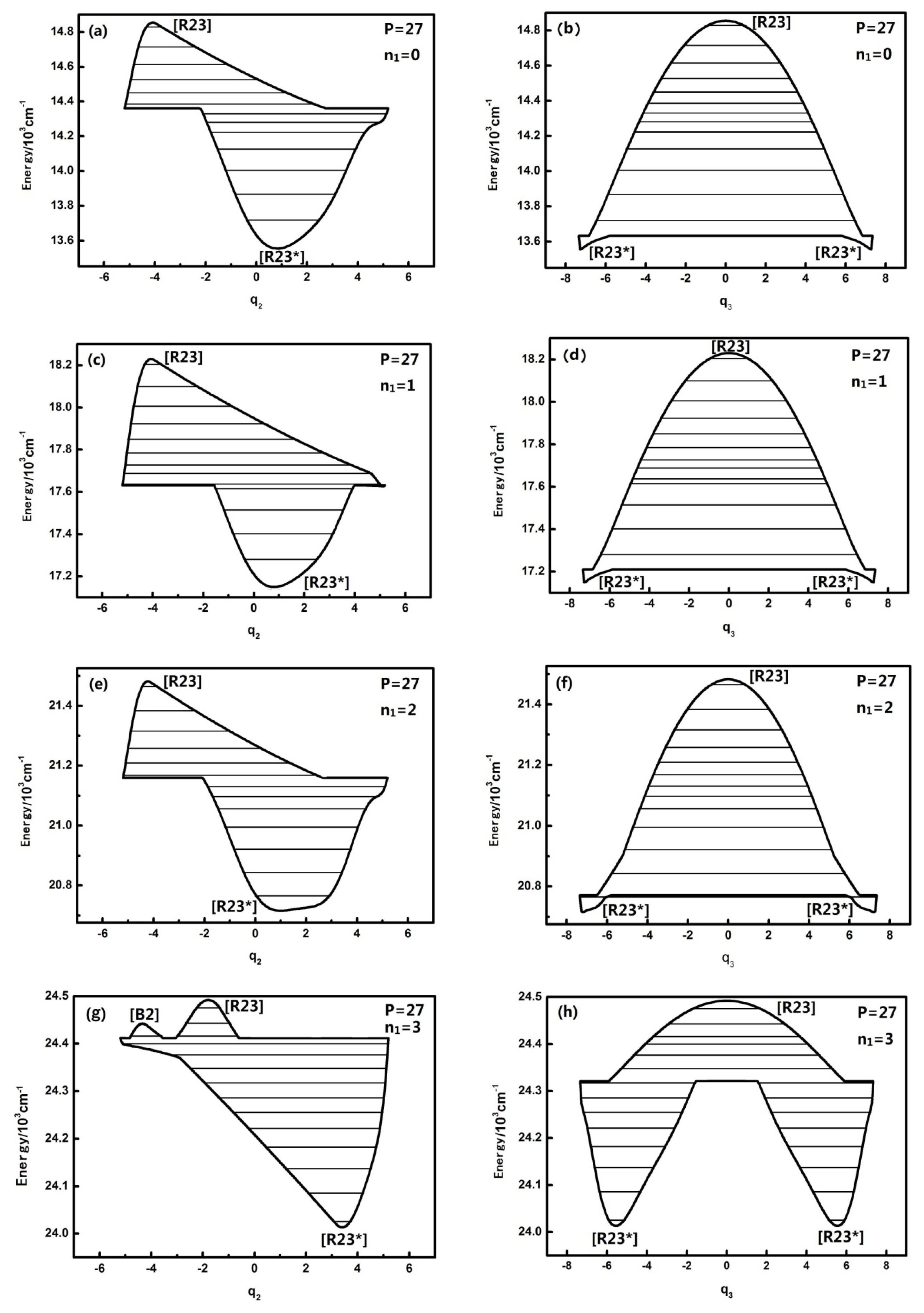
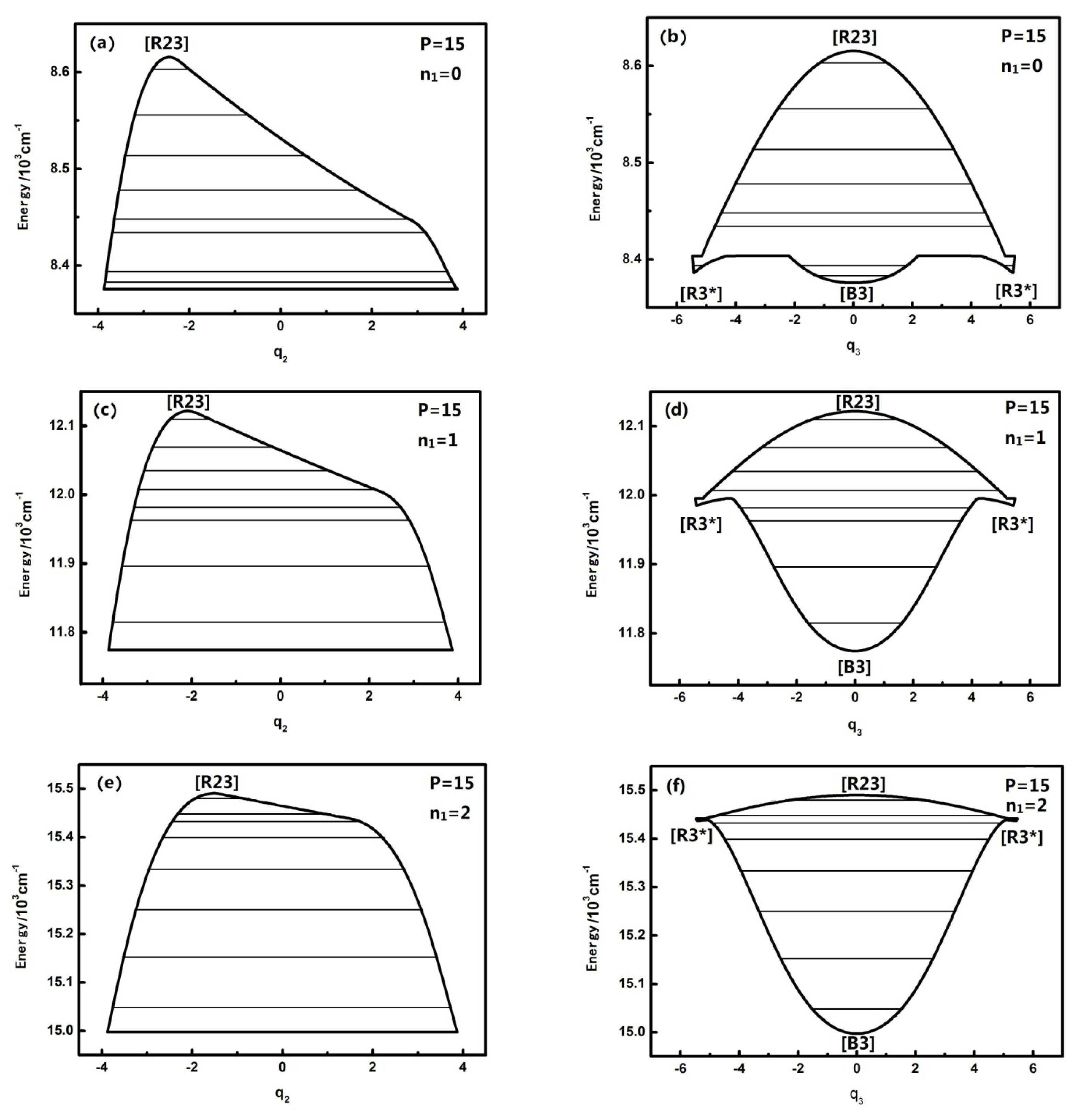
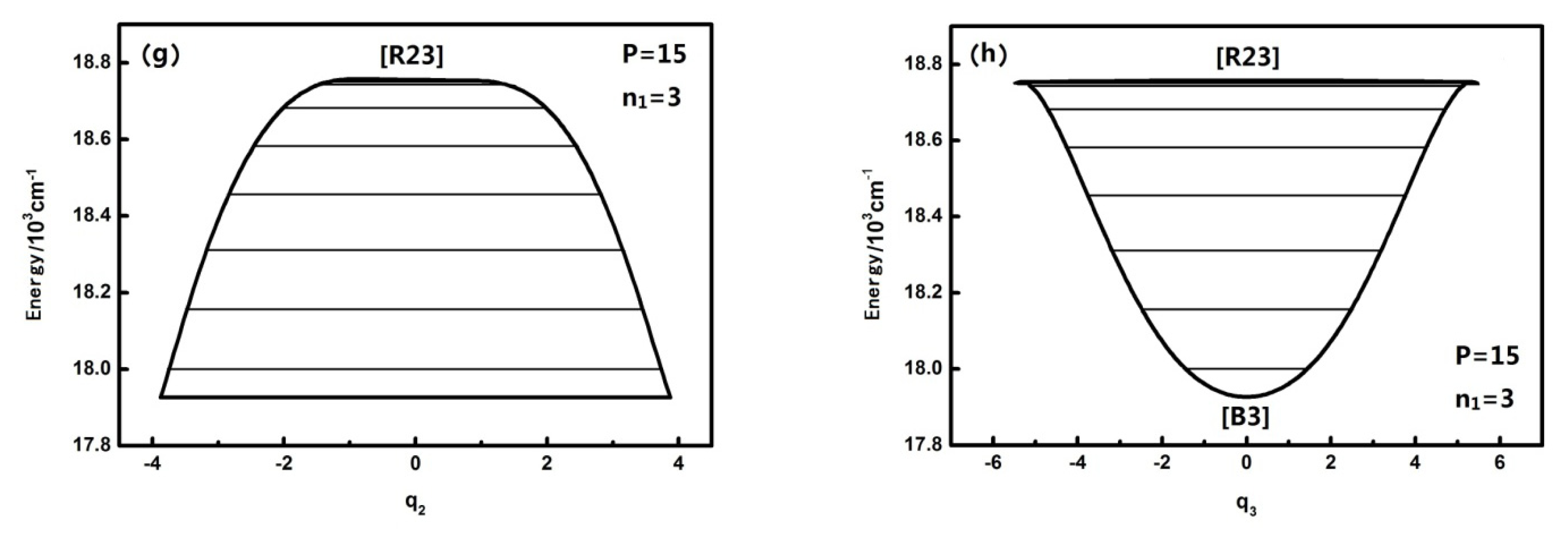
3.2. The Nature and Quantal Environment of the Level of a Specific Polyad Number (P = 27)
The qualitatively different quantum number n
1 of the HOBr system corresponding to dynamical potentials and characteristics are discussed in the previous section. Next we will discuss quantitatively the dynamical potentials of the specific
P numbers with the analysis of the phase space trajectories and the action integral of every energy level. For instance, the dynamical potentials of HOBr with
P = 27 are shown in
Figure 3a,b in
q2 and
q3 coordinates, where the horizontal lines show the energy levels sharing the designated P. The reason why the case of
P = 27 is singled out for discussion is that this case is quite representative and possesses the most fruitful characteristics. The dynamic potentials obtained when
n1 is equal to three are as follows:
The energy is with respect to the ground state which is 2764.0 cm
−1 above the bottom of the potential well, and the points in
Figure 3 designated by [B], [R], [R*] are stable fixed points where both ∂
H/∂
qi and ∂
H/∂
pi (
i = 2 or 3) are zero [
10–
13]. It is noted that [R23] is stable for the upper realm of the dynamical potential because the potential curve is inverse (upside down) and low energy regions have a positive potential at the bottom, which indicates that [R23*] is a stable fixed point. Similarly, in
Figure 3a, [B2] is also a stable fixed point. Generally, dynamical potentials are considered to be able to find all of the fixed points by the two coordinates, and it is easy to find fixed points from intuitive geometry with the dynamics potential, which provides a convenient way to study the dynamics of a system.
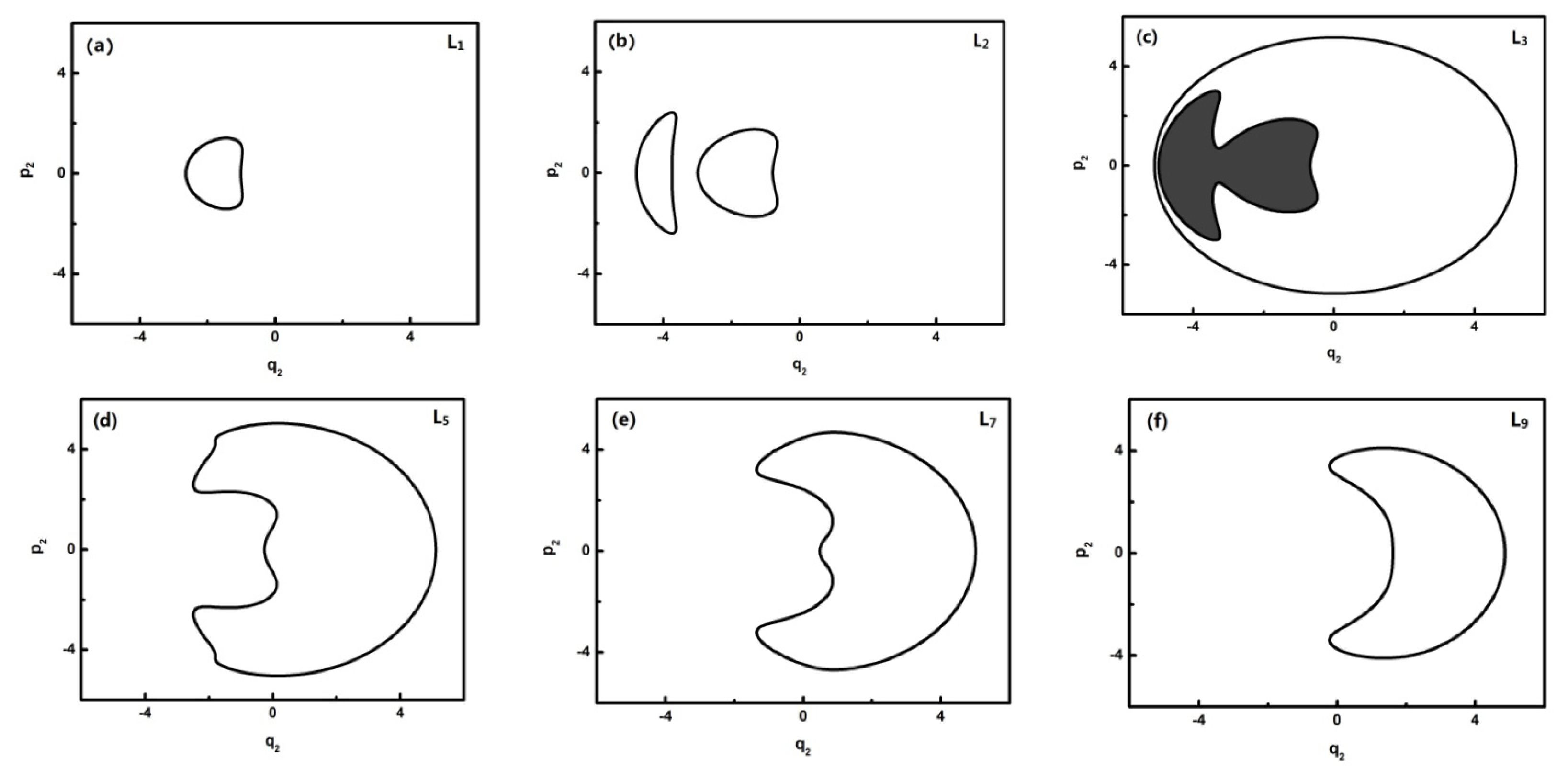
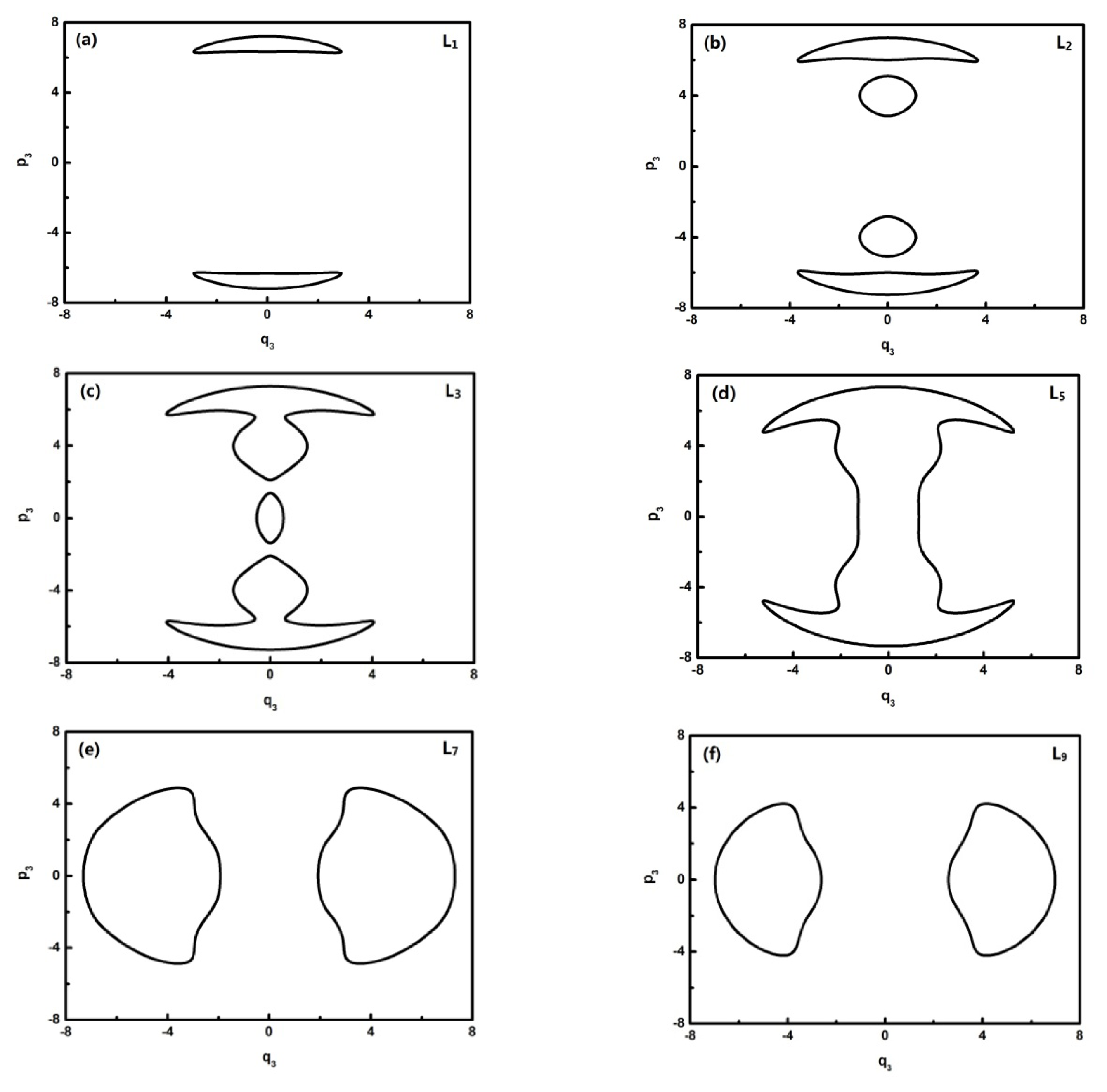
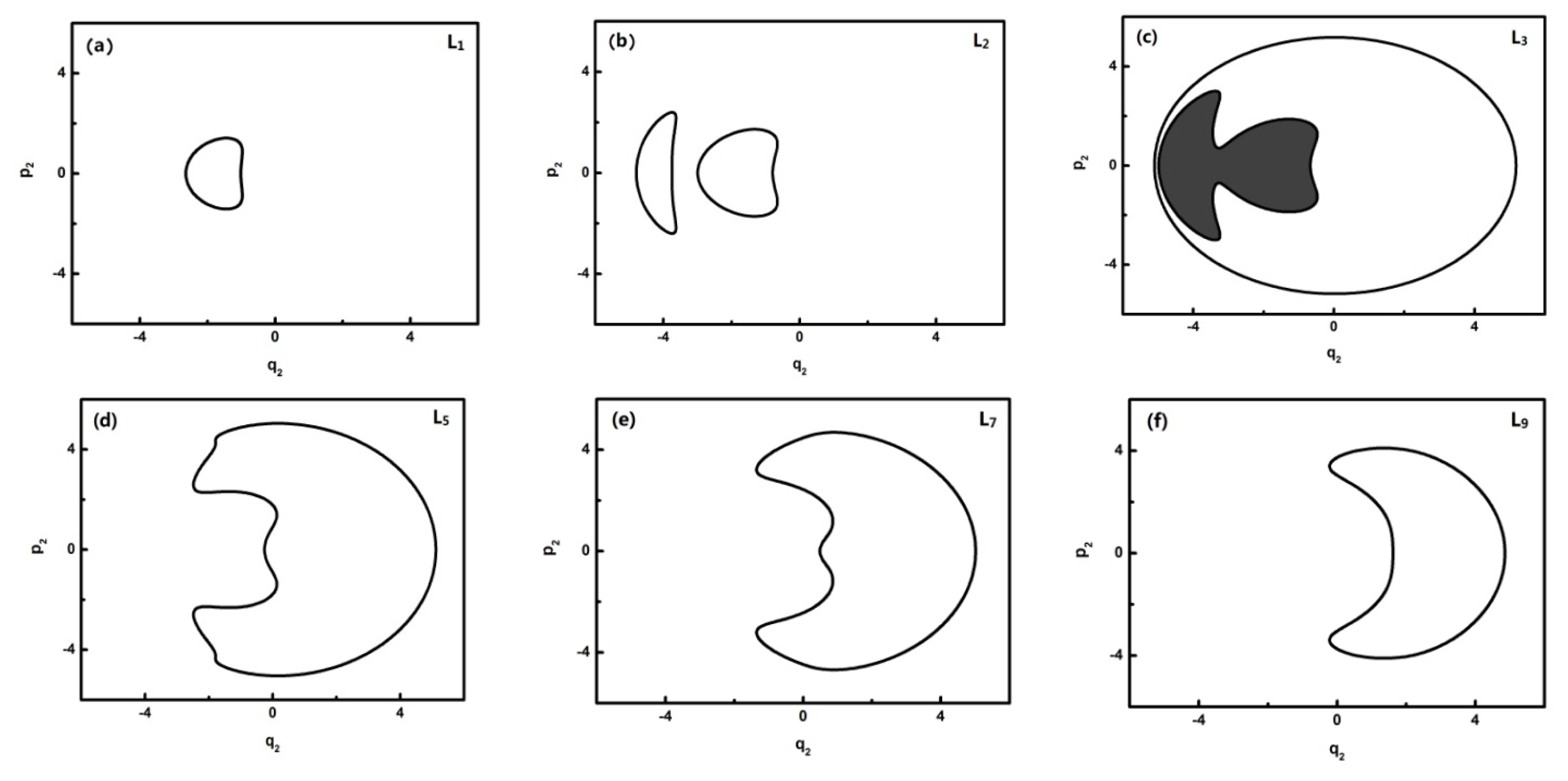
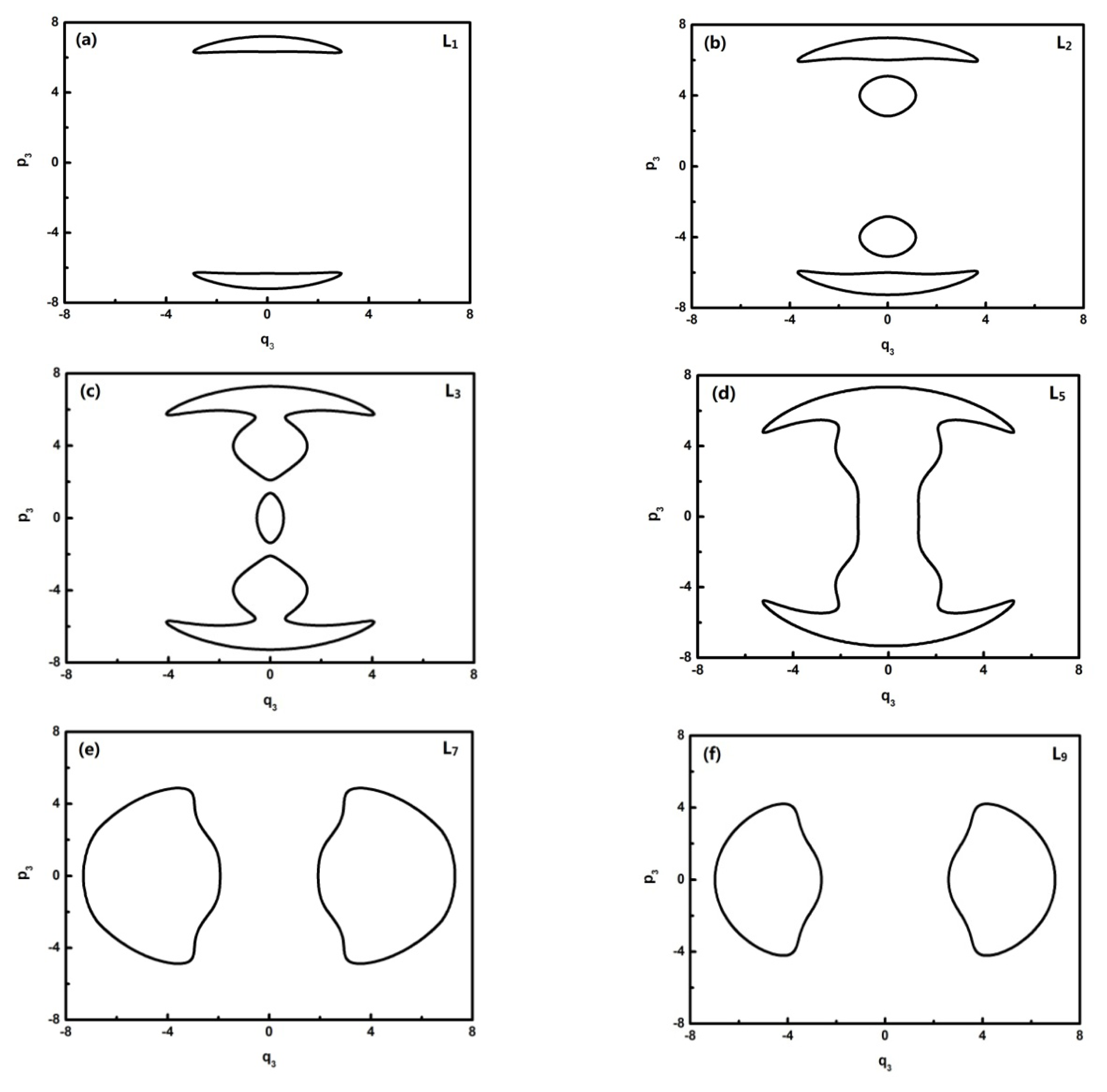
To further quantitatively analyze the features of related energy levels, the representative trajectories of phase space in
pi −
qi of each energy level is obtained. From the geometric properties of the pattern, the trajectory of phase space can be classified into different sets. It is found that the trajectories of L0–L1, L2, L3, L4–L8, L9–L13 in
q2 coordinates constitute a class and the ones of the L0–L1, L2, L3, L4–L6, L7–L13 corresponding levels, respectively, constitute a class in
q3 coordinates. They are partly shown in
Figures 4 and
5:
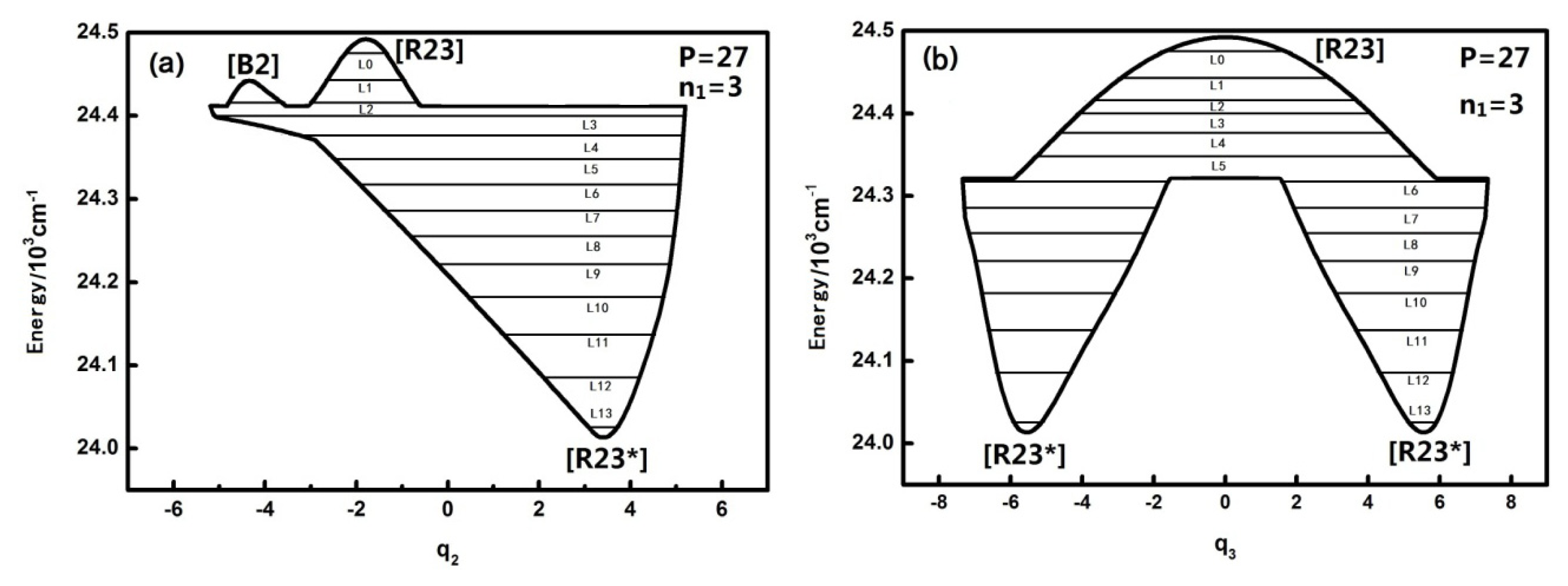
In
Figure 4, L0–L2 are in inverted Morse potentials and the area of the trajectory of the phase space increases with the reduction of energy. L3–L13 are in a positive Morse potential and the area of the trajectory of the phase space decreases with the reduction of energy. It is also found that the trajectory of the phase space of L2 is divided into two separate trajectories, of which L2 is located at the double-well potential in the dynamical potential shown in
Figure 3.
In
Figure 5, L0–L6 are in inverted Morse potentials and the area of the phase diagram increases with the reduction of energy. L7–L13 are in the positive Morse potential and the area of the phase diagram decreases with the reduction of energy. The trajectories of the phase space of L0–L1 are composed of two symmetrical parts (the upper one and lower one) because there is a positive and negative direction of momentum, and particularly, the trajectory of the phase space of L2 is composed of four symmetrical parts due to a multiplied effect of positive/negative directions of momentum on the energy level located in the double-well potential (
Figure 3a). There is some difference in the phase space of L3, and it is shown that the trajectory is composed of three parts caused by an approximate harmonic vibration. Similarly, there are two parts of the trajectory in the phase space of L7–L13 because they are located in the two potential wells. Through the above-mentioned analysis, we found that the trajectories of the phase space can more intuitively reflect the dynamical characteristics of the vibration mode.
In order to study the quantal environment of each energy level, the calculation of the action integral of the phase space trajectories (quantum number) is needed and the corresponding formula is as follows:
Table 2 shows the action integrals for the levels corresponding to
P = 27, calculated from the trajectories of the phase space in (
q2,
p2) and (
q3,
p3). From the action integrals calculated from (
q2,
p2), the levels can be grouped into two subsets: L0 to L2 and L3 to L13. For the former ones, the action integral increases almost in a constant with the decline of energy levels due to the inverse-Morse well where they are located, and for the latter ones [
15], the action integral decreases (also almost in a constant) with the decline of energy levels due to the positive Morse well where they are located. From the action integrals calculated from (
q3,
p3), the levels can be grouped into two subsets, also with constant increment (decrement) in their action integrals: L0 to L6 and L7 to L13. For the former ones, the action integral increases with the decline of energy levels due to the inverse-Morse well where they are located, but with minor deviation on L2, and for the latter ones, the action integral decreases with the decline of energy levels due to the positive Morse wells where they are located. The constant action integral increment/decrement demonstrates the compatibility of our classical treatment with the quantized levels and also shows that the levels stay in various dynamical environments, which are defined essentially by the classical fixed points though these energies belong to the same Polyad number. In addition, from the view of the (
q2,
p2) space, the difference between adjacent energy levels’ action integrals is almost always the same (approximately equal to one, but the L1 is an exception and should be further studied in the future). This value is equal to two if in the view of (
q2,
p2) because
P = 2
n2 +
n3, which is also shown in
Table 2. From the above results, it is elucidated that the action integral differential of adjacent energy levels nearly doubles that of (
q2,
p2), which corresponds exactly with the system of 2:1 Fermi resonance. The dynamics potential is actually the same as the trajectory connecting each fixed point, depicting the restrictive conditions in which individual energy levels are located.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}