Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
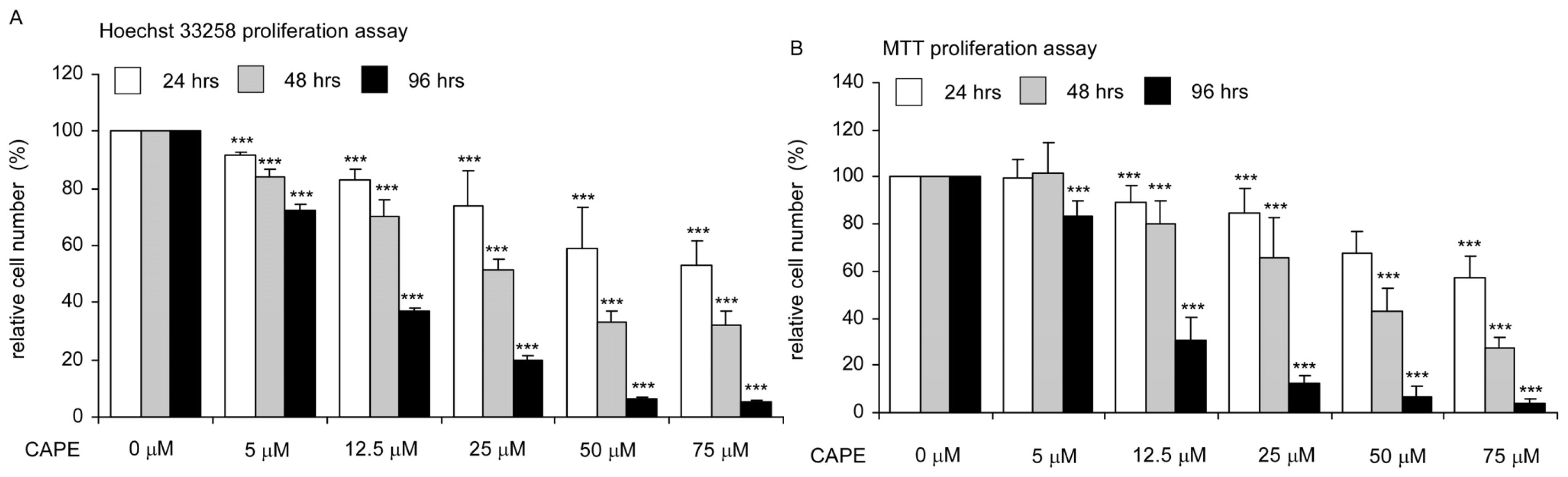
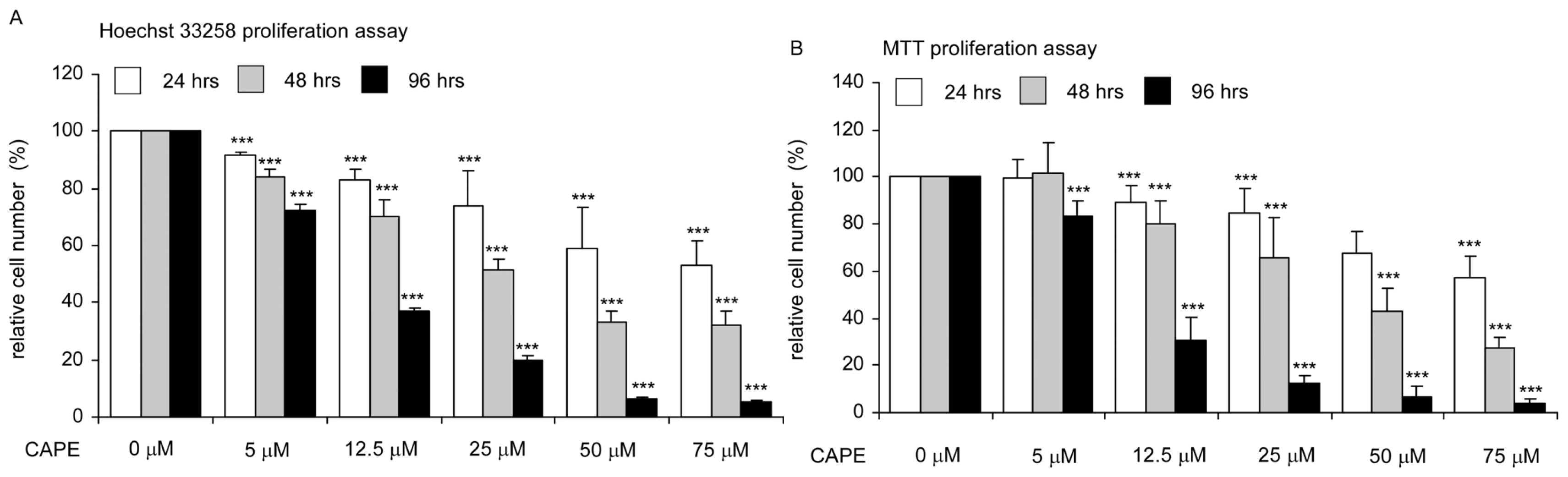
2.1. CAPE Treatment Suppressed the Proliferation and Survival of TW2.6 Human Oral Cancer Cells
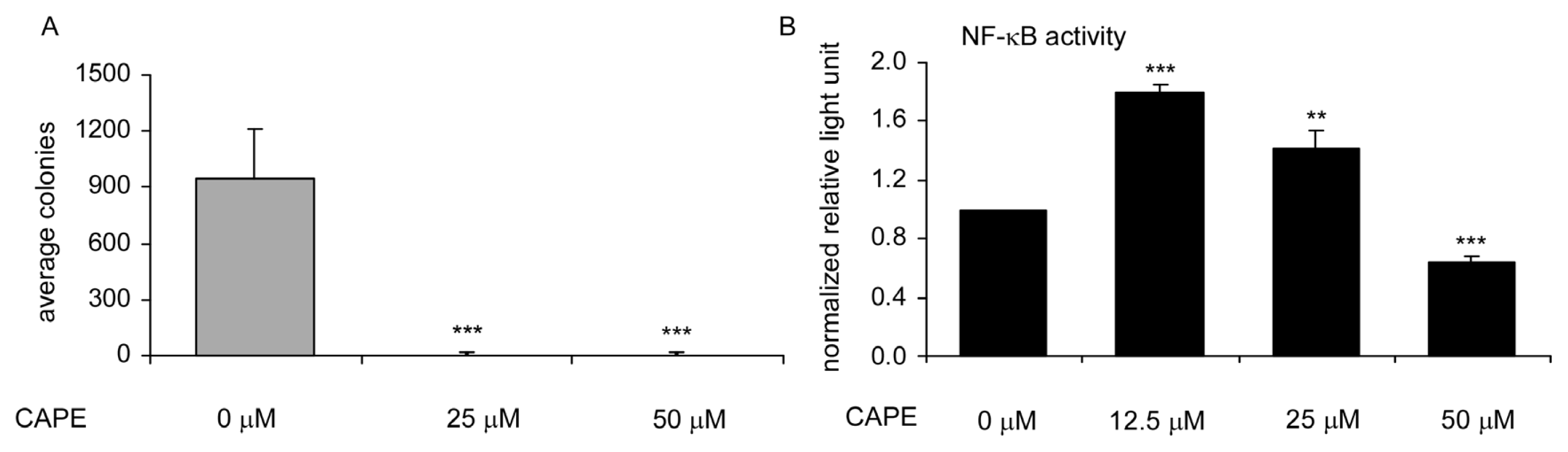
2.2. CAPE Treatment Suppressed TW2.6 Cells Soft Agar Colony Formation and NF-κB Activity in TW2.6 Cells
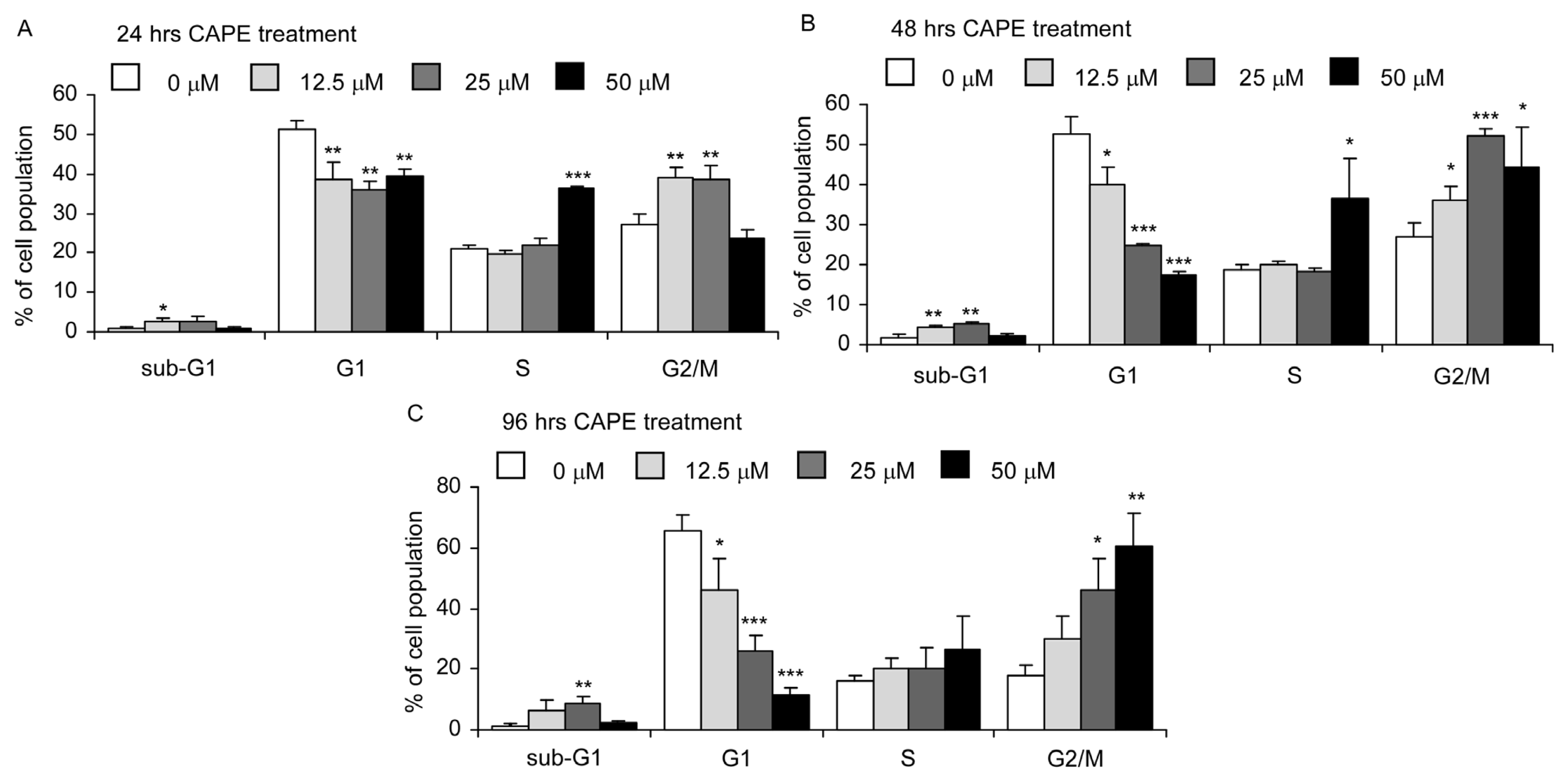
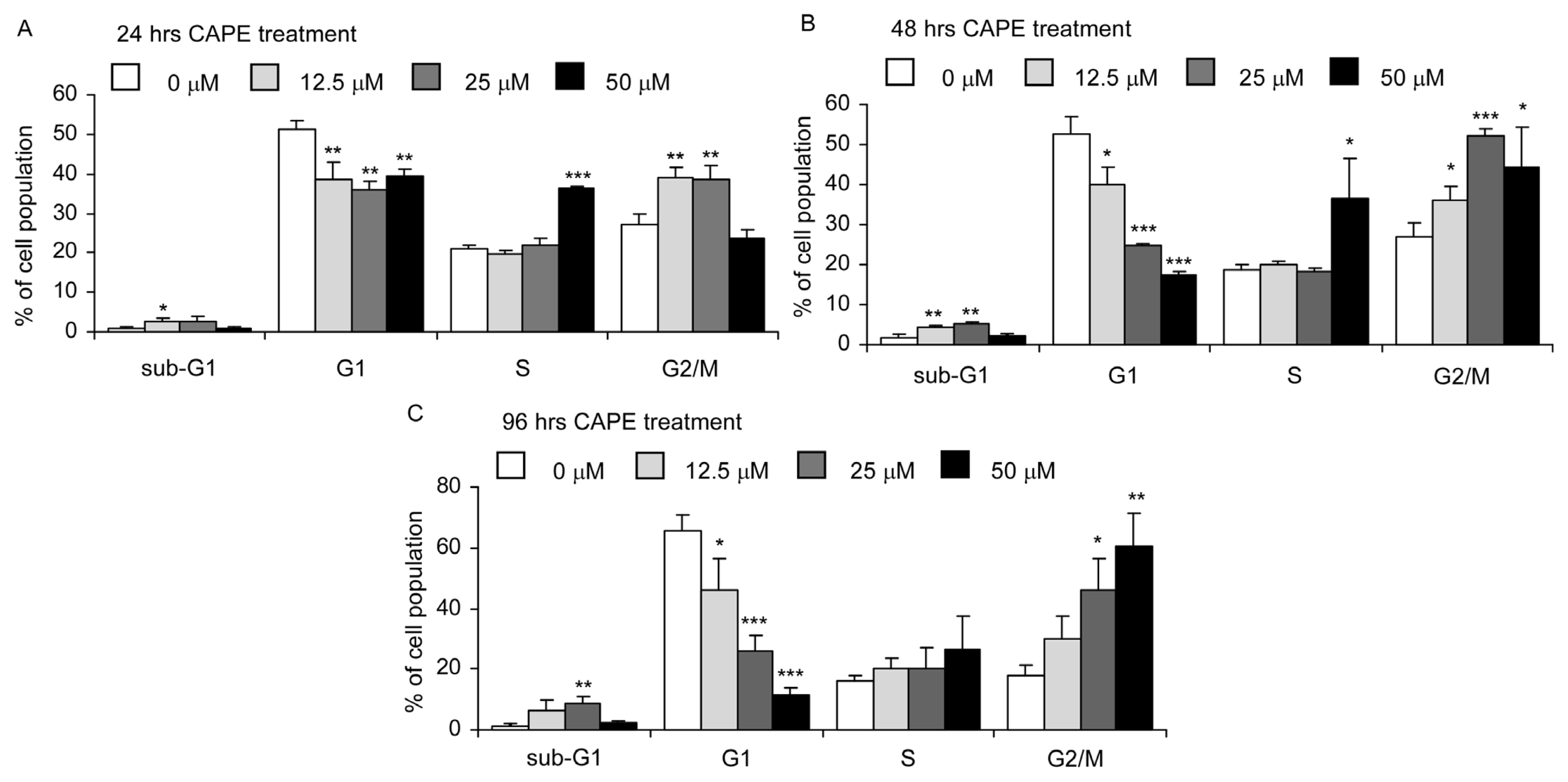
2.3. CAPE Treatment Caused Dysregulation of Cell Cycle
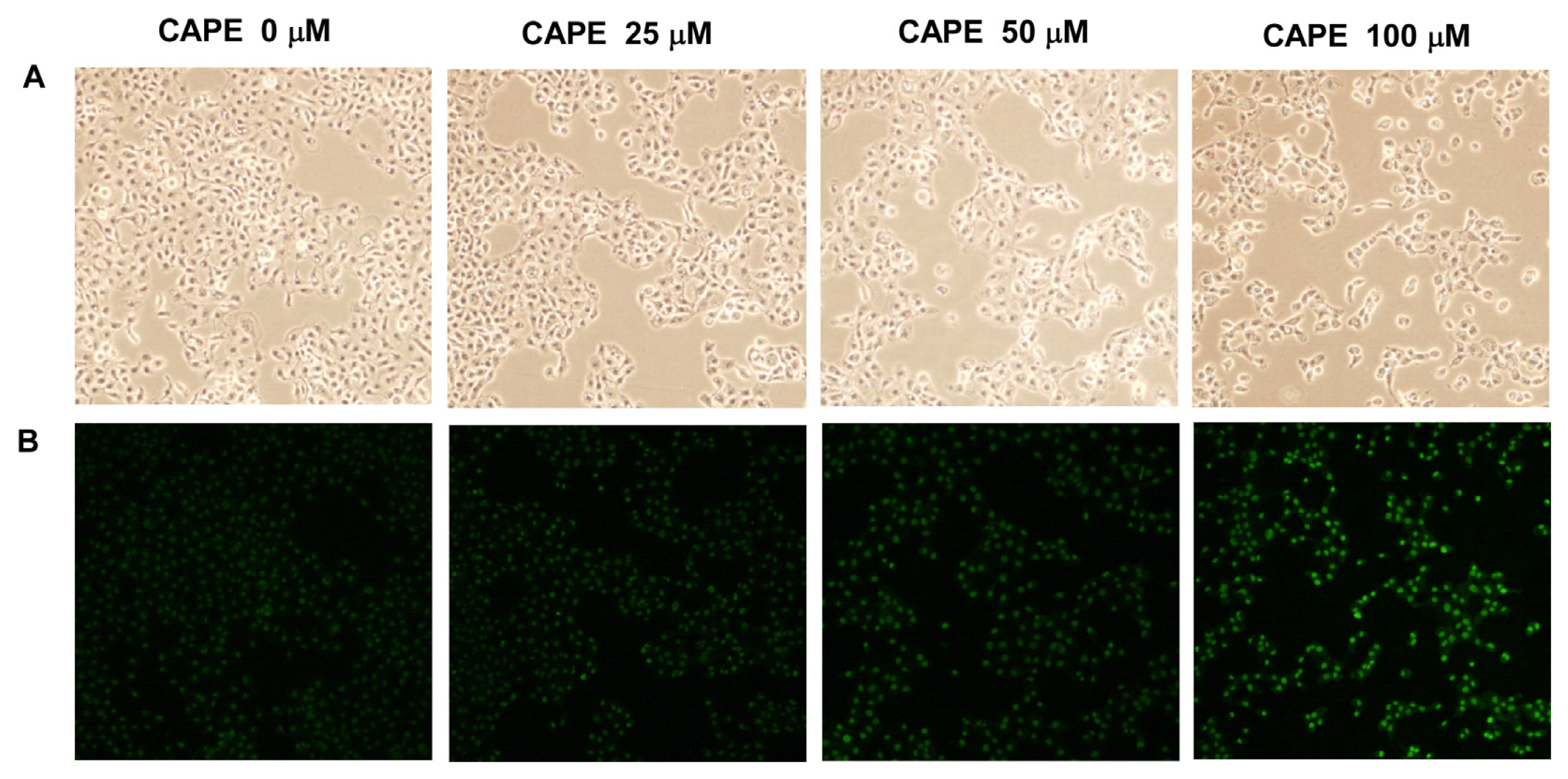
2.4. CAPE Treatment Induced Apoptosis in TW2.6 Cells
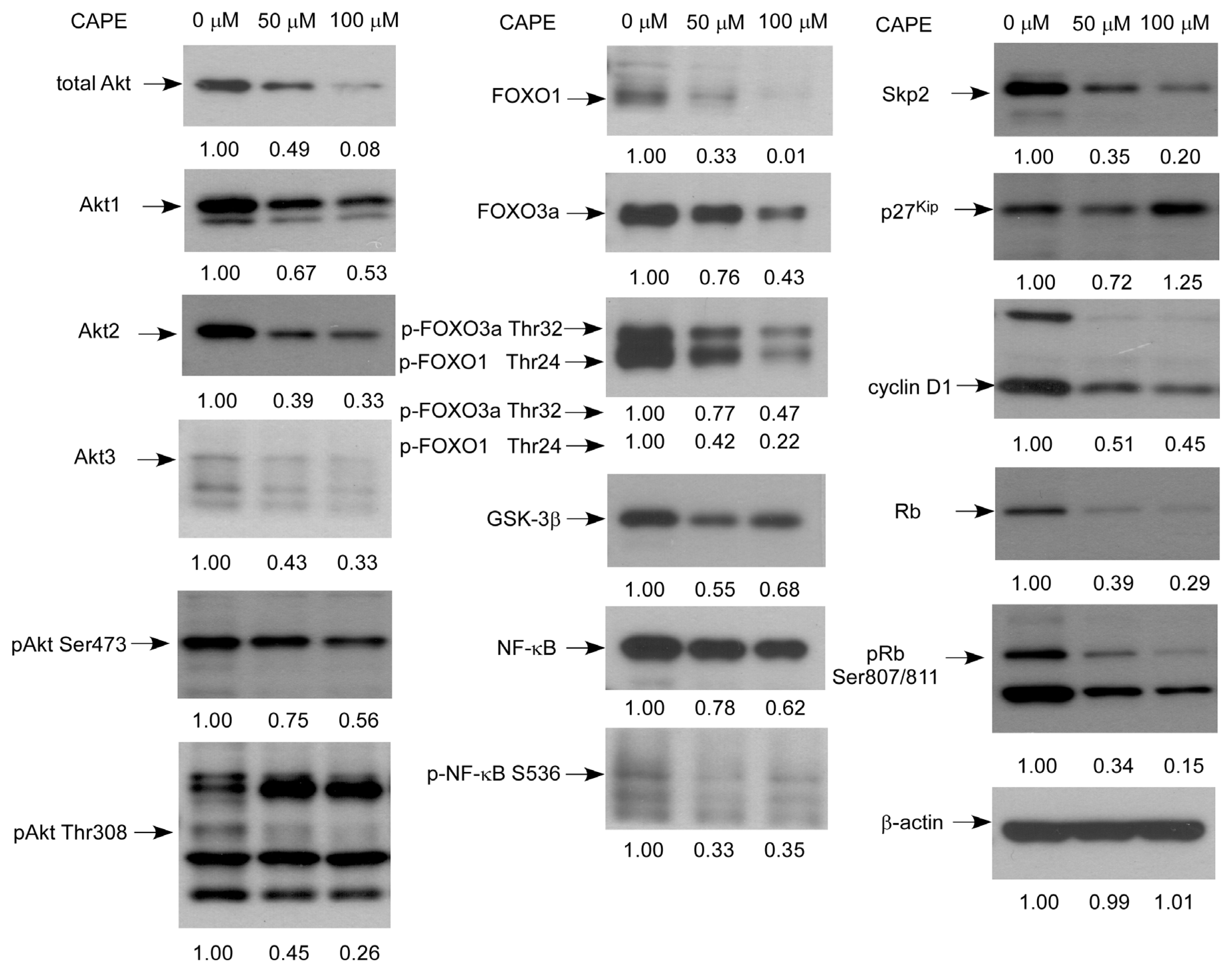
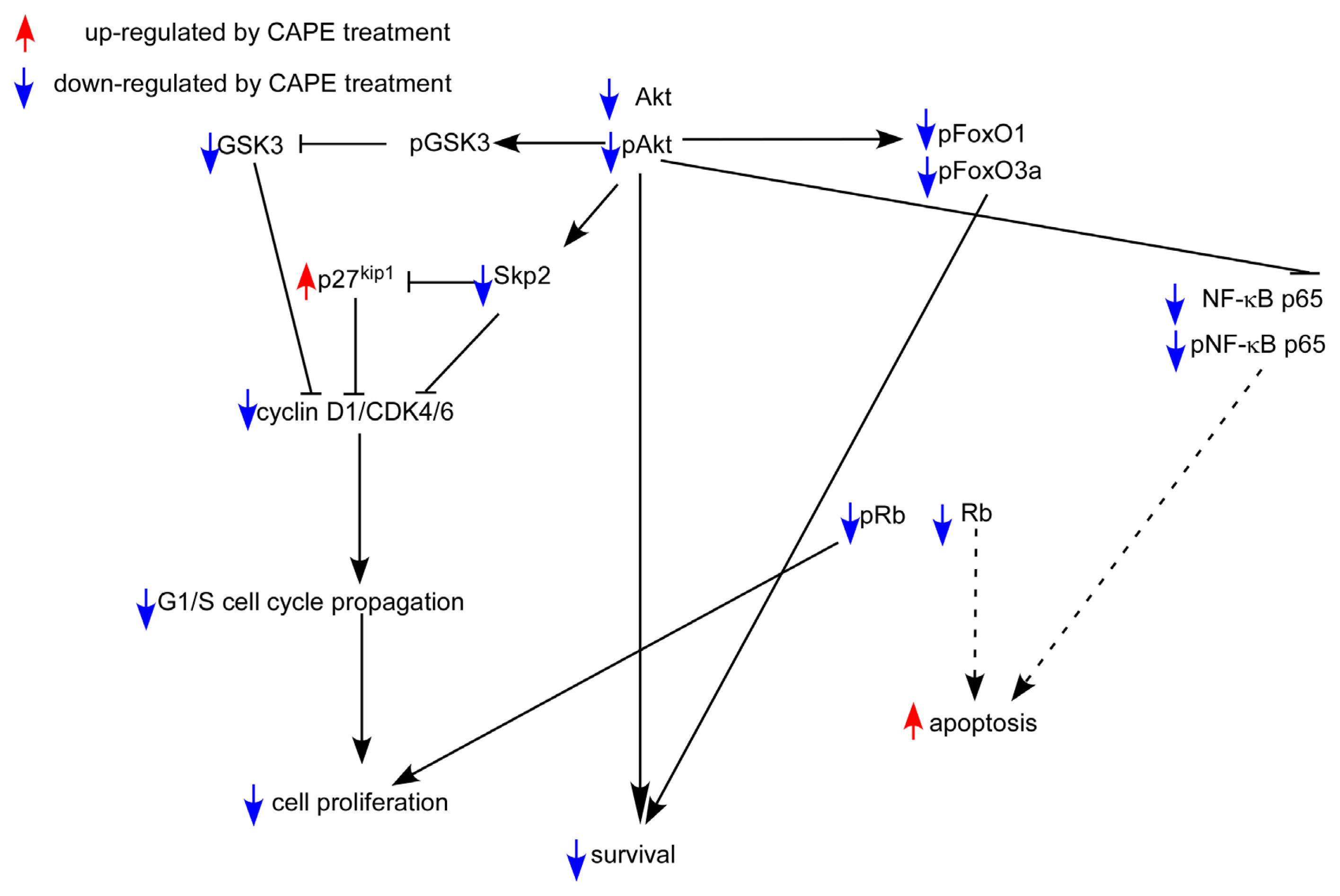
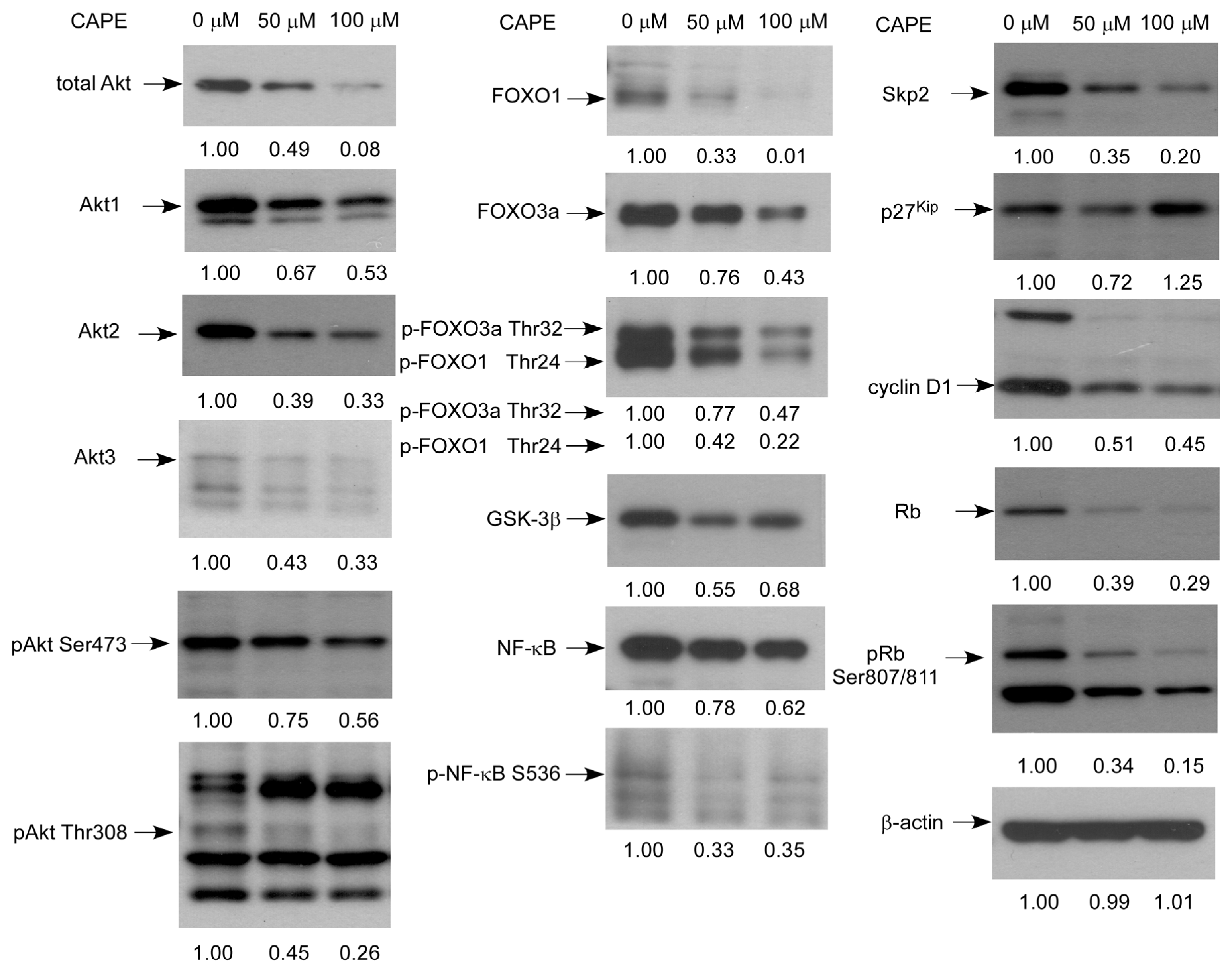
2.5. CAPE Caused a Reduction in Abundance of Signaling Proteins Regulating Cell Cycle and Akt Activity
2.6. Overexpression of Akt1 or Akt2 Rescued Growth Inhibition Caused by CAPE Treatment
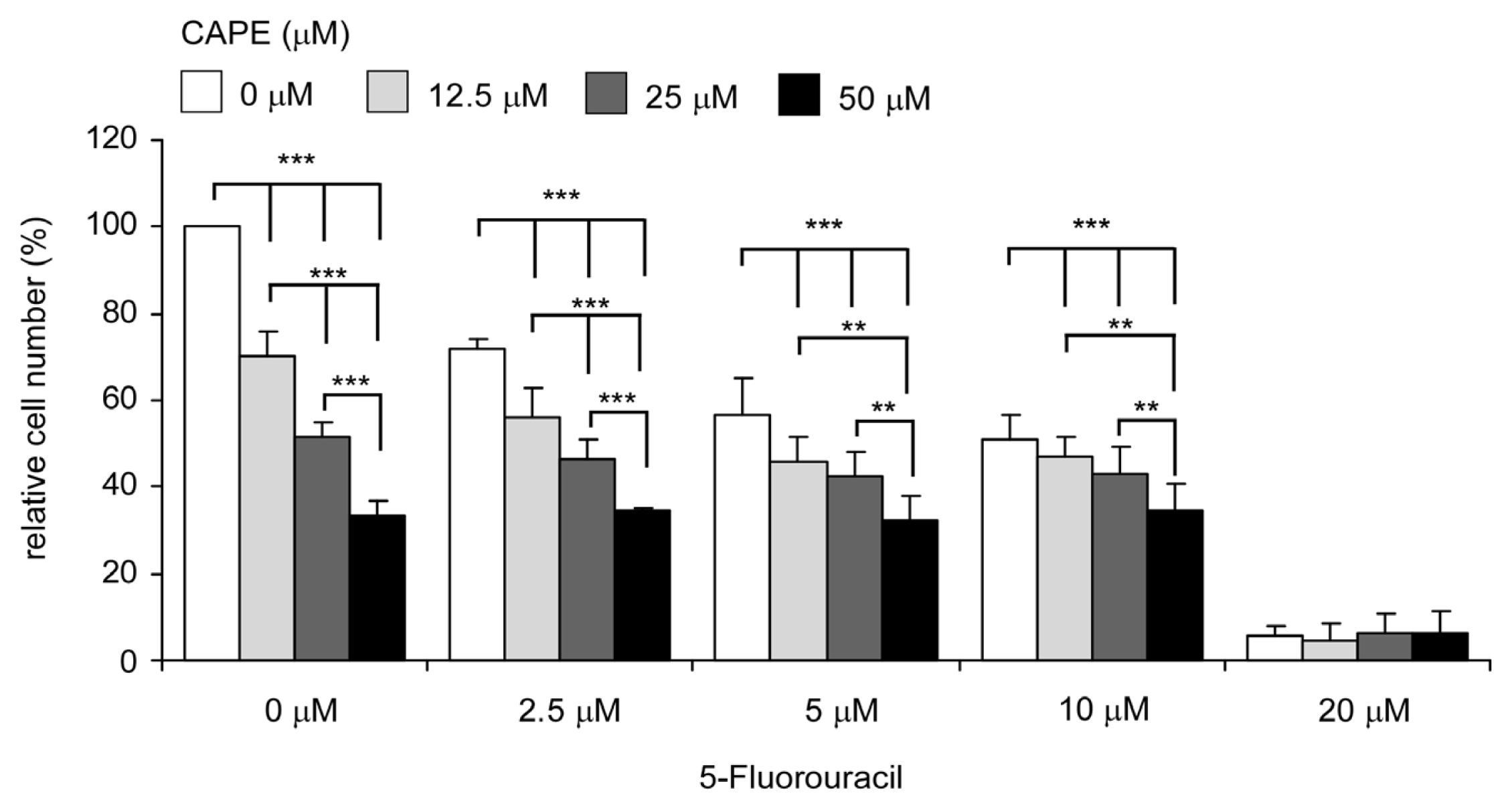
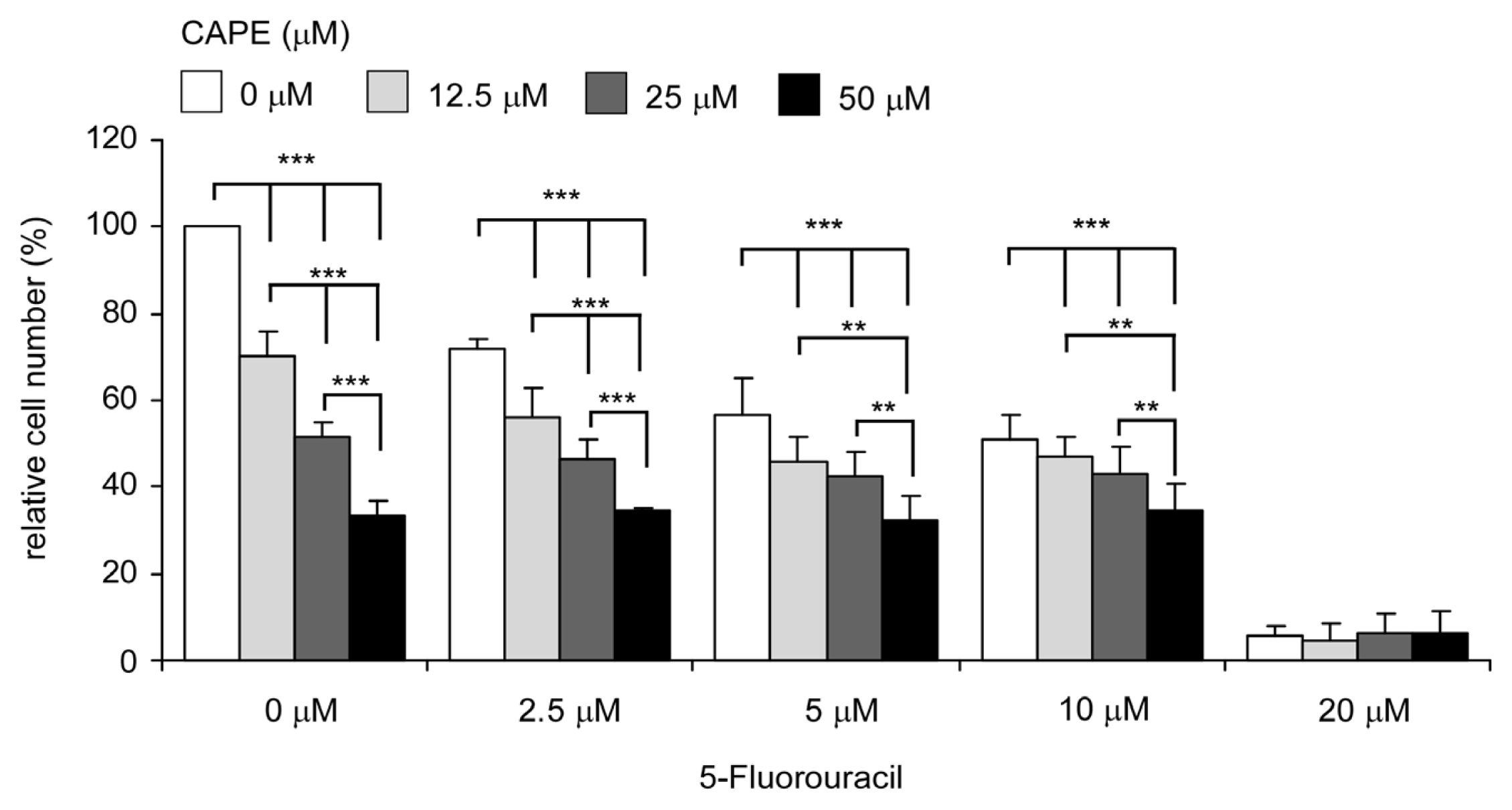
2.7. Co-Treatment of CAPE with Chemotherapeutic Drug 5-fluorouracil Suppressed Proliferation of TW2.6 Cells More Efficiently
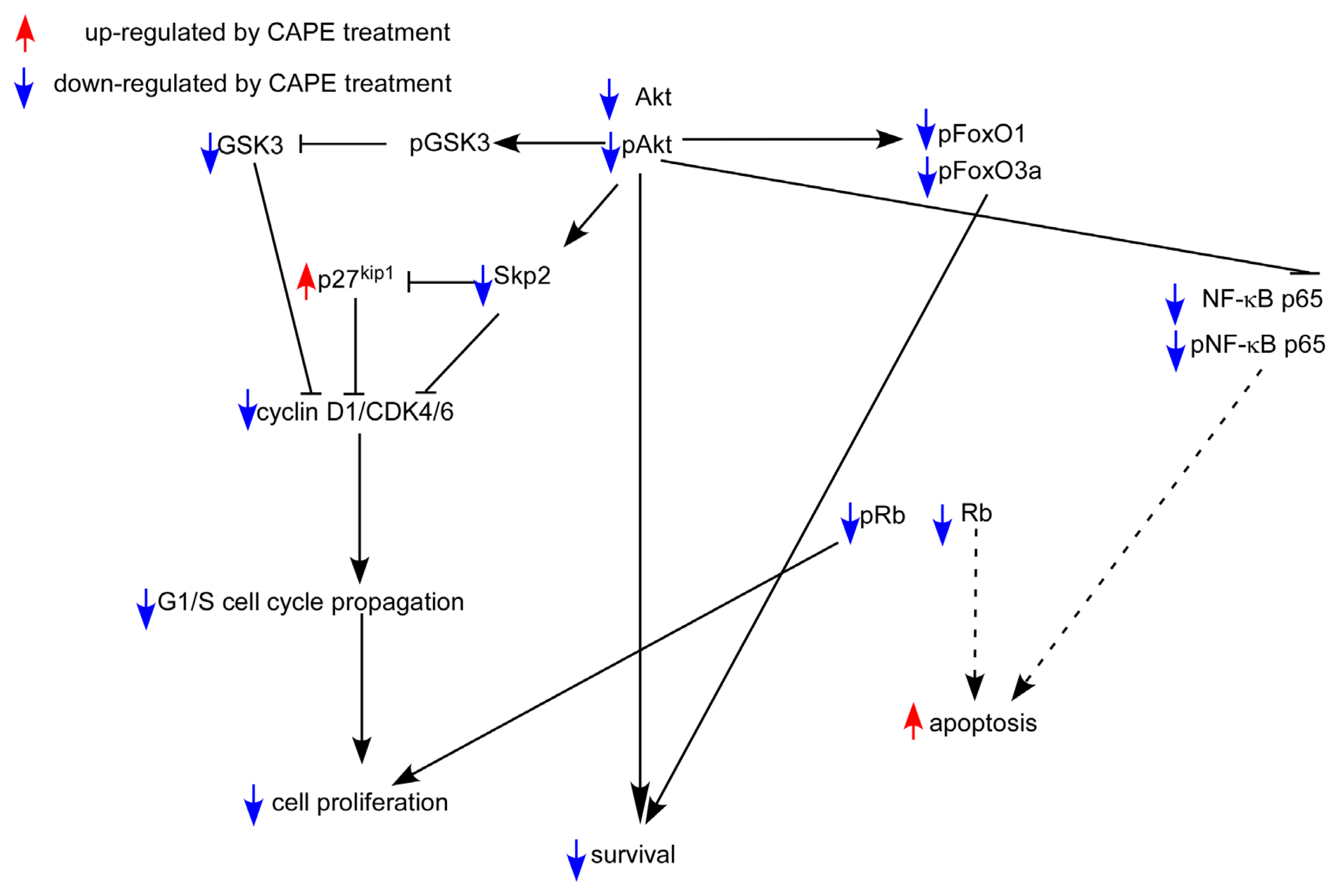
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Cell Culture
4.3. Hoechst Dye 33258-Based Cell Proliferation Assay
4.4. Cell Viability Assay
4.5. Soft Agar Colony Formation Assay
4.6. Luciferase-Reporter Assay
4.7. Flow Cytometric Analysis
4.8. Western Blotting Analysis
4.9. Overexpression of Akt1 and Akt2
4.10. TUNEL Assay
4.11. Data Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar]
- Mascolo, M.; Siano, M.; Ilardi, G.; Russo, D.; Merolla, F.; de Rosa, G.; Staibano, S. Epigenetic disregulation in oral cancer. Int. J. Mol. Sci 2012, 13, 2331–2353. [Google Scholar]
- Sharma, A.; Mendez, E.; Yueh, B.; Lohavanichbutr, P.; Houck, J.; Doody, D.R.; Futran, N.D.; Upton, M.P.; Schwartz, S.M.; Chen, C. Human papillomavirus-positive oral cavity and oropharyngeal cancer patients do not have better quality-of-life trajectories. Otolaryngol. Head. Neck Surg 2012, 146, 739–745. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Chen, Y.J.; Chang, J.T.; Liao, C.T.; Wang, H.M.; Yen, T.C.; Chiu, C.C.; Lu, Y.C.; Li, H.F.; Cheng, A.J. Head and neck cancer in the betel quid chewing area: Recent advances in molecular carcinogenesis. Cancer Sci 2008, 99, 1507–1514. [Google Scholar]
- Petersen, P.E. Oral cancer prevention and control—The approach of the World Health Organization. Oral Oncol 2009, 45, 454–460. [Google Scholar]
- Kok, S.H.; Hong, C.Y.; Lin, S.K.; Lee, J.J.; Chiang, C.P.; Kuo, M.Y. Establishment and characterization of a tumorigenic cell line from areca quid and tobacco smoke-associated buccal carcinoma. Oral Oncol 2007, 43, 639–647. [Google Scholar]
- Bhimani, R.S.; Troll, W.; Grunberger, D.; Frenkel, K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res 1993, 53, 4528–4533. [Google Scholar]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett 2011, 308, 43–53. [Google Scholar]
- Watabe, M.; Hishikawa, K.; Takayanagi, A.; Shimizu, N.; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J. Biol. Chem 2004, 279, 6017–6026. [Google Scholar]
- Chuu, C.P.; Lin, H.P.; Ciaccio, M.F.; Kokontis, J.M.; Hause, R.J., Jr; Hiipakka, R.A.; Liao, S.; Jones, R.B. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prev. Res. (Phila.) 2012, 5, 788–797. [Google Scholar]
- Lin, H.P.; Jiang, S.S.; Chuu, C.P. Caffeic acid phenethyl ester causes p21 induction, akt signaling reduction, and growth inhibition in PC-3 human prostate cancer cells. PLoS One 2012, 7, e31286. [Google Scholar]
- Lin, H.P.; Lin, C.Y.; Liu, C.C.; Su, L.C.; Huo, C.; Kuo, Y.Y.; Tseng, J.C.; Hsu, J.M.; Chen, C.K.; Chuu, C.P. Caffeic Acid phenethyl ester as a potential treatment for advanced prostate cancer targeting akt signaling. Int. J. Mol. Sci 2013, 14, 5264–5283. [Google Scholar]
- Liu, C.C.; Hsu, J.M.; Kuo, L.K.; Chuu, C.P. Caffeic acid phenethyl ester as an adjuvant therapy for advanced prostate cancer. Med. Hypotheses 2013, 80, 617–619. [Google Scholar]
- Chen, M.F.; Wu, C.T.; Chen, Y.J.; Keng, P.C.; Chen, W.C. Cell killing and radiosensitization by caffeic acid phenethyl ester (CAPE) in lung cancer cells. J. Radiat. Res. (Tokyo) 2004, 45, 253–260. [Google Scholar]
- Lin, H.P.; Kuo, L.K.; Chuu, C.P. Combined treatment of curcumin and small molecule inhibitors suppresses proliferation of A549 and H1299 human non-small-cell lung cancer cells. Phytother. Res 2011, 26, 122–126. [Google Scholar]
- Hung, M.W.; Shiao, M.S.; Tsai, L.C.; Chang, G.G.; Chang, T.C. Apoptotic effect of caffeic acid phenethyl ester and its ester and amide analogues in human cervical cancer ME180 cells. Anticancer Res 2003, 23, 4773–4780. [Google Scholar]
- Lee, Y.T.; Don, M.J.; Hung, P.S.; Shen, Y.C.; Lo, Y.S.; Chang, K.W.; Chen, C.F.; Ho, L.K. Cytotoxicity of phenolic acid phenethyl esters on oral cancer cells. Cancer Lett 2005, 223, 19–25. [Google Scholar]
- Celli, N.; Dragani, L.K.; Murzilli, S.; Pagliani, T.; Poggi, A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J. Agric. Food Chem 2007, 55, 3398–3407. [Google Scholar]
- Hu, J.; Nakano, H.; Sakurai, H.; Colburn, N.H. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis 2004, 25, 1991–2003. [Google Scholar]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol 2007, 8, 49–62. [Google Scholar]
- Nakayama, H.; Ikebe, T.; Beppu, M.; Shirasuna, K. High expression levels of nuclear factor kappaB, IkappaB kinase alpha and Akt kinase in squamous cell carcinoma of the oral cavity. Cancer 2001, 92, 3037–3044. [Google Scholar]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol 1999, 1, 193–199. [Google Scholar]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol 1999, 9, 661–664. [Google Scholar]
- Murphree, A.L.; Benedict, W.F. Retinoblastoma: Clues to human oncogenesis. Science 1984, 223, 1028–1033. [Google Scholar]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar]
- Lew, D.J.; Dulic, V.; Reed, S.I. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 1991, 66, 1197–1206. [Google Scholar]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar]
- Coffer, P.J.; Jin, J.; Woodgett, J.R. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998, 335 Pt 1, 1–13. [Google Scholar]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol 1997, 7, 261–269. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar]
- Li, M.; Wang, X.; Meintzer, M.K.; Laessig, T.; Birnbaum, M.J.; Heidenreich, K.A. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol. Cell Biol 2000, 20, 9356–9363. [Google Scholar]
- Rossig, L.; Badorff, C.; Holzmann, Y.; Zeiher, A.M.; Dimmeler, S. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem 2002, 277, 9684–9689. [Google Scholar]
- Welcker, M.; Singer, J.; Loeb, K.R.; Grim, J.; Bloecher, A.; Gurien-West, M.; Clurman, B.E.; Roberts, J.M. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 2003, 12, 381–392. [Google Scholar]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998, 12, 3499–3511. [Google Scholar]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem 1999, 274, 17179–17183. [Google Scholar]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar]
- Zhang, H.; Pan, Y.; Zheng, L.; Choe, C.; Lindgren, B.; Jensen, E.D.; Westendorf, J.J.; Cheng, L.; Huang, H. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res 2011, 71, 3257–3267. [Google Scholar]
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol 2000, 2, E65–E67. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar]
- Li, M.H.; Ito, D.; Sanada, M.; Odani, T.; Hatori, M.; Iwase, M.; Nagumo, M. Effect of 5-fluorouracil on G1 phase cell cycle regulation in oral cancer cell lines. Oral Oncol 2004, 40, 63–70. [Google Scholar]
- Akyol, S.; Ginis, Z.; Armutcu, F.; Ozturk, G.; Yigitoglu, M.R.; Akyol, O. The potential usage of caffeic acid phenethyl ester (CAPE) against chemotherapy-induced and radiotherapy-induced toxicity. Cell Biochem. Funct 2012, 30, 438–443. [Google Scholar]
- Yagmurca, M.; Erdogan, H.; Iraz, M.; Songur, A.; Ucar, M.; Fadillioglu, E. Caffeic acid phenethyl ester as a protective agent against doxorubicin nephrotoxicity in rats. Clin. Chim. Acta 2004, 348, 27–34. [Google Scholar]
- Fadillioglu, E.; Oztas, E.; Erdogan, H.; Yagmurca, M.; Sogut, S.; Ucar, M.; Irmak, M.K. Protective effects of caffeic acid phenethyl ester on doxorubicin-induced cardiotoxicity in rats. J. Appl. Toxicol 2004, 24, 47–52. [Google Scholar]
- Irmak, M.K.; Fadillioglu, E.; Sogut, S.; Erdogan, H.; Gulec, M.; Ozer, M.; Yagmurca, M.; Gozukara, M.E. Effects of caffeic acid phenethyl ester and alpha-tocopherol on reperfusion injury in rat brain. Cell Biochem. Funct 2003, 21, 283–289. [Google Scholar]
- Iraz, M.; Ozerol, E.; Gulec, M.; Tasdemir, S.; Idiz, N.; Fadillioglu, E.; Naziroglu, M.; Akyol, O. Protective effect of caffeic acid phenethyl ester (CAPE) administration on cisplatin-induced oxidative damage to liver in rat. Cell Biochem. Funct 2006, 24, 357–361. [Google Scholar]
- Yilmaz, H.R.; Sogut, S.; Ozyurt, B.; Ozugurlu, F.; Sahin, S.; Isik, B.; Uz, E.; Ozyurt, H. The activities of liver adenosine deaminase, xanthine oxidase, catalase, superoxide dismutase enzymes and the levels of malondialdehyde and nitric oxide after cisplatin toxicity in rats: Protective effect of caffeic acid phenethyl ester. Toxicol. Ind. Health 2005, 21, 67–73. [Google Scholar]
- Oktem, F.; Yilmaz, H.R.; Ozguner, F.; Olgar, S.; Ayata, A.; Uzare, E.; Uz, E. Methotrexate-induced renal oxidative stress in rats: The role of a novel antioxidant caffeic acid phenethyl ester. Toxicol. Ind. Health 2006, 22, 241–247. [Google Scholar]
- Ozyurt, H.; Sogut, S.; Yildirim, Z.; Kart, L.; Iraz, M.; Armutcu, F.; Temel, I.; Ozen, S.; Uzun, A.; Akyol, O. Inhibitory effect of caffeic acid phenethyl ester on bleomycine-induced lung fibrosis in rats. Clin. Chim. Acta 2004, 339, 65–75. [Google Scholar]
- Albukhari, A.A.; Gashlan, H.M.; El-Beshbishy, H.A.; Nagy, A.A.; Abdel-Naim, A.B. Caffeic acid phenethyl ester protects against tamoxifen-induced hepatotoxicity in rats. Food Chem. Toxicol 2009, 47, 1689–1695. [Google Scholar]
- Kokontis, J.M.; Hay, N.; Liao, S. Progression of LNCaP prostate tumor cells during androgen deprivation: Hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol 1998, 12, 941–953. [Google Scholar]
- Kokontis, J.M.; Hsu, S.; Chuu, C.P.; Dang, M.; Fukuchi, J.; Hiipakka, R.A.; Liao, S. Role of androgen receptor in the progression of human prostate tumor cells to androgen independence and insensitivity. Prostate 2005, 65, 287–298. [Google Scholar]
- Chuu, C.P.; Lin, H.P. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer Res 2010, 30, 3643–3648. [Google Scholar]
- Chuu, C.P.; Kokontis, J.M.; Hiipakka, R.A.; Fukuchi, J.; Lin, H.P.; Lin, C.Y.; Huo, C.; Su, L.C.; Liao, S. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci 2011, 102, 2022–2028. [Google Scholar]
- Chuu, C.P.; Chen, R.Y.; Hiipakka, R.A.; Kokontis, J.M.; Warner, K.V.; Xiang, J.; Liao, S. The liver X receptor agonist T0901317 acts as androgen receptor antagonist in human prostate cancer cells. Biochem. Biophys. Res. Commun 2007, 357, 341–346. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kuo, Y.-Y.; Lin, H.-P.; Huo, C.; Su, L.-C.; Yang, J.; Hsiao, P.-H.; Chiang, H.-C.; Chung, C.-J.; Wang, H.-D.; Chang, J.-Y.; et al. Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling. Int. J. Mol. Sci. 2013, 14, 8801-8817. https://doi.org/10.3390/ijms14058801
Kuo Y-Y, Lin H-P, Huo C, Su L-C, Yang J, Hsiao P-H, Chiang H-C, Chung C-J, Wang H-D, Chang J-Y, et al. Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling. International Journal of Molecular Sciences. 2013; 14(5):8801-8817. https://doi.org/10.3390/ijms14058801
Chicago/Turabian StyleKuo, Ying-Yu, Hui-Ping Lin, Chieh Huo, Liang-Cheng Su, Jonathan Yang, Ping-Hsuan Hsiao, Hung-Che Chiang, Chi-Jung Chung, Horng-Dar Wang, Jang-Yang Chang, and et al. 2013. "Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling" International Journal of Molecular Sciences 14, no. 5: 8801-8817. https://doi.org/10.3390/ijms14058801
APA StyleKuo, Y.-Y., Lin, H.-P., Huo, C., Su, L.-C., Yang, J., Hsiao, P.-H., Chiang, H.-C., Chung, C.-J., Wang, H.-D., Chang, J.-Y., Chen, Y.-W., & Chuu, C.-P. (2013). Caffeic Acid Phenethyl Ester Suppresses Proliferation and Survival of TW2.6 Human Oral Cancer Cells via Inhibition of Akt Signaling. International Journal of Molecular Sciences, 14(5), 8801-8817. https://doi.org/10.3390/ijms14058801