Combining Coarse-Grained Protein Models with Replica-Exchange All-Atom Molecular Dynamics


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
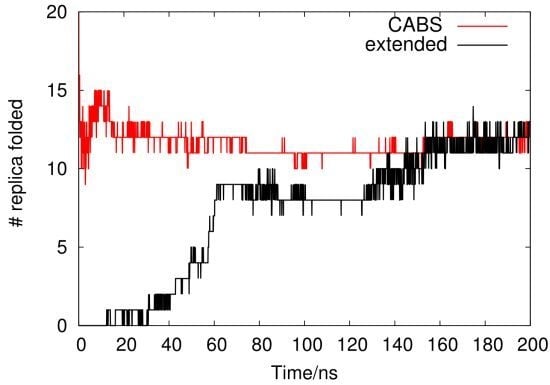
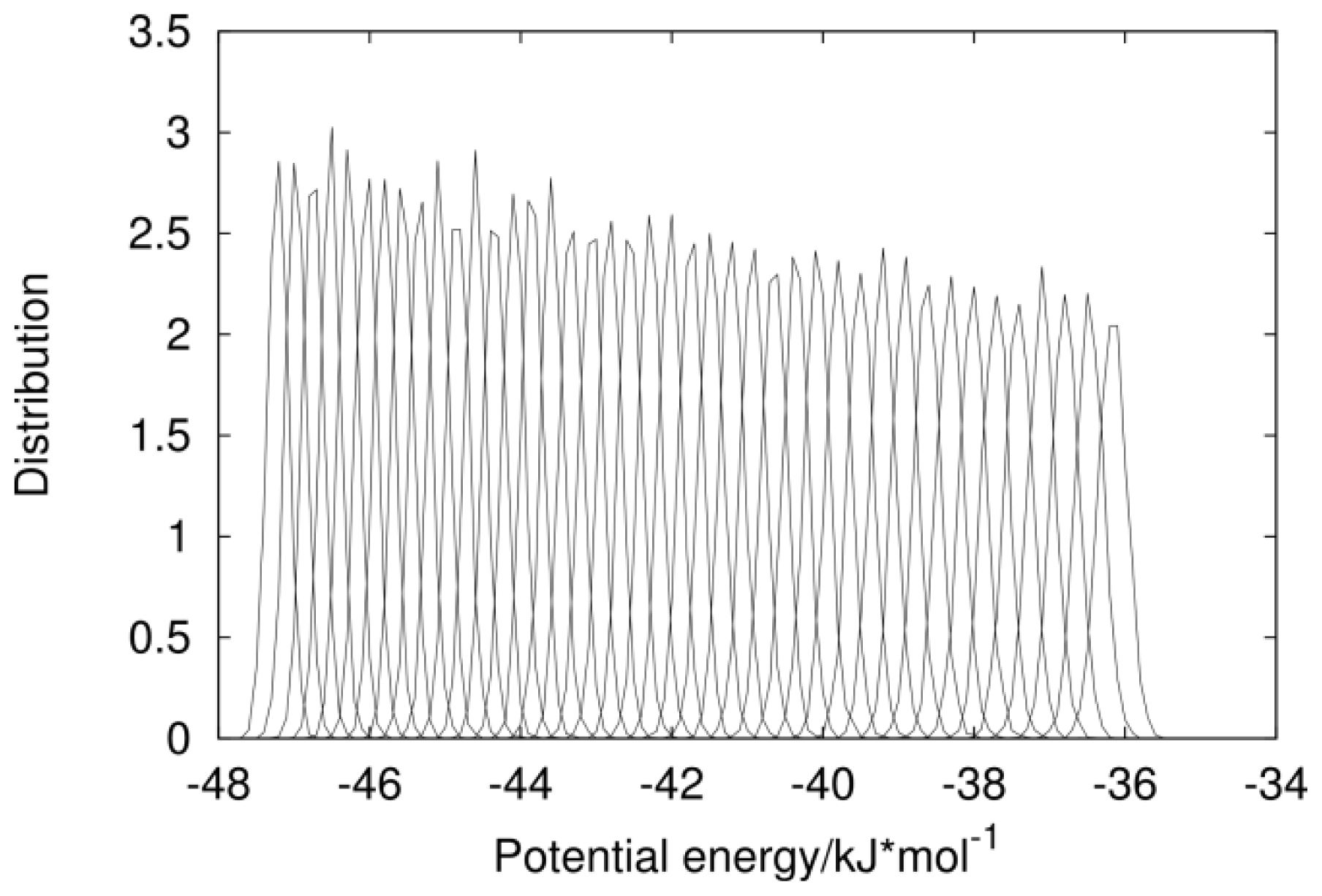
2.1. Analysis of REMD Simulation Convergence
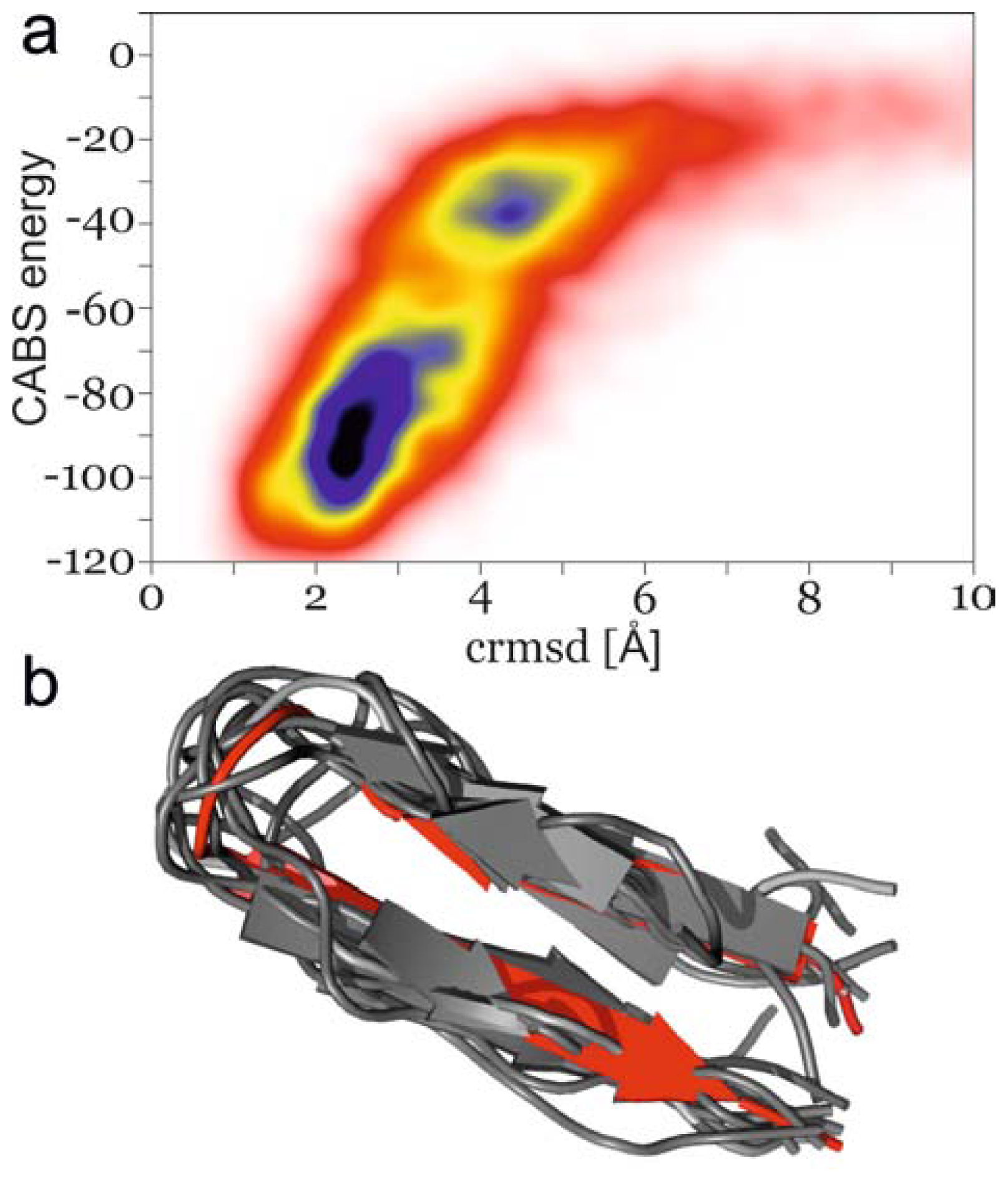
2.2. Secondary Structure Propagation at 300 K
3. Methods
- Simulation #1 was conducted with 42 replicas (initialized with CABS output models as described above) with the replica exchange trial every 1 ps. Simulation time was 200 ns per replica. We used a dodecahedron simulation box containing 2087 water molecules and Na+ and Cl− ions at a concentration of 0.15 M. Periodic boundary conditions were applied. Forty two temperature replicas were distributed in a range of 280–562 K. The OPLS-AA [18] force field was used with the spc [62] model for water. Bonds were constrained using the LINCS [63] algorithm.
- Simulation #2 was conducted in the same way as #1 using the extended peptide as a starting conformation.The same procedures (#1 and #2) were carried out using AMBER99sb [66] (two simulations). The total simulation time was 33.6 μs.
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Kolinski, A.; Bujnicki, J.M. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins 2005, 61, 84–90. [Google Scholar]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu. Rev. Phys. Chem 2007, 58, 57–83. [Google Scholar]
- Kouza, M.; Hu, C.K.; Zung, H.; Li, M.S. Protein mechanical unfolding: Importance of non-native interactions. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef]
- Malolepsza, E.; Boniecki, M.; Kolinski, A.; Piela, L. Theoretical model of prion propagation: A misfolded protein induces misfolding. Proc. Natl. Acad. Sci. USA 2005, 102, 7835–7840. [Google Scholar]
- Kmiecik, S.; Jamroz, M.; Kolinski, A. Multiscale Approach to Protein Folding Dynamics. In Multiscale Approaches to Protein Modeling; Kolinski, A., Ed.; Springer: New York, NY, USA, 2011; pp. 281–293. [Google Scholar]
- Shakhnovich, E. Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem. Rev 2006, 106, 1559–1588. [Google Scholar]
- Liwo, A.; He, Y.; Scheraga, H.A. Coarse-grained force field: General folding theory. PCCP 2011, 13, 16890–16901. [Google Scholar]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol 2004, 51, 349–371. [Google Scholar]
- Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol 2005, 15, 144–150. [Google Scholar]
- Kmiecik, S.; Kolinski, A. Characterization of protein-folding pathways by reduced-space modeling. Proc. Natl. Acad. Sci. USA 2007, 104, 12330–12335. [Google Scholar]
- Kmiecik, S.; Kolinski, A. Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys. J 2008, 94, 726–736. [Google Scholar]
- Kmiecik, S.; Kolinski, A. Simulation of chaperonin effect on protein folding: A shift from nucleation-condensation to framework mechanism. J. Am. Chem. Soc 2011, 133, 10283–10289. [Google Scholar]
- Kmiecik, S.; Gront, D.; Kouza, M.; Kolinski, A. From coarse-grained to atomic-level characterization of protein dynamics: Transition state for the folding of B domain of protein A. J. Phys. Chem. B 2012, 116, 7026–7032. [Google Scholar]
- Jamroz, M.; Orozco, M.; Kolinski, A.; Kmiecik, S. Consistent view of protein fluctuations from all-atom molecular dynamics and coarse-grained dynamics with knowledge-based force-field. J. Chem. Theory. Comput 2013, 9, 119–125. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 1988, 110, 1657–1666. [Google Scholar]
- Boczko, E.M.; Brooks, C.L. First-principles calculation of the folding free–energy of a 3–helix bundle protein. Science 1995, 269, 393–396. [Google Scholar]
- Berg, B.A.; Neuhaus, T. Multicanonical algorithms for 1st order phase-transitions. Phys. Lett. B 1991, 267, 249–253. [Google Scholar]
- Berne, B.J.; Straub, J.E. Novel methods of sampling phase space in the simulation of biological systems. Curr. Opin. Struct. Biol 1997, 7, 181–189. [Google Scholar]
- Hansmann, U.H.E. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett 1997, 281, 140–150. [Google Scholar]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- Pokarowski, P.; Kolinski, A.; Skolnick, J. A minimal physically realistic protein-like lattice model: Designing an energy landscape that ensures all-or-none folding to a unique native state. Biophys. J 2003, 84, 1518–1526. [Google Scholar]
- Kouza, M.; Hu, C.K.; Li, M.S. New force replica exchange method and protein folding pathways probed by force-clamp technique. J Chem Phys 2008, 128, 045103. [Google Scholar]
- Gront, D.; Kolinski, A.; Skolnick, J. A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics. J. Chem. Phys 2001, 115, 1569–1574. [Google Scholar]
- Kouza, M.; Hansmann, U.H.E. Velocity scaling for optimizing replica exchange molecular dynamics. J. Chem. Phys 2011, 134, 044124. [Google Scholar]
- Chaudhury, S.; Olson, M.A.; Tawa, G.; Wallqvist, A.; Lee, M.S. Efficient conformational sampling in explicit solvent using a hybrid replica exchange molecular dynamics method. J. Chem. Theory. Comput 2012, 8, 677–687. [Google Scholar]
- Nguyen, P.; Stock, G.; Mittag, E.; Hu, C.-K.; Li, M. Free energy landscape and folding mechanism of a β-hairpin in explicit water: A replica exchange molecular dynamics study. Proteins 2005, 61, 795–808. [Google Scholar]
- Okur, A.; Roe, D.R.; Cui, G.; Hornak, V.; Simmerling, C. Improving convergence of replica-exchange simulations through coupling to a high-temperature structure reservoir. J. Chem. Theory. Comput 2007, 3, 557–568. [Google Scholar]
- Roitberg, A.E.; Okur, A.; Simmerling, C. Coupling of replica exchange simulations to a non-boltzmann structure reservoir. J. Phys. Chem. B 2007, 111, 2415–2418. [Google Scholar]
- Schlick, T. Molecular dynamics-based approaches for enhanced sampling of long-time, large-scale conformational changes in biomolecules. F1000 Biol. Rep. 2009, 1. [Google Scholar] [CrossRef]
- Meli, M.; Morra, G.; Colombo, G. Investigating the mechanism of peptide aggregation: Insights from mixed monte carlo-molecular dynamics simulations. Biophys. J 2008, 94, 4414–4426. [Google Scholar]
- De Mori, G.M.S.; Micheletti, C.; Colombo, G. All-atom folding simulations of the villin headpiece from stochastically selected coarse-grained structures. J. Phys. Chem. B 2004, 108, 12267–12270. [Google Scholar]
- Colombo, G.; Micheletti, C. Protein folding simulations: combining coarse-grained models and all-atom molecular dynamics. Theor. Chem. Acc 2006, 116, 75–86. [Google Scholar]
- Thorpe, I.F.; Zhou, J.; Voth, G.A. Peptide folding using multiscale coarse-grained models. J. Phys. Chem. B 2008, 112, 13079–13090. [Google Scholar]
- Chang, C.-E.A.; Trylska, J.; Tozzini, V.; Andrew Mccammon, J. Binding pathways of ligands to HIV-1 Protease: Coarse-grained and atomistic simulations. Chem. Biol. Drug Des 2007, 69, 5–13. [Google Scholar]
- Best, R.B.; Mittal, J. Free-energy landscape of the GB1 hairpin in all-atom explicit solvent simulations with different force fields: Similarities and differences. Proteins 2011, 79, 1318–1328. [Google Scholar]
- Kolinski, A.; Ilkowski, B.; Skolnick, J. Dynamics and thermodynamics of beta-hairpin assembly: Insights from various simulation techniques. Biophys. J 1999, 77, 2942–2952. [Google Scholar]
- García, A.; Sanbonmatsu, K. Exploring the energy landscape ofβ a hairpin in explicit solvent. Proteins 2001, 42, 345–354. [Google Scholar]
- Lwin, T.Z.; Luo, R. Force field influences in β-hairpin folding simulations. Protein Sci 2006, 15, 2642–2655. [Google Scholar]
- Shao, Q.; Yang, L.; Gao, Y.Q. Structure change of beta-hairpin induced by turn optimization: An enhanced sampling molecular dynamics simulation study. J. Chem. Phys 2011, 135, 235104–235110. [Google Scholar]
- Bhattacharya, A.; Best, R.B.; Mittal, J. Smoothing of the GB1 hairpin folding landscape by interfacial confinement. Biophys. J 2012, 103, 596–600. [Google Scholar]
- Cao, Z.; Wang, J. A comparative study of two different force fields on structural and thermodynamics character of H1 peptide via molecular dynamics simulations. J. Biomol. Struct. Dyn 2010, 27, 651–661. [Google Scholar]
- Blanco, F.; Rivas, G.; Serrano, L. A short linear peptide that folds into a native stable β-hairpin in aqueous solution. Nat. Struct. Mol. Biol 1994, 1, 584–590. [Google Scholar]
- Du, D.; Zhu, Y.; Huang, C.-Y.; Gai, F. Understanding the key factors that control the rate of beta-hairpin folding. Proc. Natl. Acad. Sci. USA 2004, 101, 15915–15920. [Google Scholar]
- Lewandowska, A.; Ołdziej, S.; Liwo, A.; Scheraga, H.A. Mechanism of formation of the C-terminal beta-hairpin of the B3 domain of the immunoglobulin-binding protein G from Streptococcus. IV. Implication for the mechanism of folding of the parent protein. Biopolymers 2010, 93, 469–480. [Google Scholar]
- Skwierawska, A.; Makowska, J.; Oldziej, S.; Liwo, A.; Scheraga, H.A. Mechanism of formation of the C-terminal beta-hairpin of the B3 domain of the immunoglobulin binding protein G from Streptococcus. I. Importance of hydrophobic interactions in stabilization of beta-hairpin structure. Proteins 2009, 75, 931–953. [Google Scholar]
- Skwierawska, A.; Zmudzinska, W.; Oldziej, S.; Liwo, A.; Scheraga, H.A. Mechanism of formation of the C-terminal beta-hairpin of the B3 domain of the immunoglobulin binding protein G from Streptococcus. II. Interplay of local backbone conformational dynamics and long-range hydrophobic interactions in hairpin formation. Proteins 2009, 76, 637–654. [Google Scholar]
- Munoz, V.; Thompson, P.A.; Hofrichter, J.; Eaton, W.A. Folding dynamics and mechanism of [beta]-hairpin formation. Nature 1997, 390, 196–199. [Google Scholar]
- Paschek, D.; Day, R.; Garcia, A.E. Influence of water-protein hydrogen bonding on the stability of Trp-cage miniprotein. A comparison between the TIP3P and TIP4P-Ew water models. Phys. Chem. Chem. Phys 2011, 13, 19840–19847. [Google Scholar]
- Paschek, D.; Hempel, S.; García, A.E. Computing the stability diagram of the Trp-cage miniprotein. Proc. Natl. Acad. Sci. USA 2008, 105, 17754–17759. [Google Scholar]
- Kim, E.; Jang, S.; Pak, Y. Consistent free energy landscapes and thermodynamic properties of small proteins based on a single all-atom force field employing an implicit solvation. J. Chem. Phys 2007, 127, 145104. [Google Scholar]
- Garcia, A.E.; Paschek, D. Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J. Am. Chem. Soc 2007, 130, 815–817. [Google Scholar]
- Zhou, R.; Berne, B.; Germain, R. The free energy landscape for β hairpin folding in explicit water. Proc. Natl. Acad. Sci. USA 2001, 98, 14931–14936. [Google Scholar]
- Best, R.B.; Mittal, J. Balance between α and β Structures in Ab initio protein folding. J. Phys. Chem. B 2010, 114, 8790–8798. [Google Scholar]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J 2011, 100, L47–L49. [Google Scholar]
- CABSfold. Available online: http://www.biocomp.chem.uw.edu.pl/CABSfold/ (accesed on 1 Febury 2013).
- Kmiecik, S.; Gront, D.; Kolinski, A. Towards the high-resolution protein structure prediction. Fast refinement of reduced models with all-atom force field. BMC Struct. Biol. 2007, 7. [Google Scholar] [CrossRef]
- Gront, D.; Kmiecik, S.; Kolinski, A. Backbone building from quadrilaterals: A fast and accurate algorithm for protein backbone reconstruction from alpha carbon coordinates. J. Comput. Chem 2007, 28, 1593–1597. [Google Scholar]
- Canutescu, A.; Shelenkov, A.; Dunbrack, R. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci 2003, 12, 2001–2014. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. Intermolecular Forces 1981, 331–342. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J Comput Chem 1997, 18, 1463–1472. [Google Scholar]
- van Gunsteren, W.F.; Berendsen, H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simul 1988, 1, 173–185. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. The Journal of Chemical Physics 1995, 103, 8577–8593. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory. Comput 2008, 4, 435–447. [Google Scholar]
- Gront, D.; Kolinski, A. BioShell-a package of tools for structural biology computations. Bioinformatics 2006, 22, 621–622. [Google Scholar]
- Gront, D.; Kolinski, A. Utility library for structural bioinformatics. Bioinformatics 2008, 24, 584–585. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.4.1; Schrodinger, LLC: Portland, OR, USA, 2010. [Google Scholar]
- Gront, D.; Kolinski, A. Efficient scheme for optimization of parallel tempering Monte Carlo method. J. Phys. Cond. Mat. 2007, 19. [Google Scholar] [CrossRef]
- Gront, D.; Kmiecik, S.; Blaszczyk, M.; Ekonomiuk, D.; Koliński, A. Optimization of protein models. WIREs Comp. Mol. Sci 2012, 2, 479–493. [Google Scholar]
- Cao, Z.; Liu, L.; Wang, J. Why the OPLS-AA force field cannot produce the beta-hairpin structure of H1 peptide in solution when comparing with the GROMOS 43A1 force field? J. Biomol. Struct. Dyn 2011, 29, 527–539. [Google Scholar]
- Seibert, M.M.; Patriksson, A.; Hess, B.; van der Spoel, D. Reproducible polypeptide folding and structure prediction using molecular dynamics simulations. J. Mol. Biol 2005, 354, 173–183. [Google Scholar]




© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wabik, J.; Kmiecik, S.; Gront, D.; Kouza, M.; Koliński, A. Combining Coarse-Grained Protein Models with Replica-Exchange All-Atom Molecular Dynamics. Int. J. Mol. Sci. 2013, 14, 9893-9905. https://doi.org/10.3390/ijms14059893
Wabik J, Kmiecik S, Gront D, Kouza M, Koliński A. Combining Coarse-Grained Protein Models with Replica-Exchange All-Atom Molecular Dynamics. International Journal of Molecular Sciences. 2013; 14(5):9893-9905. https://doi.org/10.3390/ijms14059893
Chicago/Turabian StyleWabik, Jacek, Sebastian Kmiecik, Dominik Gront, Maksim Kouza, and Andrzej Koliński. 2013. "Combining Coarse-Grained Protein Models with Replica-Exchange All-Atom Molecular Dynamics" International Journal of Molecular Sciences 14, no. 5: 9893-9905. https://doi.org/10.3390/ijms14059893