β-Cyclodextrin Inclusion Complex to Improve Physicochemical Properties of Pipemidic Acid: Characterization and Bioactivity Evaluation

Abstract
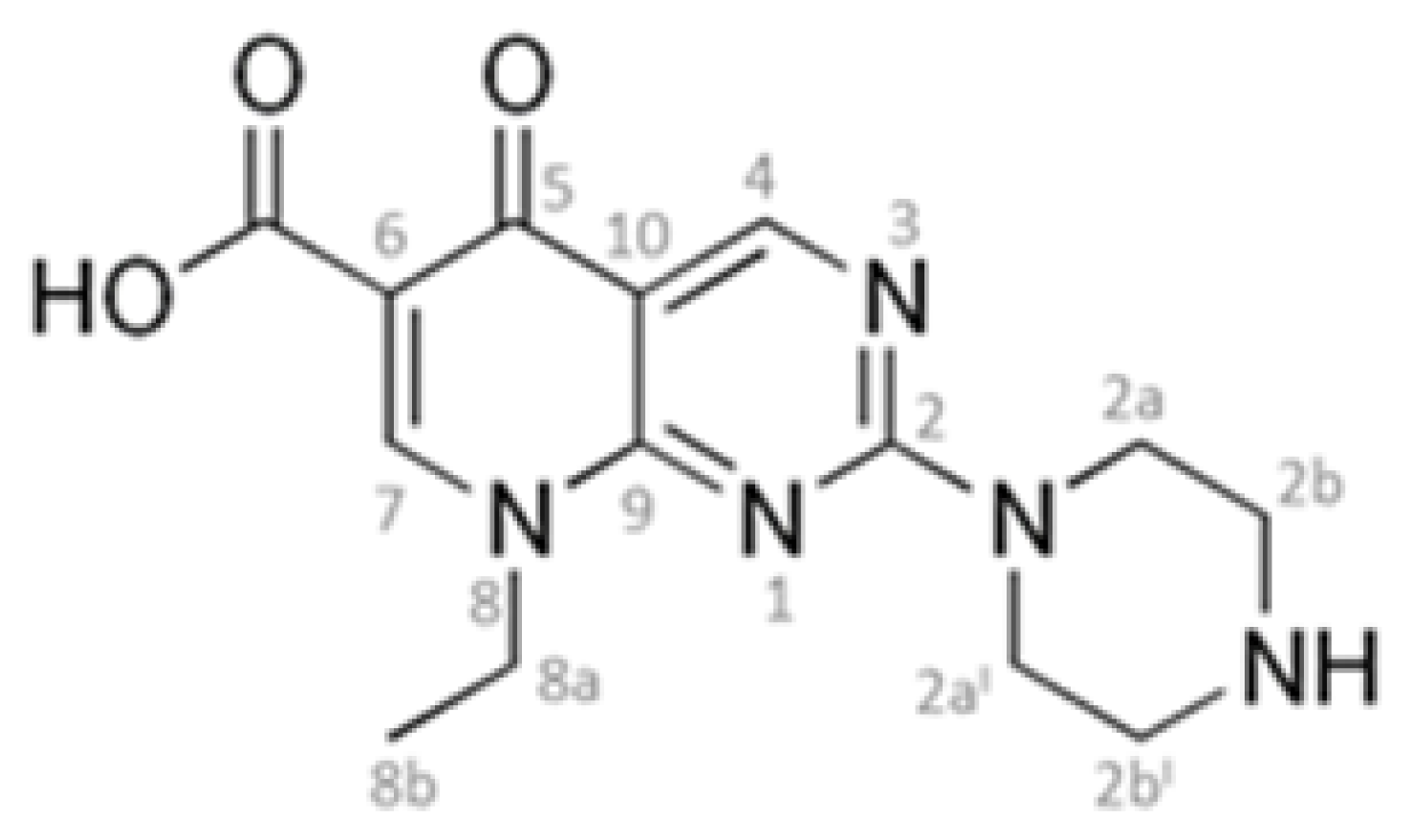
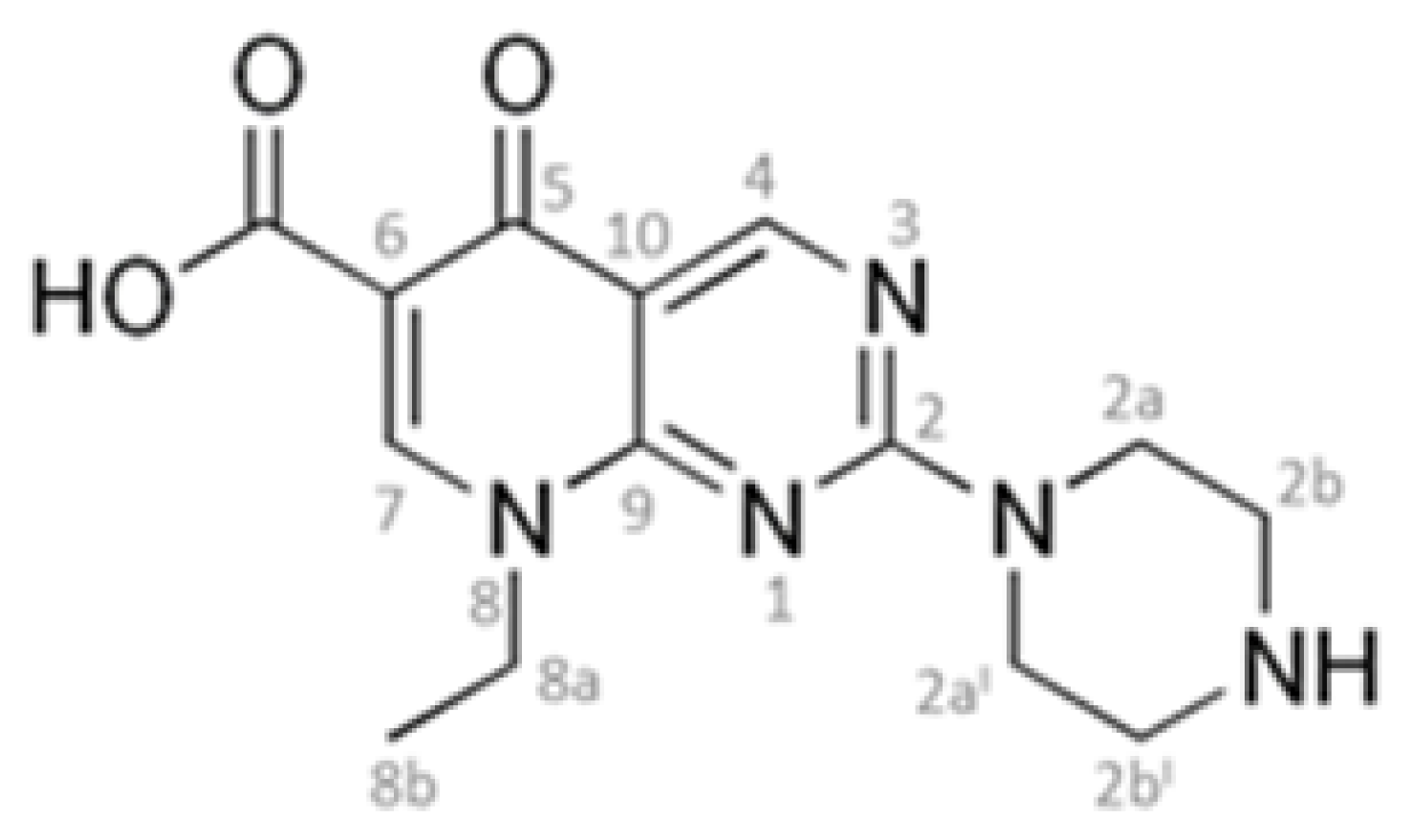
:1. Introduction
2. Results and Discussion
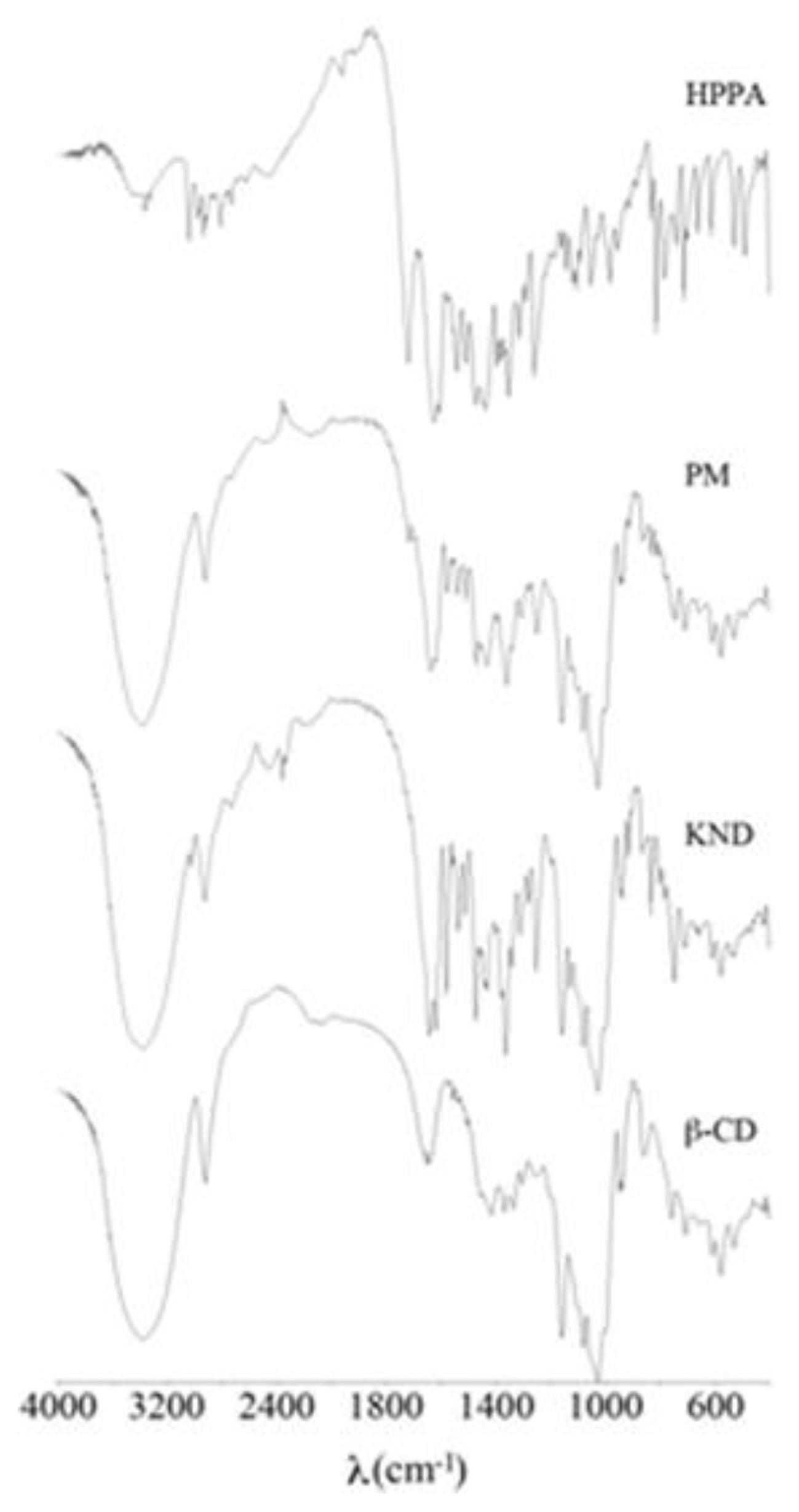
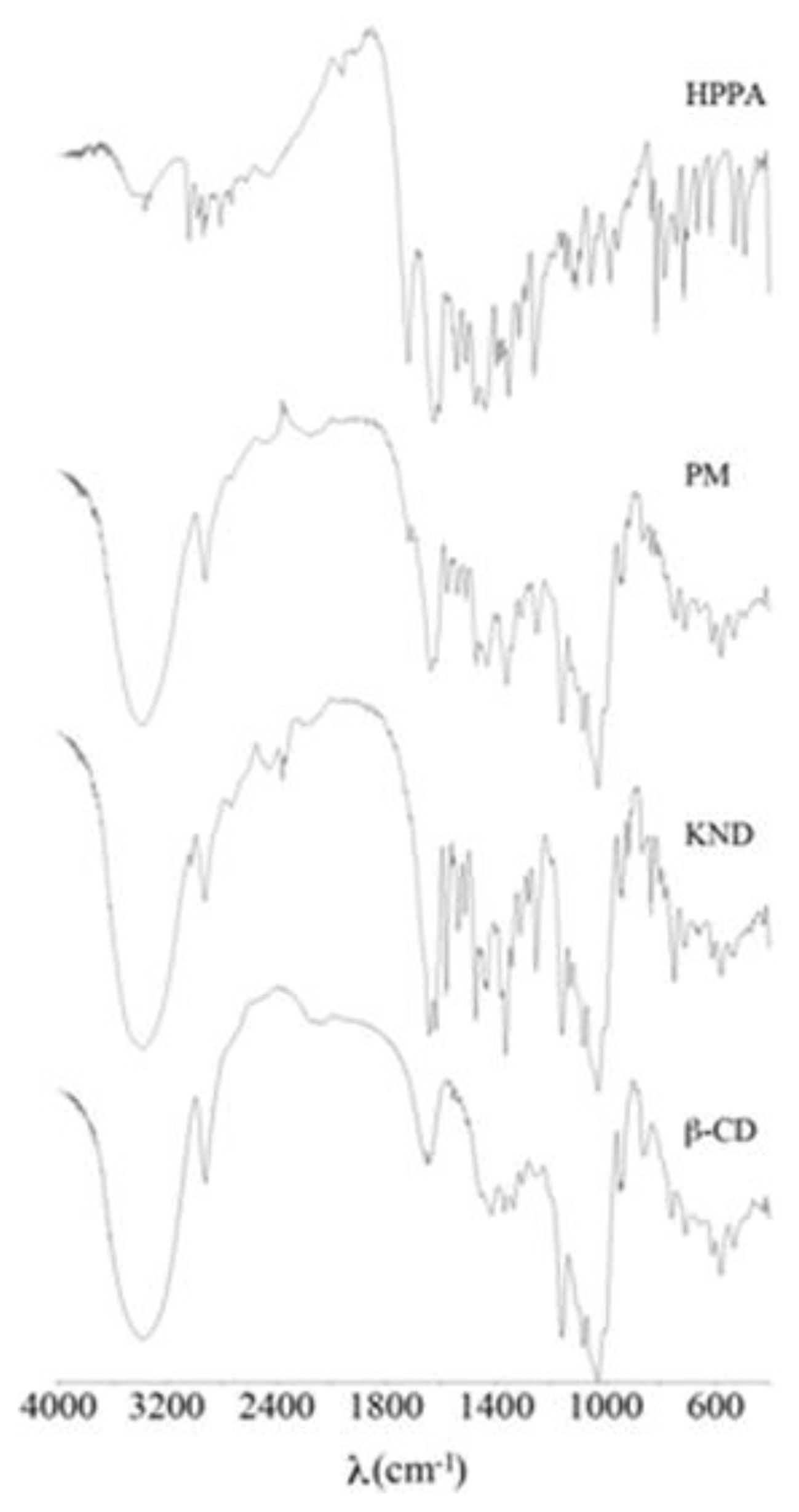
2.1. FT-IR Spectroscopy
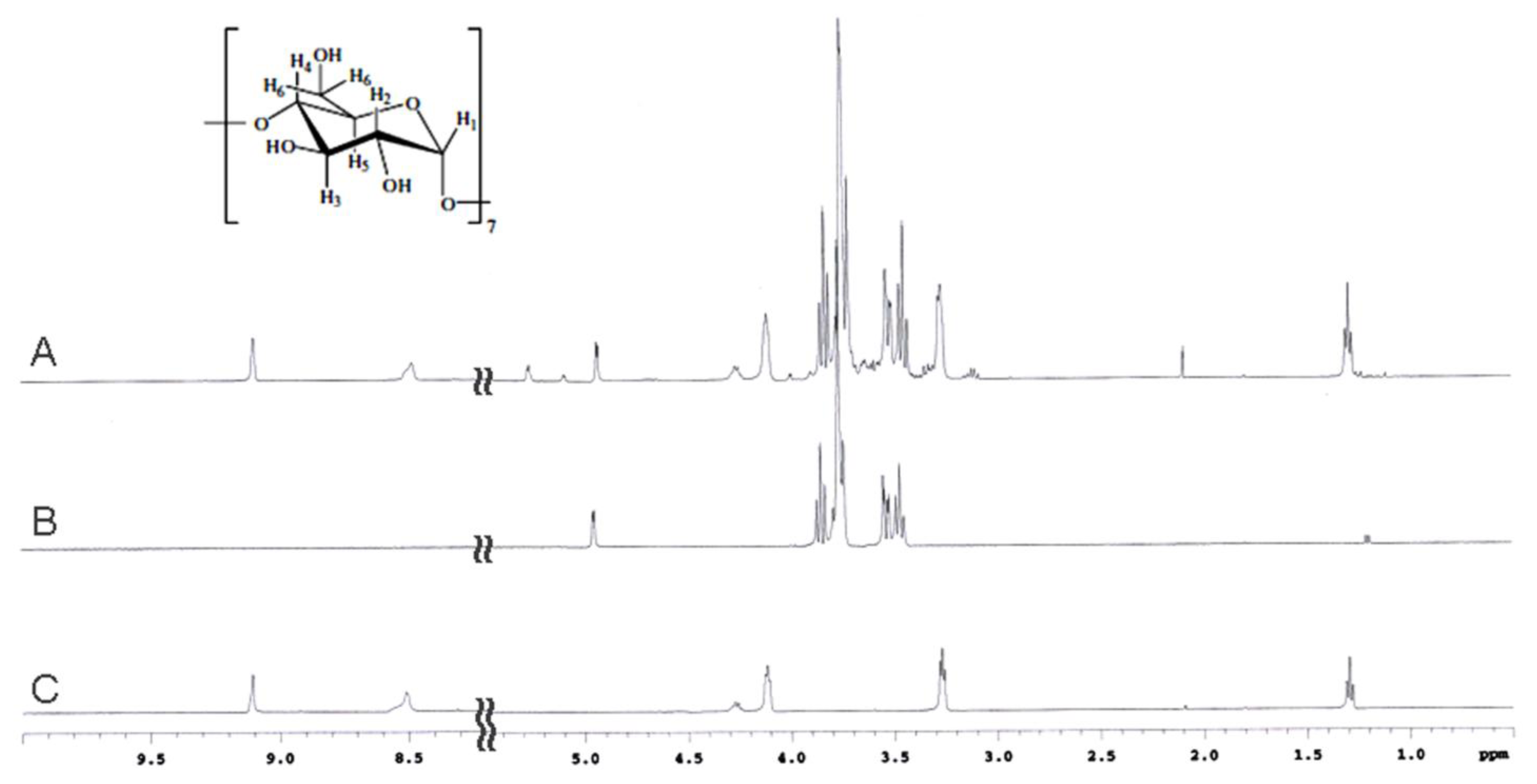
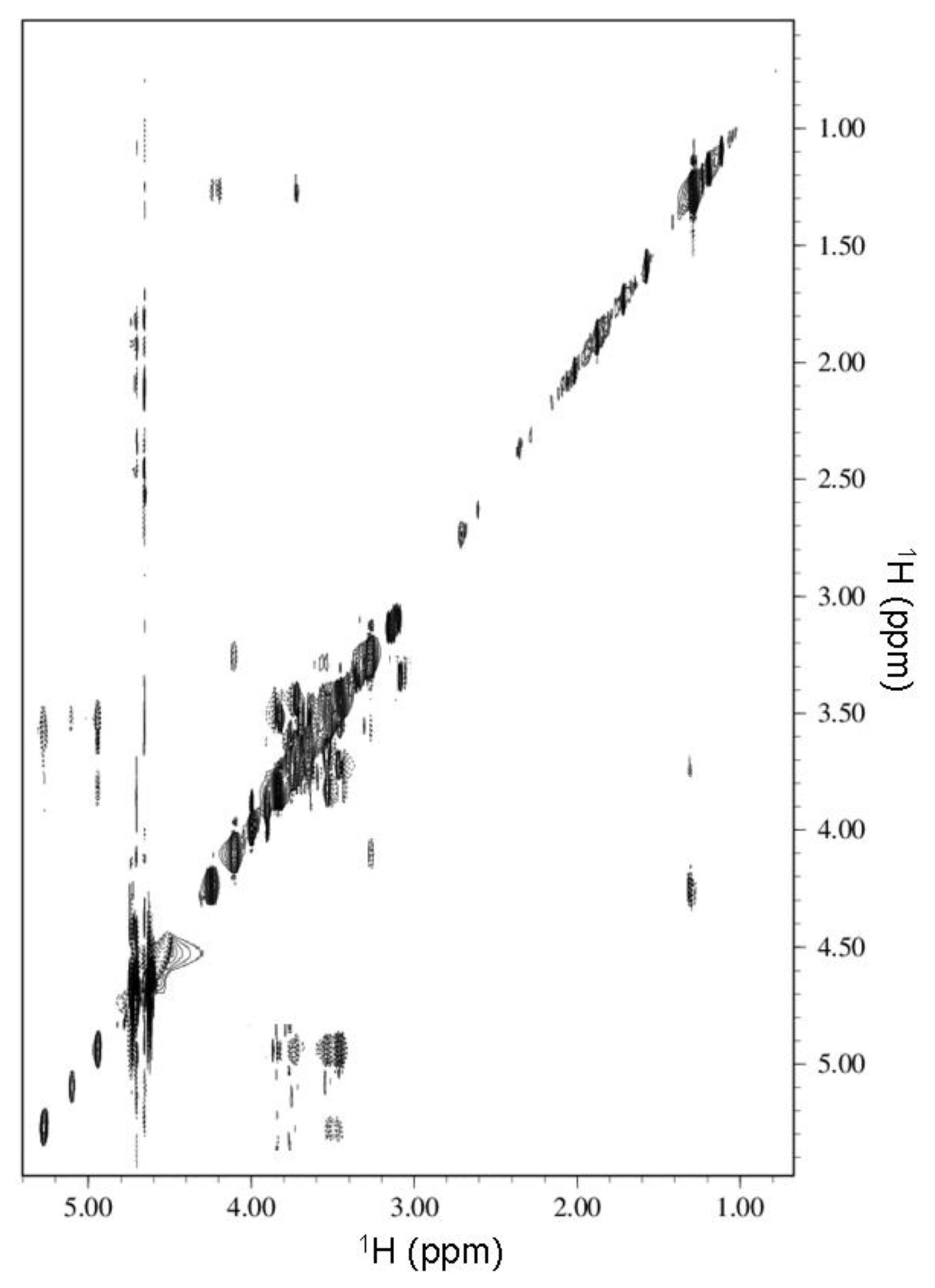
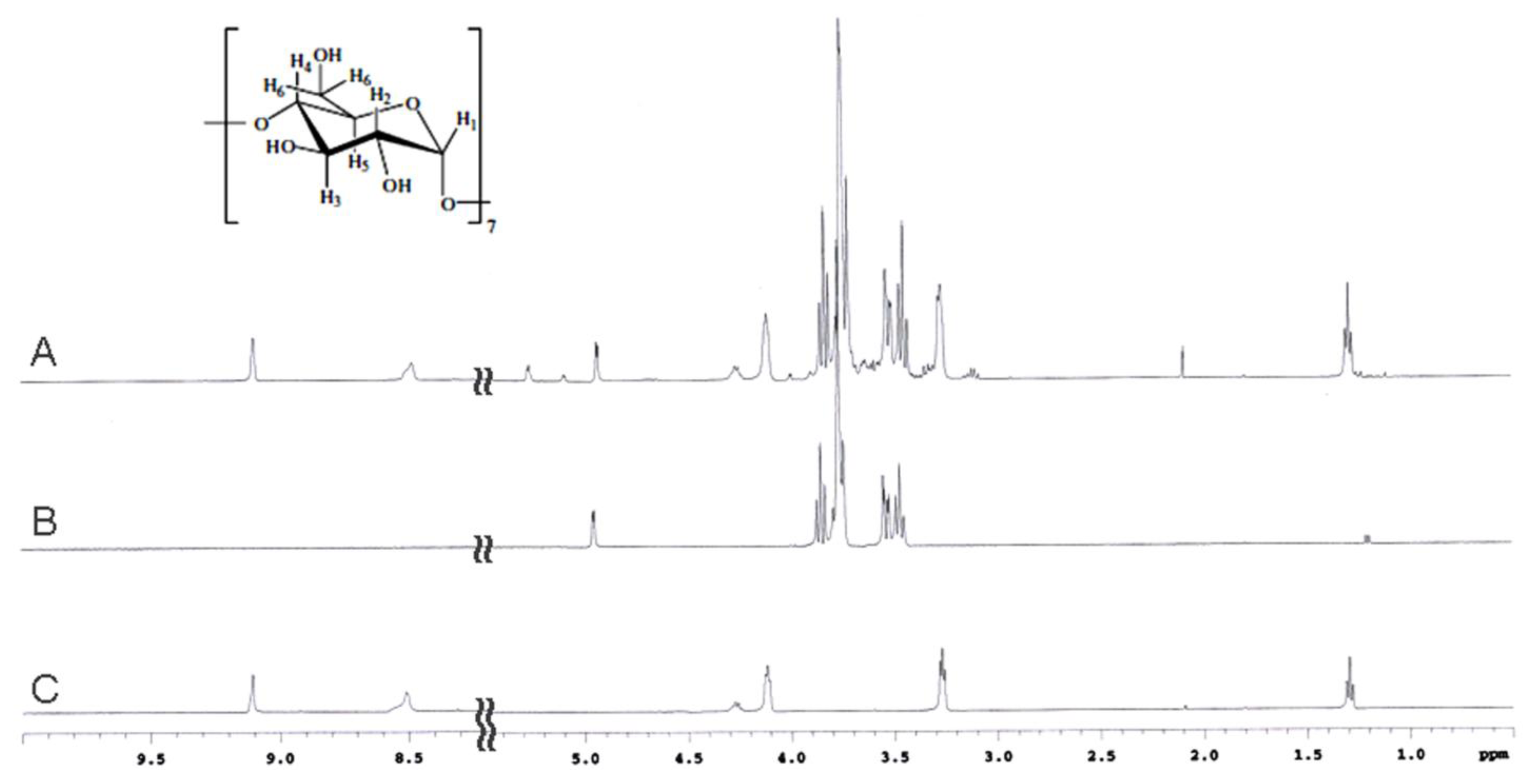
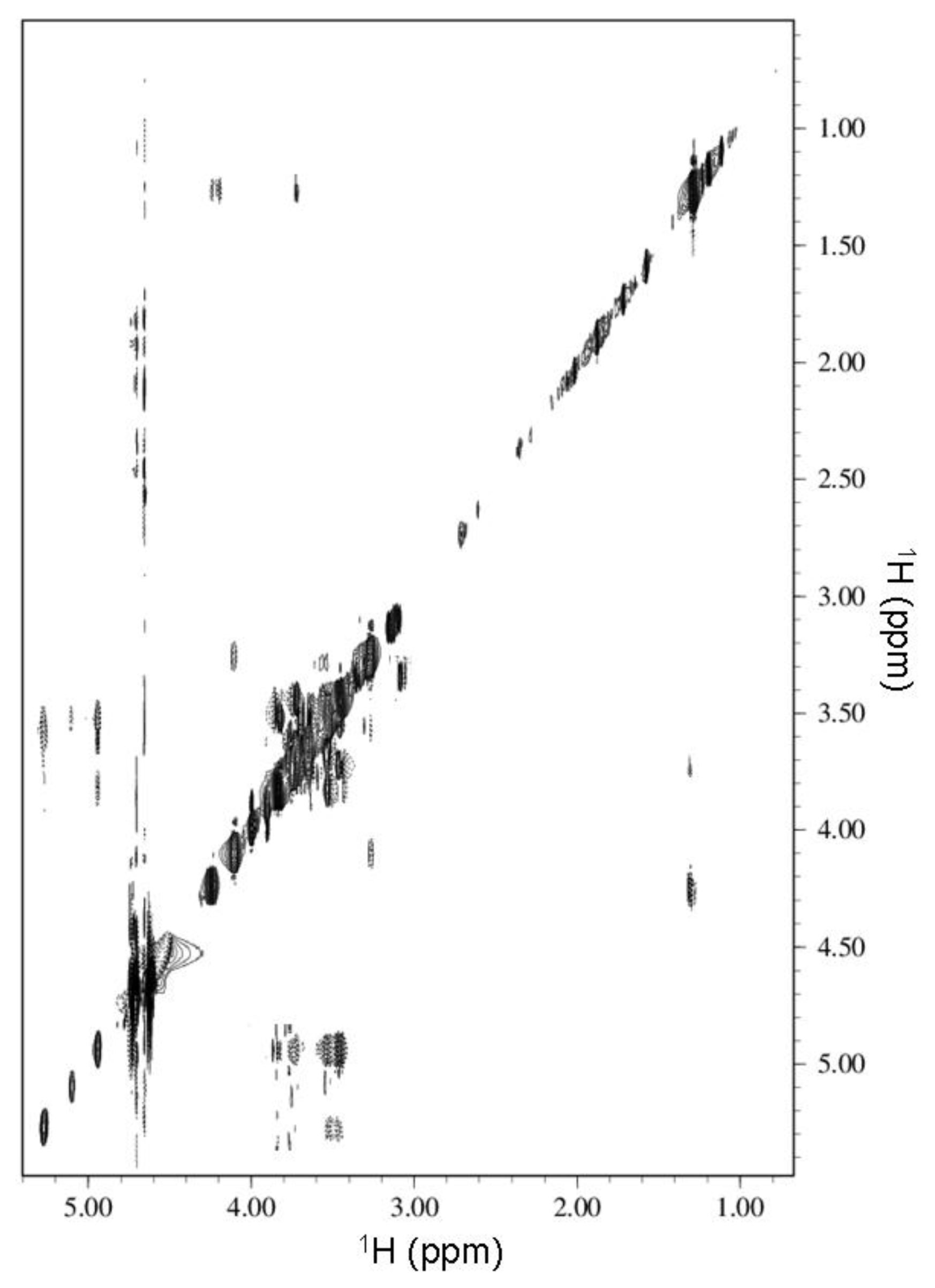
2.2. NMR Spectroscopy
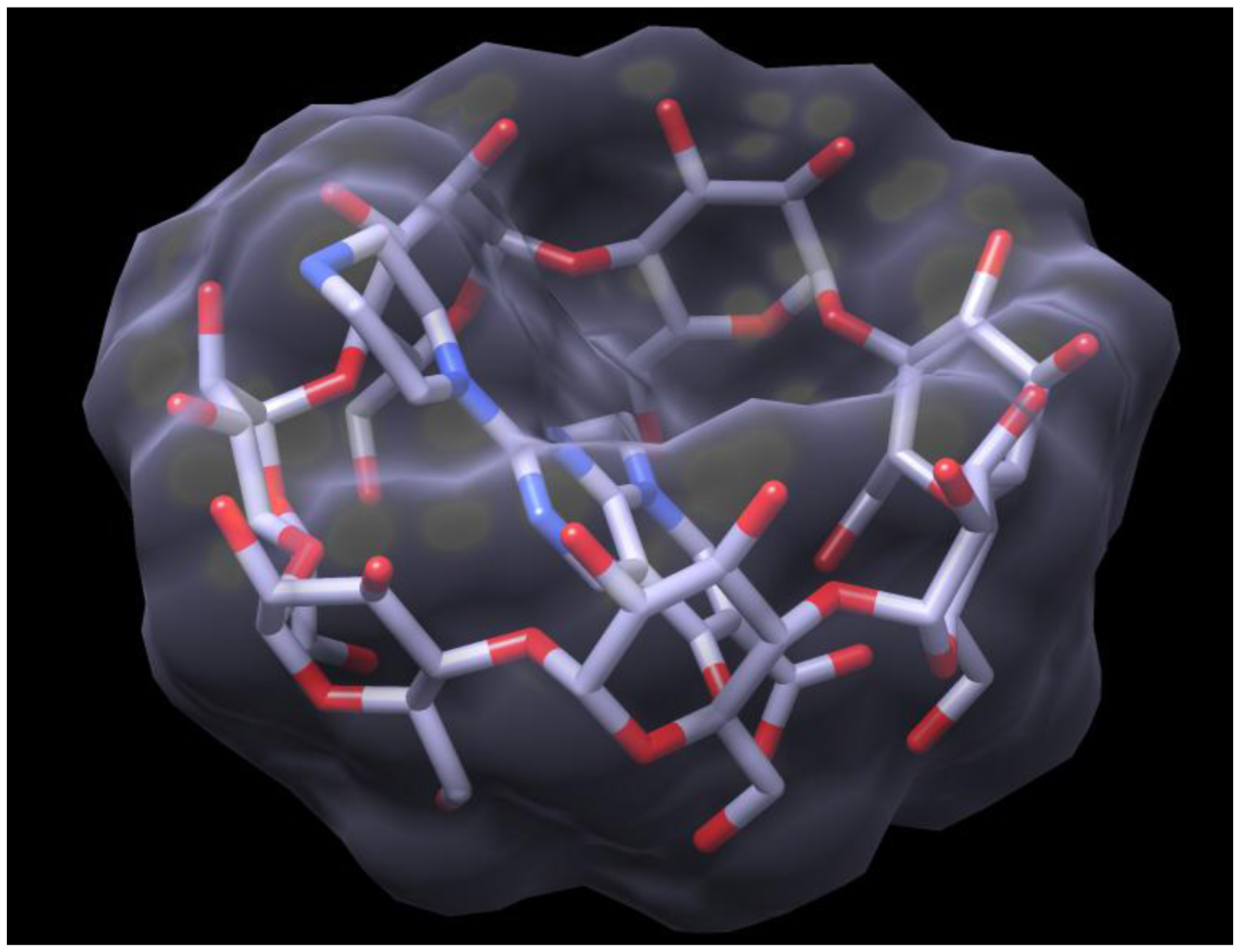
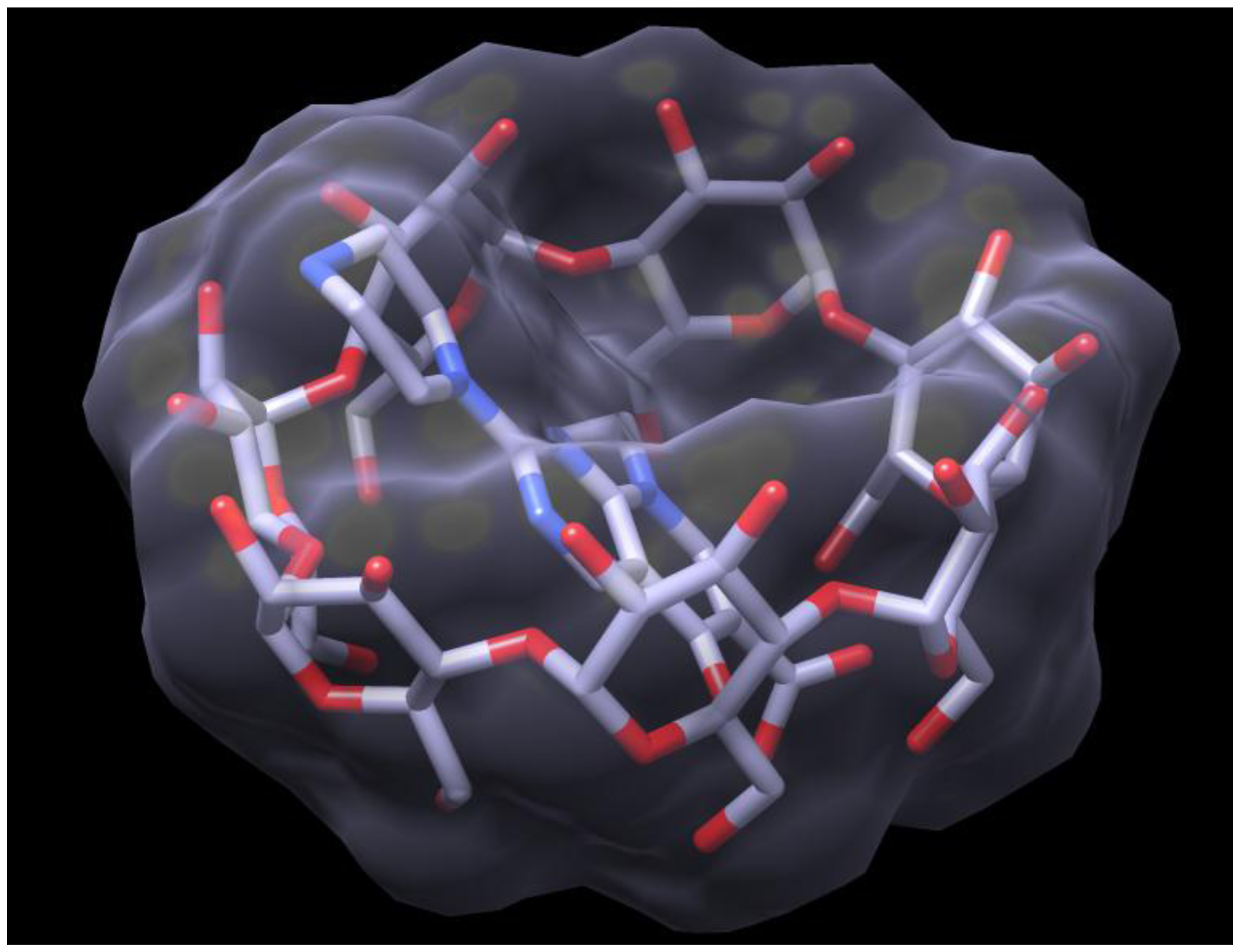
2.3. Docking of HPPA onto β-CD
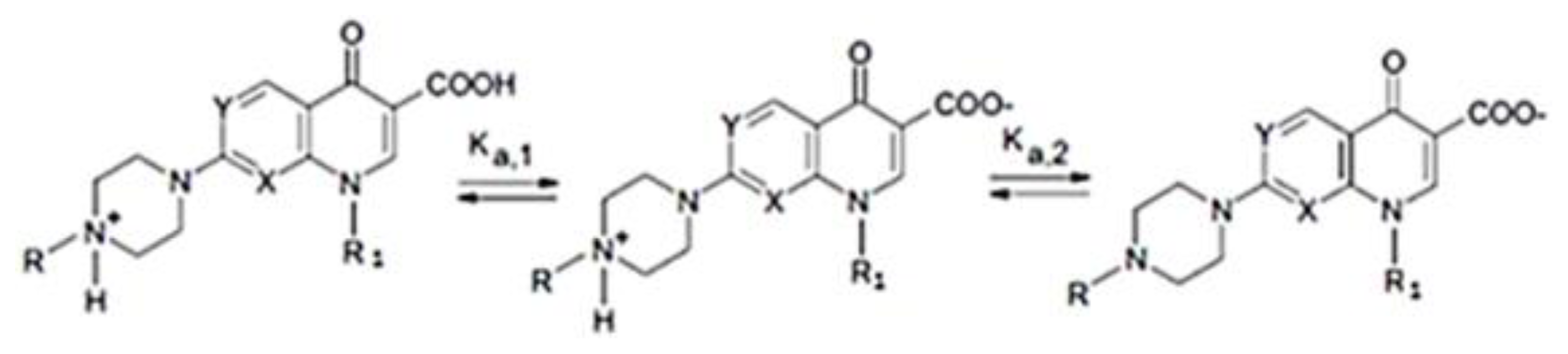
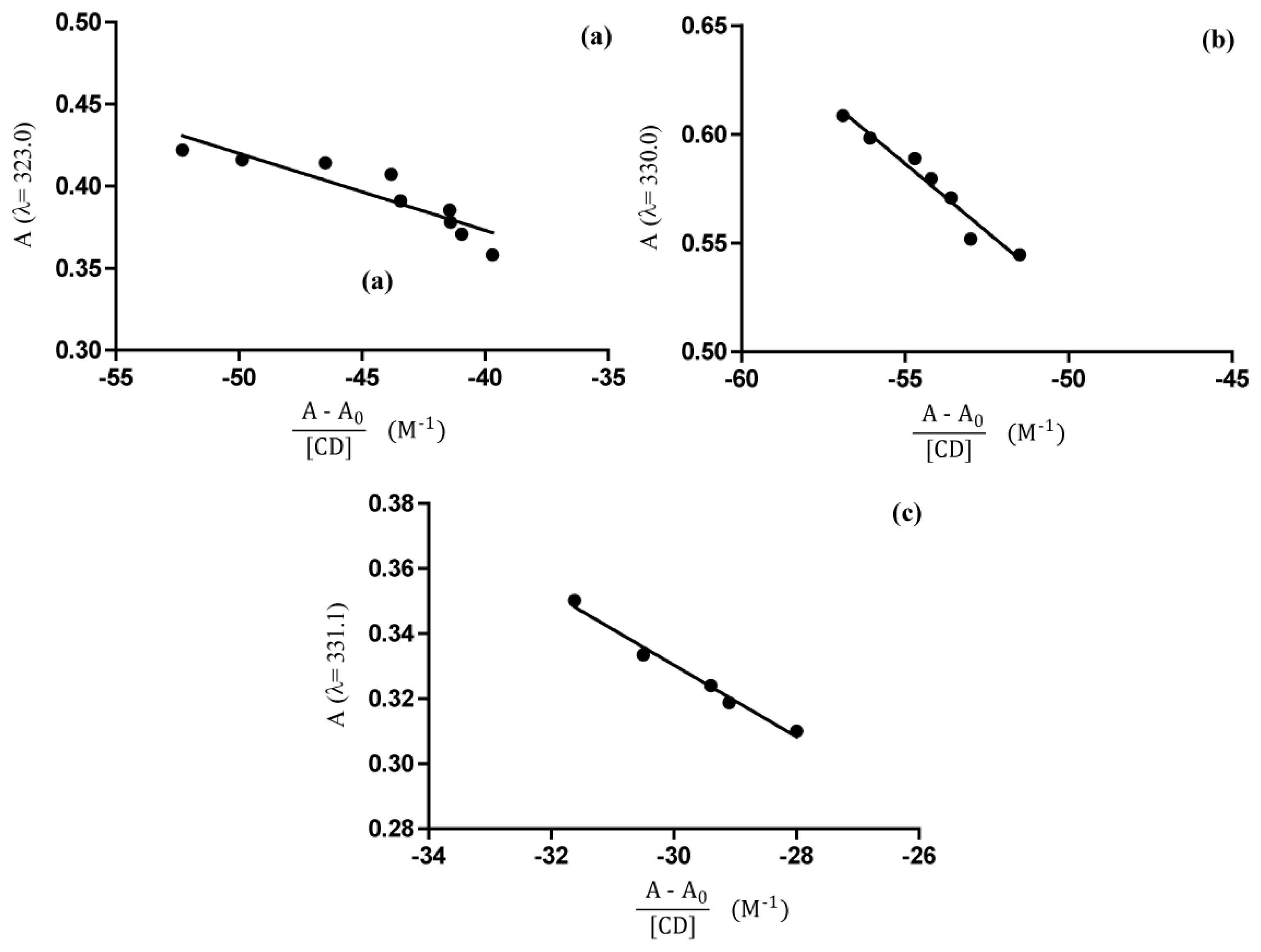
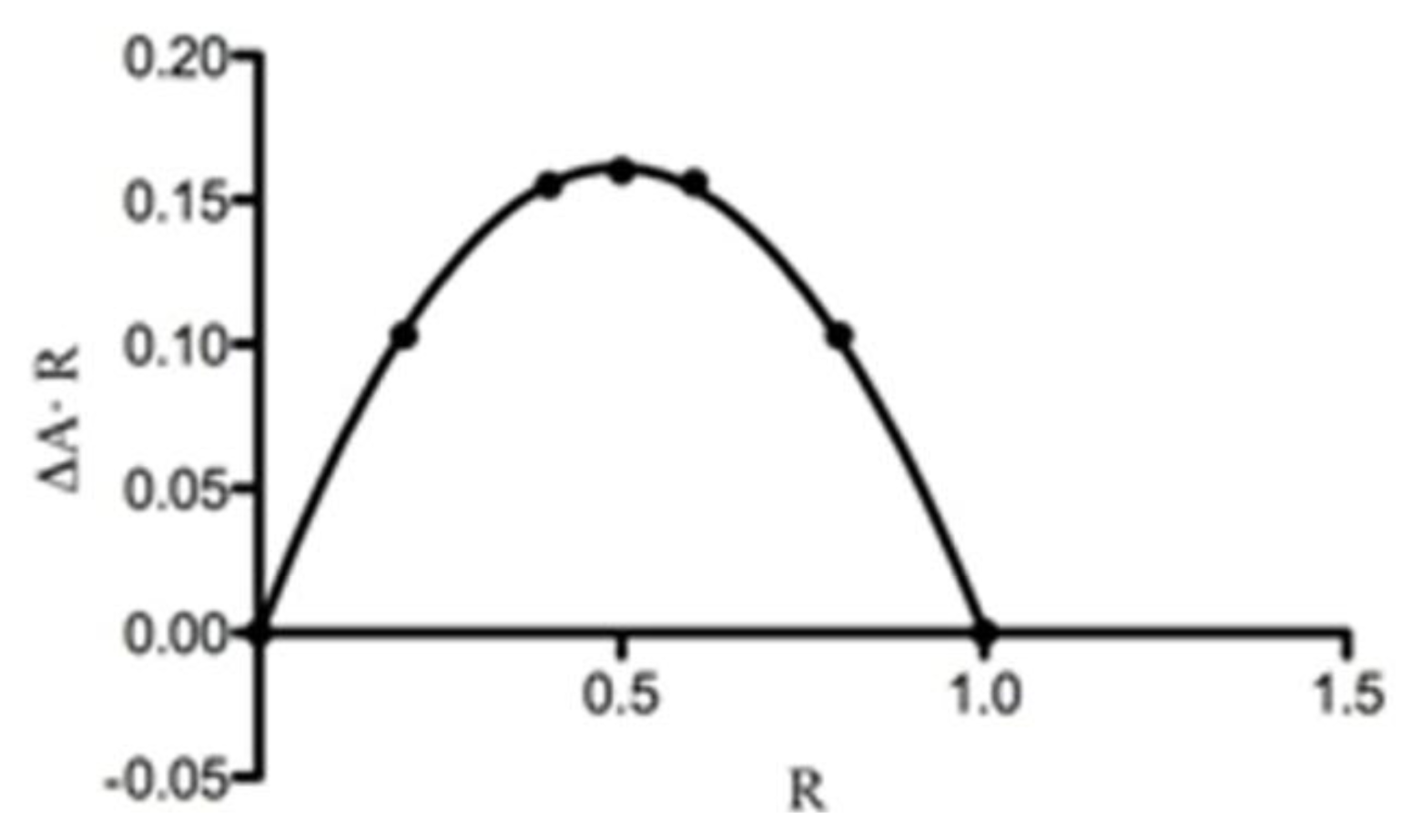
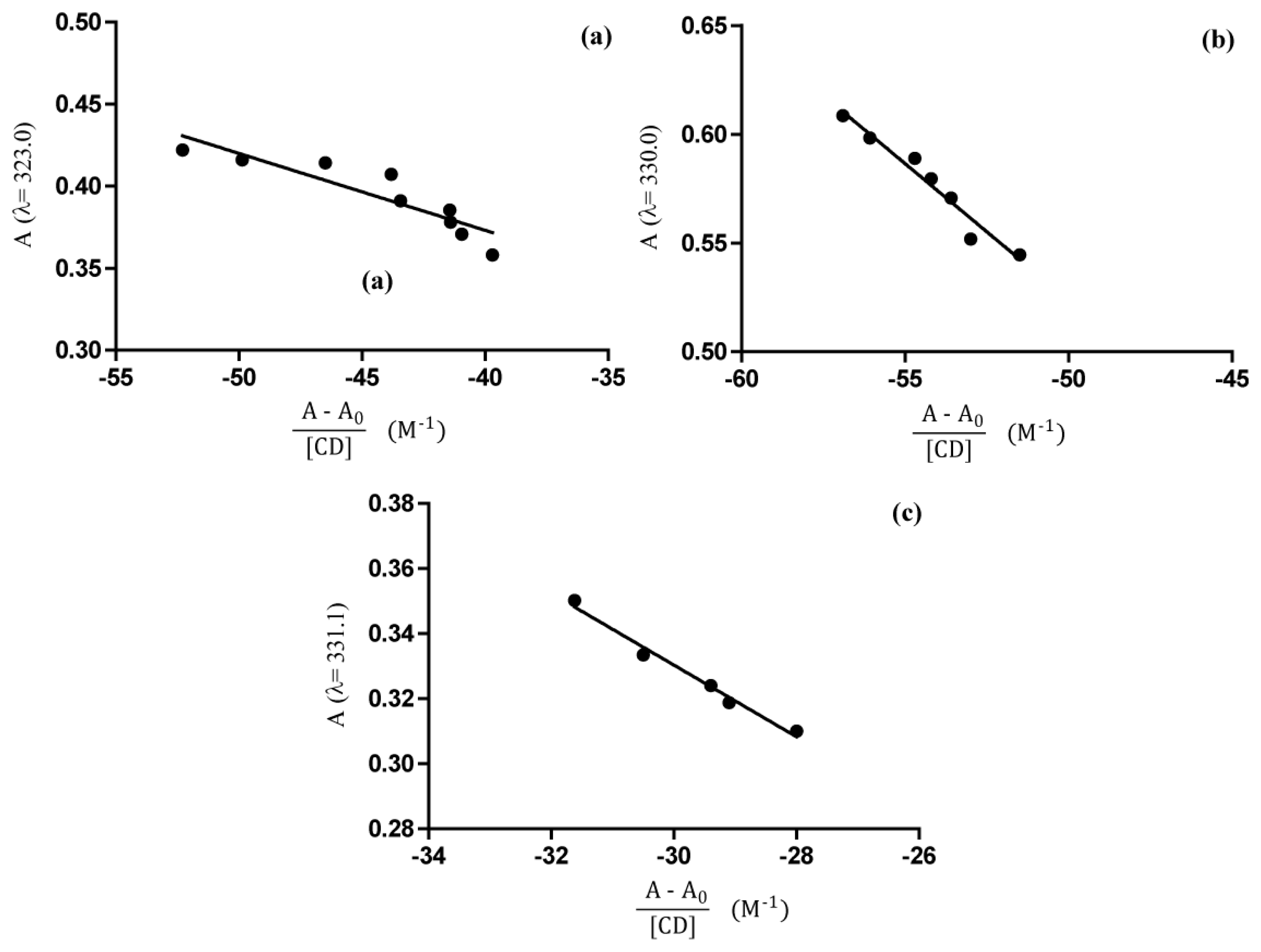
2.4. UV-Vis Spectroscopy
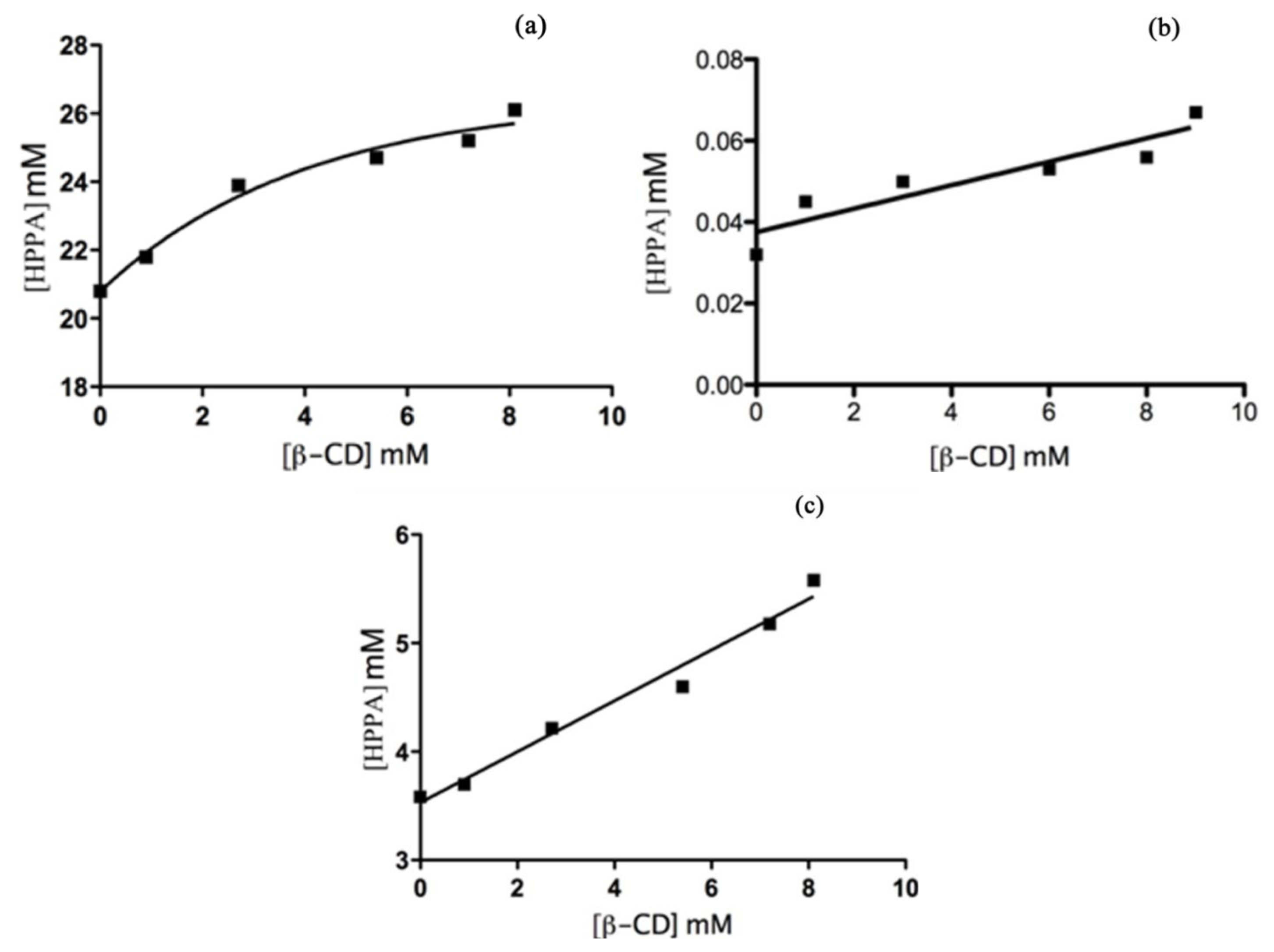
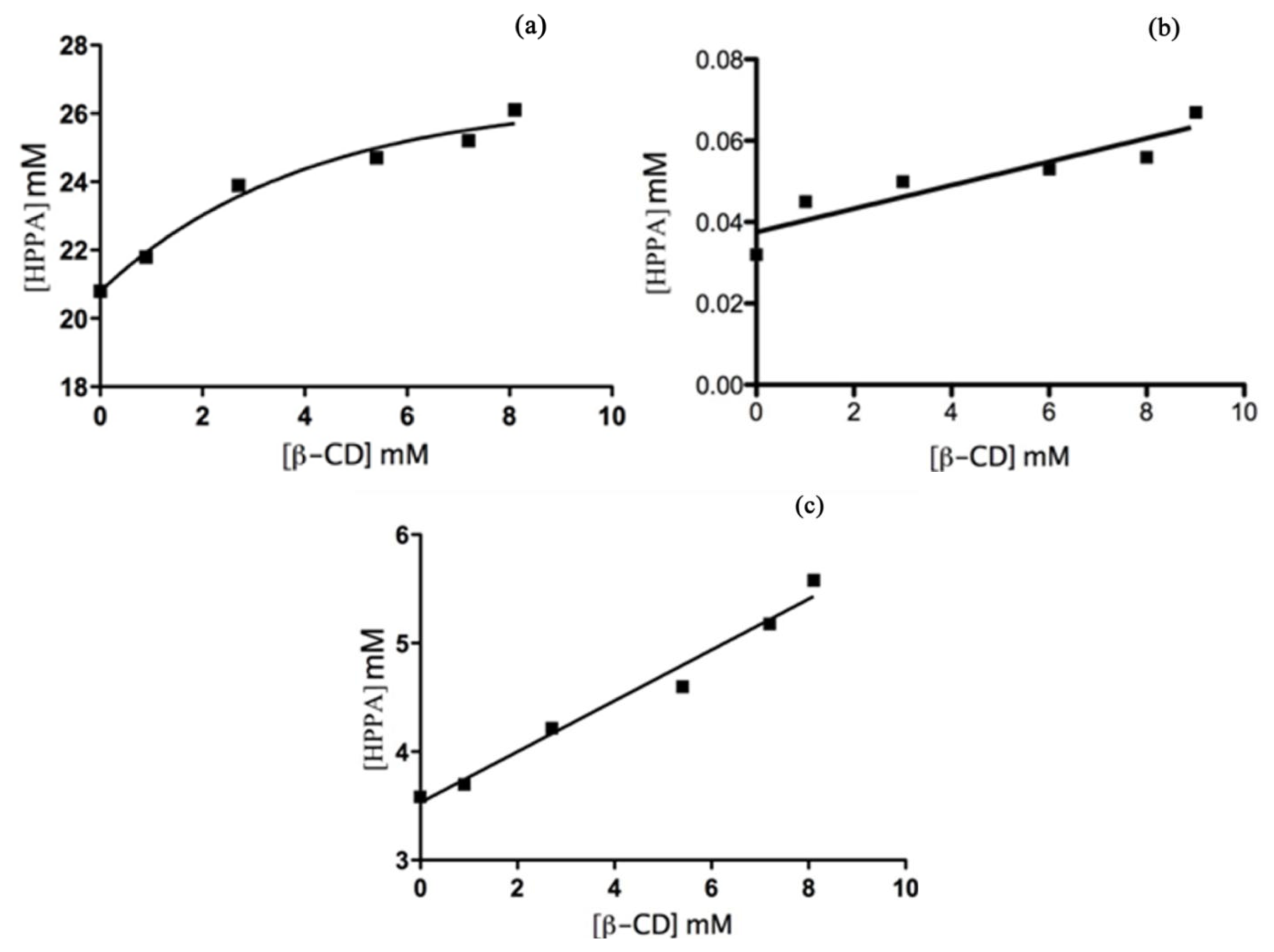
Phase Solubility Studies
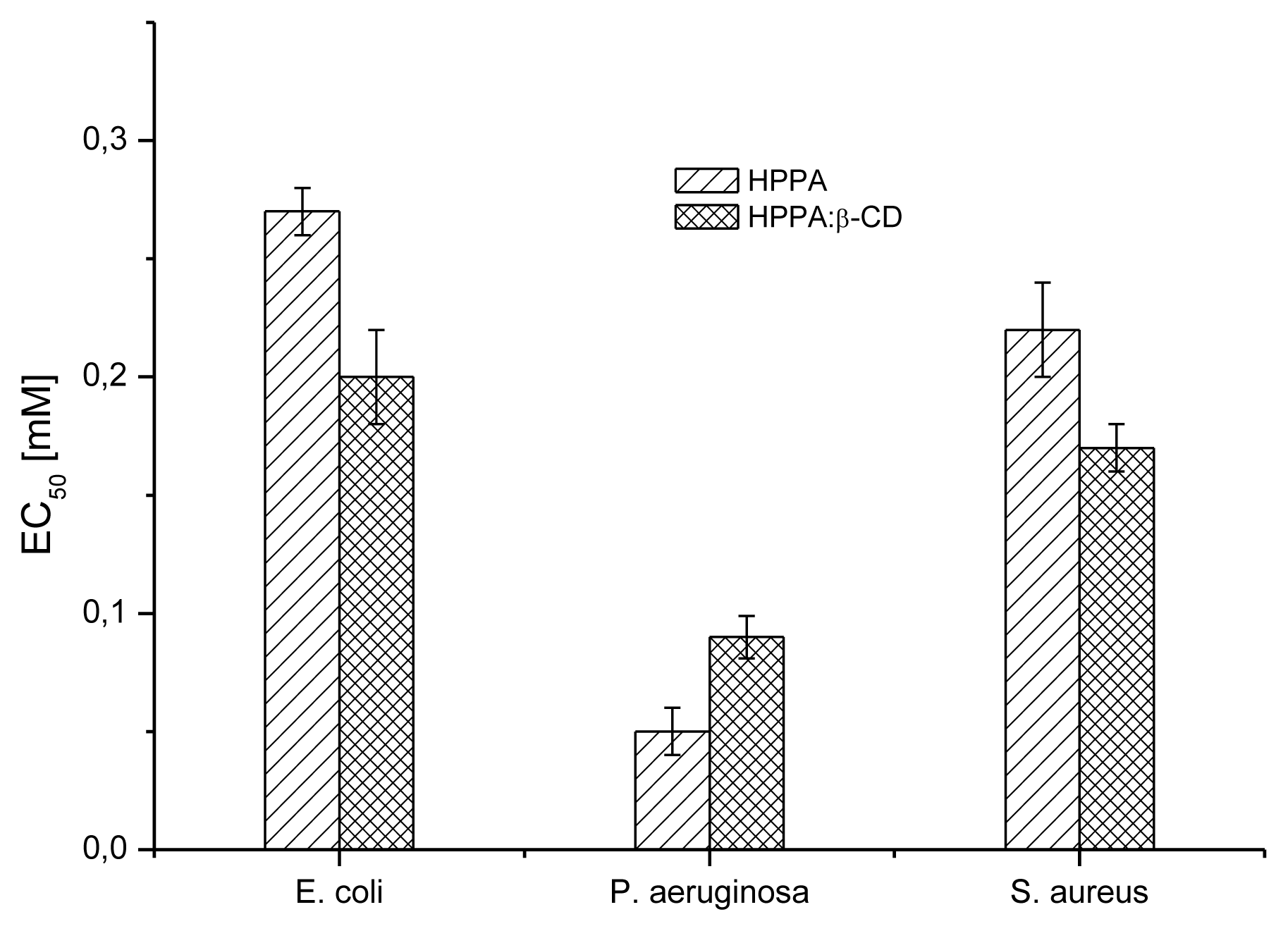
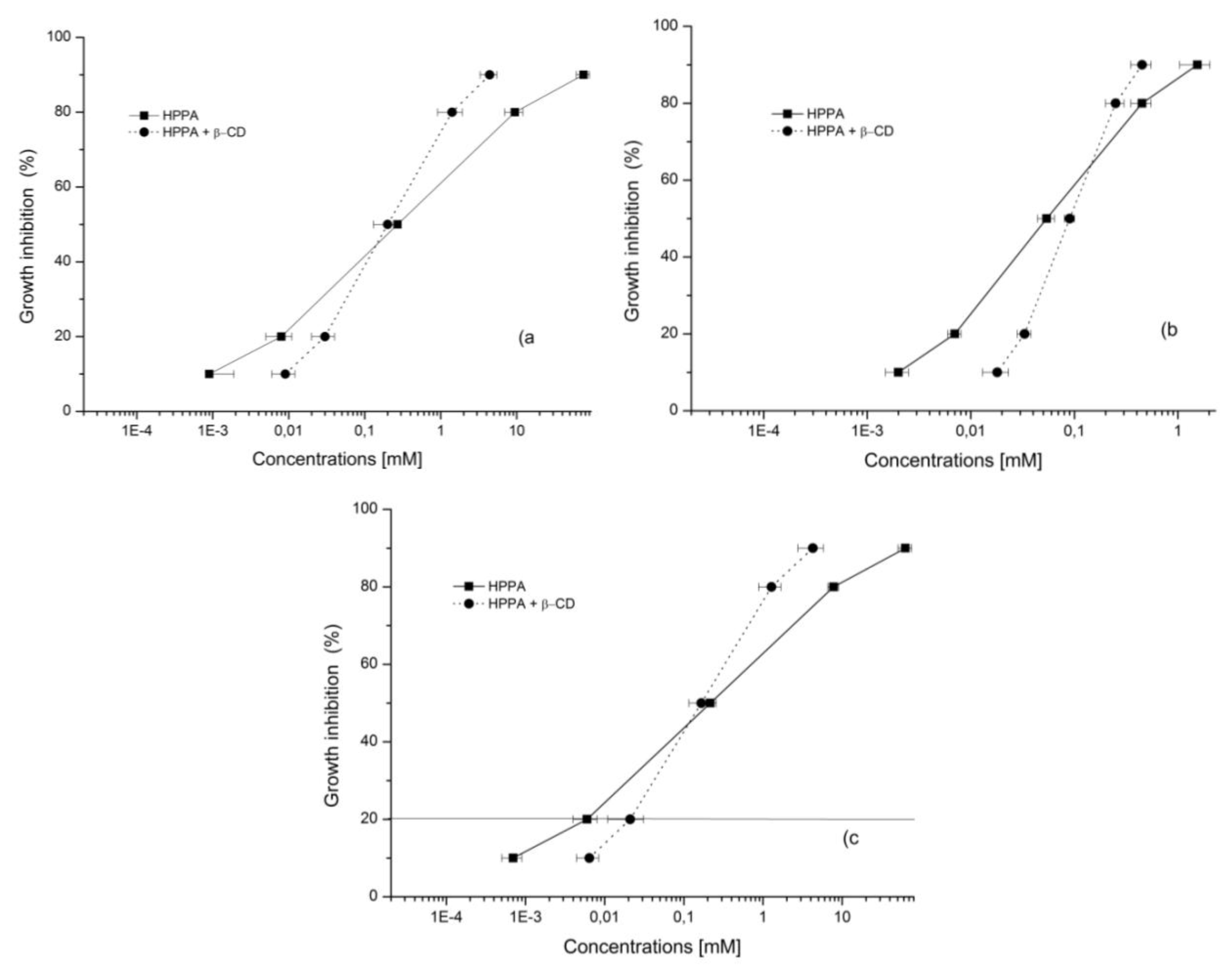
2.5. Bioactivity Evaluation
2.5.1. Microbial Susceptibility Test
2.5.2. MTT Assay
3. Experimental Section
3.1. Materials
3.2. Preparation of Solid Binary System
3.2.1. Physical Mixing Method
3.2.2. Kneading Method
3.3. Fourier Transform Infrared (FT-IR) Spectroscopy
3.4. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.5. Molecular Docking
3.6. Ultraviolet-Visible (UV-Vis) Spectroscopy
Phase Solubility Studies
3.7. Bioactivity Evaluation
3.7.1. Microbial Susceptibility Test
3.7.2. MTT-Assay
3.7.3. Data Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Matsumoto, J.; Minami, S. Pyrido[2,3-dipyrimidine antibacterial agents. 3,8-Alkyl-and 8-vinyl-5,8-dihydro-5-oxo-2-(1-piperazinyl)pyrido[2,3-d]-pyrimidine-6-carboxylic acids and their derivatives. J. Med. Chem 1975, 18, 74–79. [Google Scholar]
- Fonseca, I.; Martinex-Carrera, S.; Garcia-Blanco, S. Structure of pipemidic acid. Acta Crystallogr. Sect. C 1986, 42, 1618–1621. [Google Scholar]
- Toscano, M.A.; Serrao, A.; Ventimiglia, B.; Colosi, V.; Pennisi, M.; Morgana, R.; Bevilacqua, T. The use of pipemidic acid in urinary infections. Miner. Urol 1982, 34, 257–260. [Google Scholar]
- Hirai, K.; Ito, A.; Abe, Y.; Suzue, S.; Irikura, T.; Inoue, M.; Mitsuhashi, S. Comparative activities of AM-715 and pipemidic and nalidixic acids against experimentally induced systemic and urinary tract infections. Antimicrob. Agents Chemother 1981, 19, 188–189. [Google Scholar]
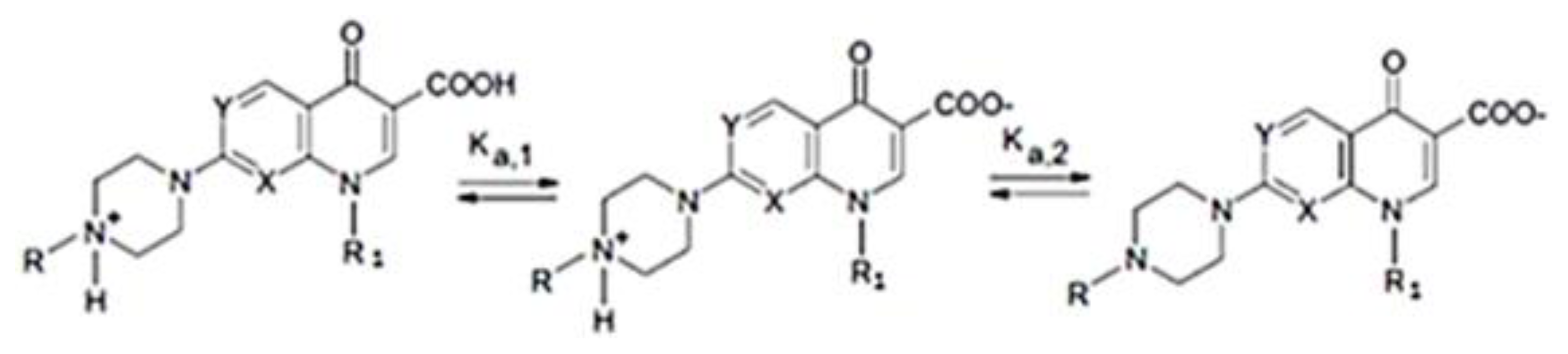
- Babić, S.; Horvat, A.J.M.; Pavlović, D.M.; Kaštelan-Macan, M. Determination of pKa values of active pharmaceutical ingredients. Trends Anal. Chem 2007, 26, 1043–1061. [Google Scholar]
- Shimizu, M.; Takase, Y.; Nakamura, S.; Katae, H.; Minami, A. Pipemidic acid: Its activities against various experimental infections. Antimicrob. Agents Chemother 1976, 9, 569–574. [Google Scholar]
- Zweerink, M.M.; Edison, A. Inhibition of micrococcus luteus DNA gyrase by norfloxacin and 10 other quinolone carboxylic acids. Antimicrob. Agents Chemother 1986, 29, 598–601. [Google Scholar]
- Efthimiadou, E.K.; Sanakis, Y.; Katsaros, N.; Karaliota, A.; Psomas, G. Transition metal complexes with the quinolone antibacterial agent pipemidic acid: Synthesis, characterization and biological activity. Polyhedron 2007, 26, 1148–1158. [Google Scholar]
- Zhang, C.L.; Wang, Y. Aqueous solubilities for ofloxacin, norfloxacin, lomefloxacin, ciprofloxacin, pefloxacin, and pipemidic acid from (293.15 to 323.15) K. J. Chem. Eng. Data 2008, 53, 1295–1297. [Google Scholar]
- Romero, S.; Bustamante, P.; Escalera, B.; Mura, P.; Cirri, M. Influence of solvent composition on the solid phase at equilibrium with saturated solutions of quinolones in different solvent mixtures. J. Pharm. Biomed. Anal 2004, 35, 715–726. [Google Scholar]
- Szejtli, J. Cyclodextrin Technology; Kluwer Academic: Dordecht, The Netherlands, 1989; pp. 8–72. [Google Scholar]
- Duchene, D.; Vaution, C.; Glomot, F. Cyclodextrin, their value in pharmaceutical technology. Drug Dev. Ind. Pharm 1988, 12, 2193–2215. [Google Scholar]
- Singh, R.; Bharti, N.; Madan, J.; Hiremath, S.N. Characterization of cyclodextrin inclusion complexes—A review. J. Pharm. Sci. Technol 2010, 2, 171–183. [Google Scholar]
- Pires, M.A.S.; dos Santos, R.A.S.; Sinisterra, R.D. Pharmaceutical composition of hydrochlorothiazide:β-cyclodextrin: Preparation by three different methods, physico-chemical characterization and in vivo diuretic activity evaluation. Molecules 2011, 16, 4482–4499. [Google Scholar]
- Wang, S.W.; Pan, S.L.; Huang, Y.C.; Guh, J.H.; Chiang, P.C.; Huang, D.Y.; Kuo, S.C.; Lee, K.H.; Teng, C.M. CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer Ther 2008, 7, 350–359. [Google Scholar]
- Chen, Y.C.; Lu, P.H.; Pan, S.L.; Teng, C.M.; Kuo, S.C.; Lin, T.P.; Ho, Y.F.; Huang, Y.C.; Guh, J.H. Quinolone analogue inhibits tubulin polymerization and induces apoptosis via Cdk1-involved signaling pathways. Biochem. Pharmacol 2007, 74, 10–19. [Google Scholar]
- Menezes, P.P.; Serafini, M.R.; Santana, B.V.; Nunes, R.S.; Quintans, L.J., Jr; Silva, G.F.; Medeiros, I.A.; Marchioro, M.; Fraga, B.P.; Santos, M.R.V.; et al. Acta 2012, 548, 45–50.
- Sahoo, S.; Chakraborti, C.K.; Behera, P.K. FTIR and Raman spectroscopic investigations of a controlled release Ciprofloxacin/Carbopol940 mucoadhesive suspension. Asian J. Pharm. Clin. Res 2012, 5, 125–130. [Google Scholar]
- Aithal, K.S.; Udupa, N. Physicochemical study of ciprofloxacin with β-cyclodextrin. Pharm. Sci. 1996, 2, 451–455. [Google Scholar]
- Li, J.; Zhao, C.; Chao, J. Investigation on the inclusion behavior of Norfloxacin with 2-methyl-β-cyclodextrin. J. Inclusion Phenom. Macrocyclic Chem 2008, 62, 325–331. [Google Scholar]
- Rawat, S.; Jain, S.K. Solubility enhancement of celecoxib using β-cyclodextrin inclusion complexes. Eur. J. Pharm. Biopharm 2004, 57, 263–267. [Google Scholar]
- Job, P. Formation and stability of inorganic complexes in solution. Annal. Chim. Fr 1928, 9, 113–203. [Google Scholar]
- Huang, C.Y. Determination of binding stoichiometry by the continuous variation method: The Job plot. Methods Enzymol 1982, 87, 509–525. [Google Scholar]
- Connors, K.A. The stability of cyclodextrin complexes in solution. Chem. Rev 1997, 97, 1325–1357. [Google Scholar]
- Benesi, H.A.; Hildebrand, J.H. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J. Am. Chem. Soc 1949, 71, 2703–2707. [Google Scholar]
- Iacovino, R.; Caso, J.V.; Rapuano, F.; Russo, A.; Isidori, M.; Lavorgna, M.; Malgieri, G.; Isernia, C. Physicochemical characterization and cytotoxic activity evaluation of hydroxymethylferrocene:β-Cyclodextrin inclusion complex. Molecules 2012, 17, 6056–6070. [Google Scholar]
- Higuchi, T.; Connors, K.A. Phase solubility techniques. Anal. Chem. Instrum 1965, 4, 117–212. [Google Scholar]
- Zughul, M.B. Rigorous nonlinear regression analysis of phase solubility diagrams to obtain complex stoichiometry and true thermodynamic drug-cyclodextrin complexation parameters. J. Incl. Phenom. Macrocyclic Chem 2007, 57, 525–530. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J. Pharm. Pharmacol 2011, 63, 1119–1135. [Google Scholar]
- Durán-Merás, I.; Muñoz De La Peña, A.; Salinas López, F.; Rodríguez Cáceres, M.I. Complexation study and spectrofluorimetric determination of pipemidic acid with γ-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem 2005, 51, 137–142. [Google Scholar]
- Shimizu, M.; Takase, Y.; Nakamura, S.; Katae, H.; Minami, A.; Nakata, K.; Inoue, S.; Ishiyama, M.; Kubo, Y. Pipemidic acid, a new antibacterial agent active against pseudomonas aeruginosa: In vitro properties. Antimicrob. Agents Chemother. 1975, 8, 132–138. [Google Scholar]
- Chin, N.; Neu, H.C. Ciprofloxacin, a quinolone carboxylic acid compound active against aerobic and anaerobic bacteria. Antimicrob. Agents Chemother 1984, 25, 319–326. [Google Scholar]
- Wada, K.; Kariyama, R.; Mitsuhata, R.; Uehara, S.; Watanabe, T.; Monden, K.; Kumon, H. Experimental and clinical studies on fluoroquinolone-insusceptible Escherichia coli isolated from patients with urinary tract infections from 1994 to 2007. Acta Med. Okayama 2009, 63, 263–272. [Google Scholar]
- Yang, L.; Tao, D.; Yang, X.; Li, Y.; Guo, Y. Synthesis, characterization, and antibacterial activities of some rare earth metal complexes of Pipemidic Acid. Chem. Pharm. Bull 2003, 51, 494–498. [Google Scholar]
- Sha, J.Q.; Liang, L.Y.; Li, X.M.; Zhang, Y.; Yan, H.; Chen, G. Ligation of the quinolone antibacterial agent pipemidic acid to Keggin polyoxotungstates. Polyhedron 2011, 30, 1657–1662. [Google Scholar]
- Sha, J.Q.; Liang, L.Y.; Yan, P.F.; Li, G.M.; Wanga, C.; Mab, D.Y. Study on ligation of copper complexes of the quinolone antibacterial drugs and octamolybdates POMs. Polyhedron 2012, 31, 422–430. [Google Scholar]
- Cavanagh, J.; Fairbrother, W.; Palmer, A.G.; Skelton, N.J. Protein NMR Spectroscopy, Principles, and Practice; Academic Press, Inc.: San Diego, CA, USA, 1996. [Google Scholar]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. A 1995, 112, 275–279. [Google Scholar]
- Bartels, C.; Xia, T.; Billeter, M.; Gunter, P.; Wüthrich, K.J. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1. [Google Scholar]
- Ritchie, D.W.; Venkatraman, V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar]
- Moriwaki, C.; Costa, G.L.; Ferracini, C.N.; de Moraes, F.F.; Zanin, G.M.; Pineda, E.A.G.; Matioli, G. Enhancement of solubility of albendazole by complexation with β-Cyclodextrin. Braz. J. Chem. Eng. 2008, 25, 255–267. [Google Scholar]
- Othman, M.; San Loh, H.; Wiart, C.; Khoo, T.J.; Hon Lim, K.; Nee Ting, K. Optimal methods for evaluating antimicrobial activities from plant extracts. J. Microbiol. Methods 2011, 84, 161–166. [Google Scholar]
- Mothanna, A.; Rosli, R.; Swee-Keong, Y.; Abdul-Rahman, O.; Abdul-Manaf, A.; Noorjahan, B.A. Selective Cytotoxicity of goniothalamin against hepatoblastoma HepG2 cells. Molecules 2011, 16, 2944–2959. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Kb (M−1) UV-Vis | Kb (M−1) PSD |
|---|---|---|
| 4.6 | 215.2 | 250.8 |
| 6.8 | 79.7 | 88.5 |
| 8.6 | 90.8 | 86.7 |
| Correlation type | Shape only |
|---|---|
| FFT Mode | 3D fast life |
| Receptor range angle | 180 |
| Ligand range angle | 180 |
| Twist range | 360 |
| Distance range | 40 |
| Docking main scan | 16 |
| Docking main search | 30 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iacovino, R.; Rapuano, F.; Caso, J.V.; Russo, A.; Lavorgna, M.; Russo, C.; Isidori, M.; Russo, L.; Malgieri, G.; Isernia, C. β-Cyclodextrin Inclusion Complex to Improve Physicochemical Properties of Pipemidic Acid: Characterization and Bioactivity Evaluation. Int. J. Mol. Sci. 2013, 14, 13022-13041. https://doi.org/10.3390/ijms140713022
Iacovino R, Rapuano F, Caso JV, Russo A, Lavorgna M, Russo C, Isidori M, Russo L, Malgieri G, Isernia C. β-Cyclodextrin Inclusion Complex to Improve Physicochemical Properties of Pipemidic Acid: Characterization and Bioactivity Evaluation. International Journal of Molecular Sciences. 2013; 14(7):13022-13041. https://doi.org/10.3390/ijms140713022
Chicago/Turabian StyleIacovino, Rosa, Filomena Rapuano, Jolanda Valentina Caso, Agostino Russo, Margherita Lavorgna, Chiara Russo, Marina Isidori, Luigi Russo, Gaetano Malgieri, and Carla Isernia. 2013. "β-Cyclodextrin Inclusion Complex to Improve Physicochemical Properties of Pipemidic Acid: Characterization and Bioactivity Evaluation" International Journal of Molecular Sciences 14, no. 7: 13022-13041. https://doi.org/10.3390/ijms140713022