Effects of Ospemifene on Drug Metabolism Mediated by Cytochrome P450 Enzymes in Humans in Vitro and in Vivo

Abstract
:1. Introduction
2. Results and Discussion
2.1. CYP Inhibition in Vitro
2.2. In Vitro CYP Induction in Human Hepatocytes
2.3. Clinical Pharmacokinetics Studies
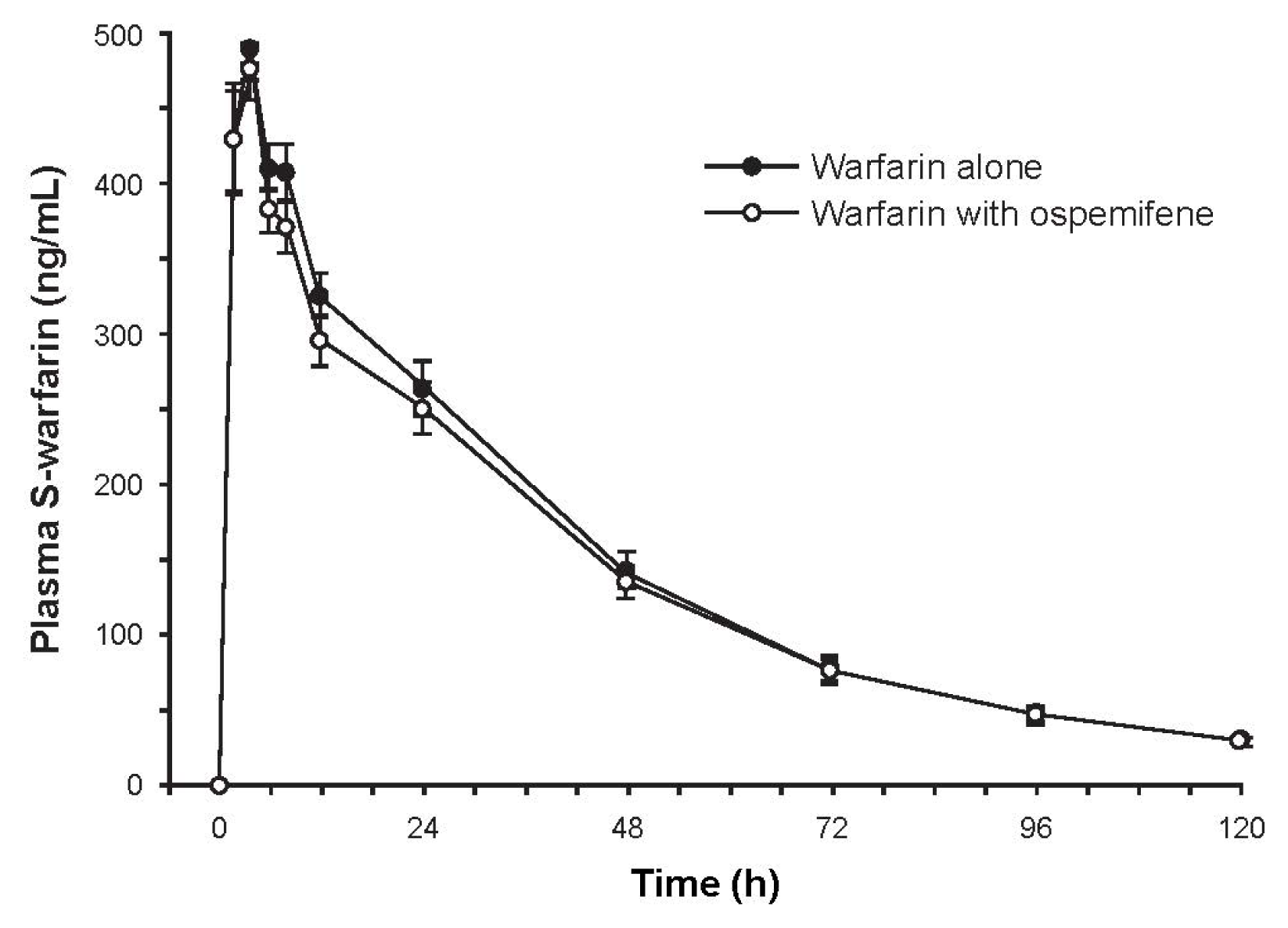
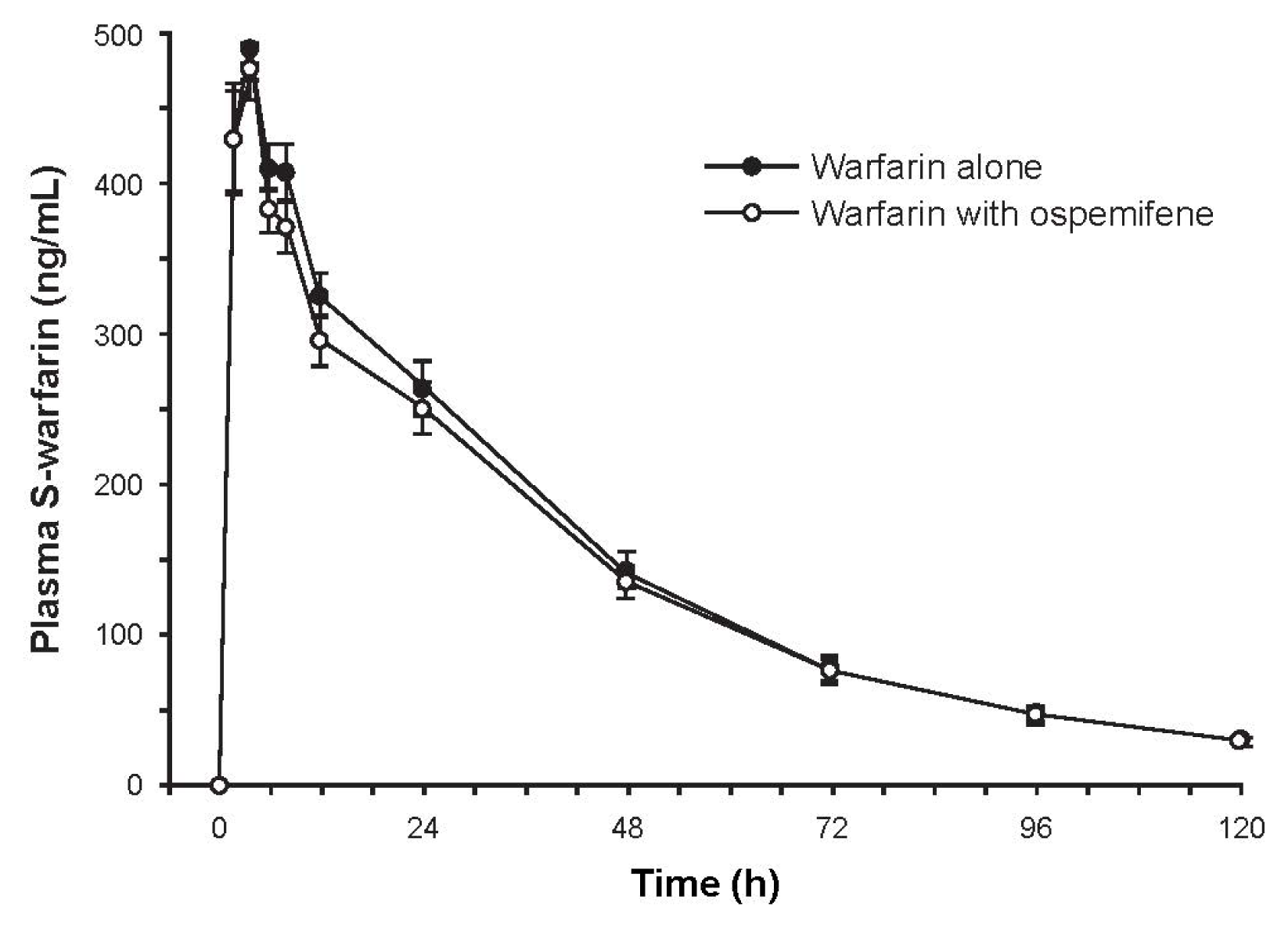
2.3.1. Study 1: The Effect of Ospemifene on Warfarin Pharmacokinetics
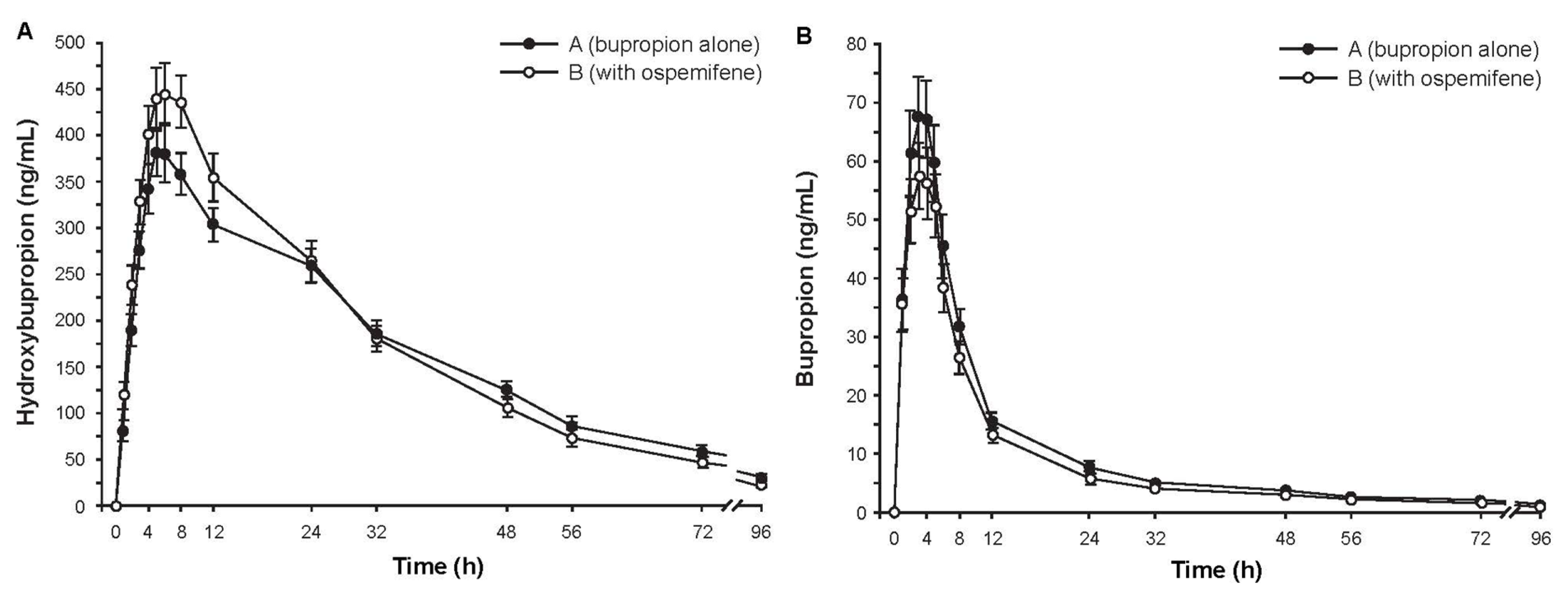
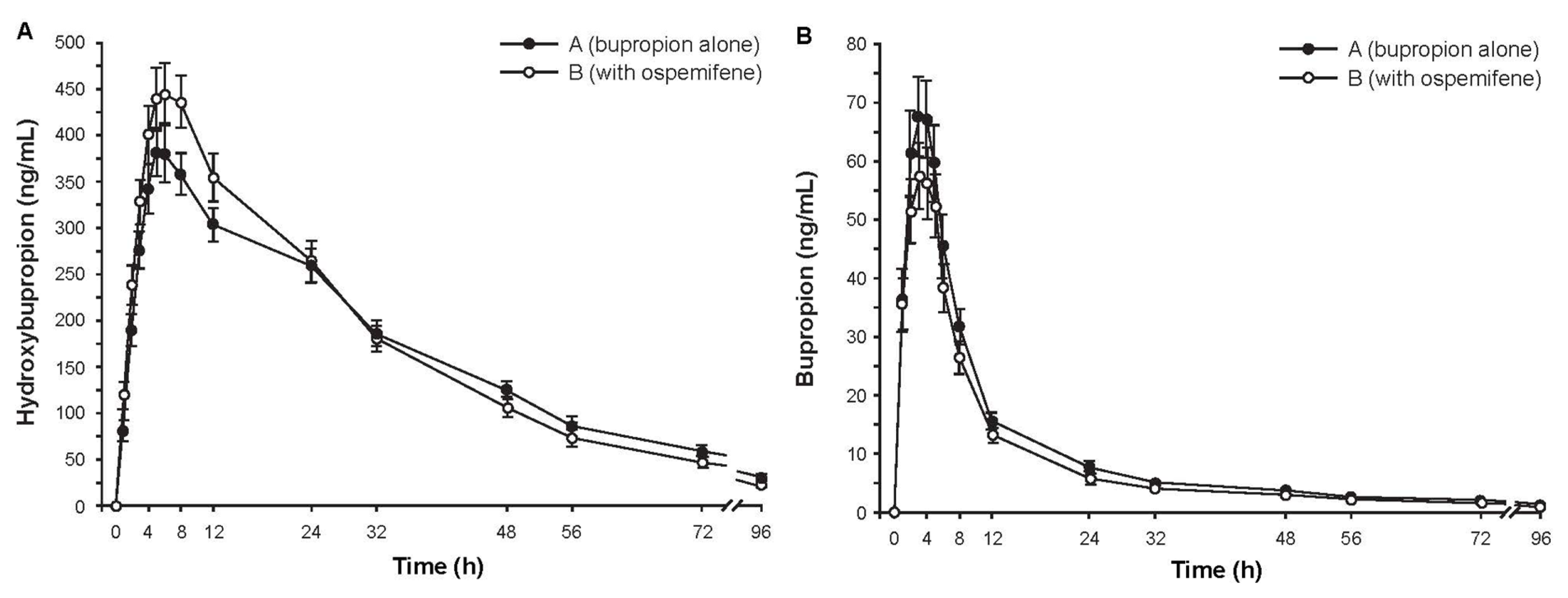
2.3.2. Study 2: The Effect of Ospemifene on Bupropion Pharmacokinetics
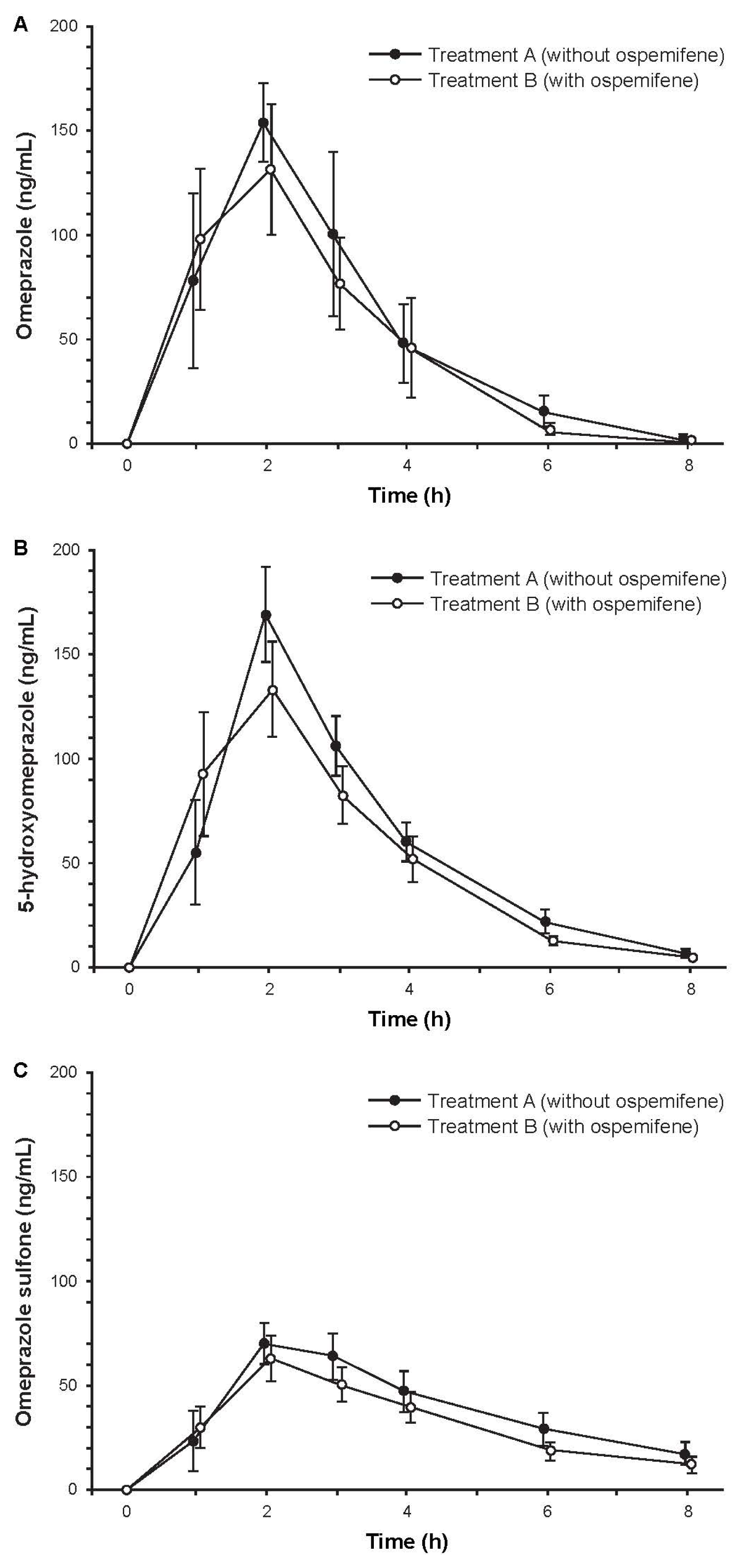
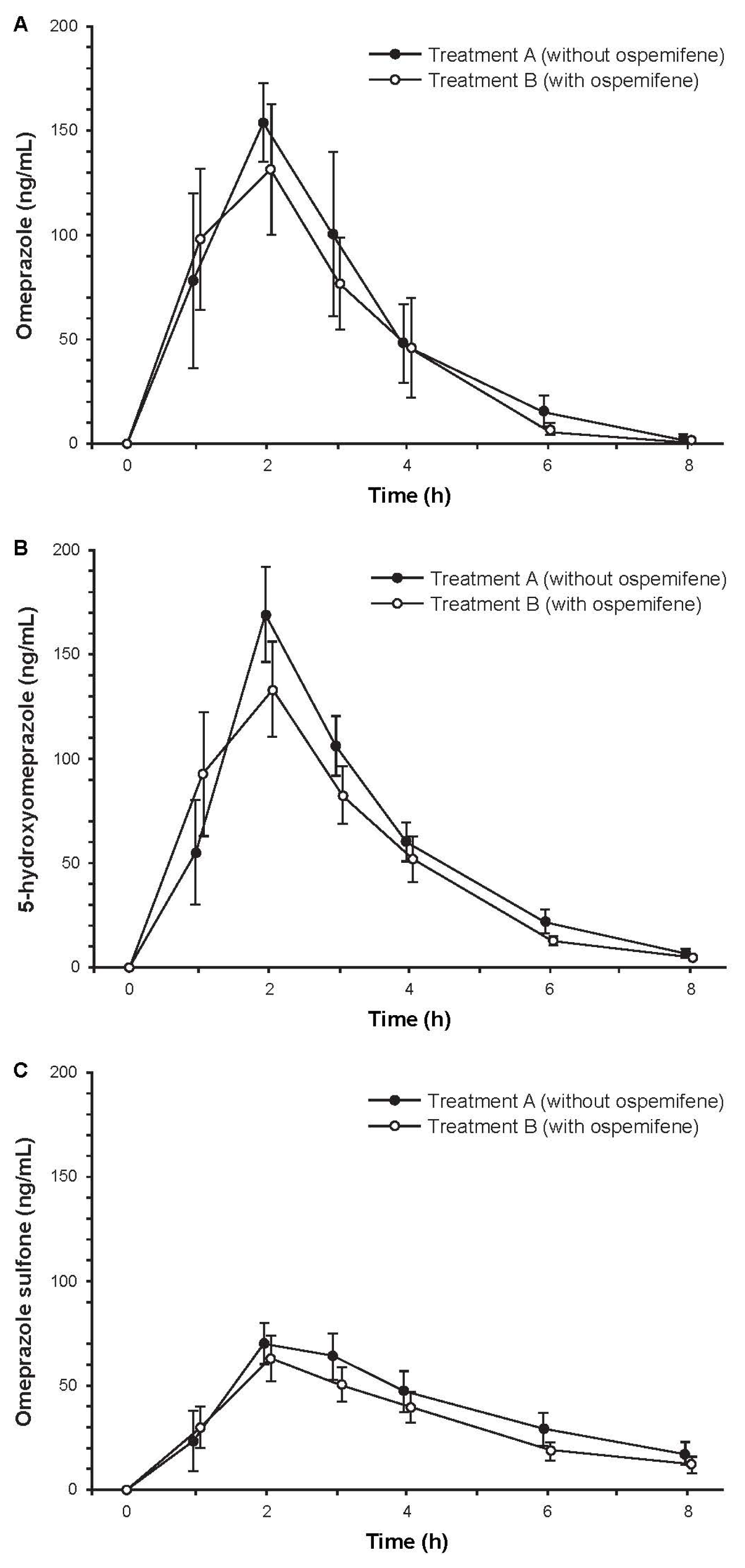
2.3.3. Study 3: The Effect of Ospemifene on Omeprazole Pharmacokinetics
2.3.4. Overall Safety
3. Experimental Section
3.1. In Vitro Metabolism
3.1.1. Reagents
3.1.2. Human Liver Microsomal Preparations
3.1.3. CYP Inhibition Assay
3.1.4. Determination of Inhibition Constants
3.1.5. Assessment of CYP Induction in Isolated Human Hepatocytes
3.2. In Vivo Drug Interaction Studies
Pharmacokinetic Parameters
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Bachmann, G.A.; Komi, J.O. The Ospemifene Study Group. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: Results from a pivotal phase 3 study. Menopause 2010, 17, 480–486. [Google Scholar]
- McCall, J.L.; DeGregorio, M.W. Pharmacologic evaluation of ospemifene. Expert Opin. Drug Metab. Toxicol 2010, 6, 773–779. [Google Scholar]
- Komi, J.; Lankinen, K.S.; Härkönen, P.; DeGregorio, M.W.; Voipio, S.; Kivinen, S.; Tuimala, R.; Vihtamäki, T.; Vihko, K.; Ylikorkala, O.; et al. Effects of ospemifene and raloxifene on hormonal status, lipids, genital tract, and tolerability in postmenopausal women. Menopause 2005, 12, 202–209. [Google Scholar]
- Rutanen, E.M.; Heikkinen, J.; Halonen, K.; Komi, J.; Lammintausta, R.; Ylikorkala, O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: A double-blind, randomized trial. Menopause 2003, 10, 433–439. [Google Scholar]
- Simon, J.A.; Lin, V.H.; Radovich, C.; Bachmann, G.A. The Ospemifene Study Group. One-year long-term safety extension study of ospemifene for the treatment of vulvar and vaginal atrophy in postmenopausal women with a uterus. Menopause 2013, 20, 418–427. [Google Scholar]
- Portman, D.J.; Bachmann, G.A.; Simon, J.A. The Ospemifene Study Group. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause 2013, 20, 623–630. [Google Scholar]
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. Pharmacokinetics of selective estrogen receptor modulators. Clin. Pharmacokinet 2003, 42, 361–372. [Google Scholar]
- Tolonen, A.; Koskimies, P.; Turpeinen, M.; Uusitalo, J.; Lammintausta, R.; Pelkonen, O. Ospemifene metabolism in humans in vitro and in vivo: Metabolite identification, quantitation, and CYP assignment of major hydroxylations. Drug Metabol. Drug Interact 2013, 14, 1–9. [Google Scholar]
- Koskimies, P.; Turunen, J.; Lammintausta, R.; Scheinin, M. Single-dose and steady-state pharmacokinetics of ospemifene, a selective estrogen receptor modulator, in postmenopausal women. Int. J. Clin. Pharm. 2013. Accepted for publication. [Google Scholar]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch. Toxicol 2008, 82, 667–715. [Google Scholar]
- European Medicines Agency, Committee for Human Medicinal Products (CHMP). Guideline on the investigation of drug interactions, CPMP/EWP/560/95. Available online: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (on accessed 26 June 2013).
- Givens, C.B.; Bullock, L.N.; Franks, A.S. Safety of concomitant tamoxifen and warfarin. Ann. Pharmacother 2009, 43, 1867–1871. [Google Scholar]
- Fareston (toremifene citrate) Label 2011; GTx, Inc. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020497s006lbl.pdf (on accessed 3 July 2013).
- Evista (raloxifene hydrochloride) Label 2007; Eli Lilly & Company. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022042lbl.pdf (on accessed 3 July 2013).
- Miller, J.W.; Skerjanec, A.; Knadler, M.P.; Ghosh, A.; Allerheiligen, S.R. Divergent effects of raloxifene HCI on the pharmacokinetics and pharmacodynamics of warfarin. Pharm. Res 2001, 18, 1024–1028. [Google Scholar]
- Turpeinen, M.; Ghiciuc, C.; Opritoui, M.; Tursas, L.; Pelkonen, O.; Pasanen, M. Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: A comparative study in vitro. Xenobiotica 2007, 37, 1367–1377. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Turpeinen, M.; Uusitalo, J.; Jalonen, J.; Pelkonen, O. Multiple P450 substrates in a single run: Rapid and comprehensive in vitro interaction assay. Eur. J. Pharm. Sci 2005, 24, 123–132. [Google Scholar]
- Tolonen, A.; Petsalo, A.; Turpeinen, M.; Uusitalo, J.; Pelkonen, O. In vitro interaction cocktail assay for nine major cytochrome P450 enzymes with 13 probe reactions and a single LC/MSMS run: Analytical validation and testing with monoclonal anti-CYP antibodies. J. Mass. Spectrom 2007, 42, 960–966. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Model reaction (probe concentration) | IC50 (μM) | ||
|---|---|---|---|---|
| Ospemifene | 4-Hydroxy-ospemifene | 4′-Hydroxy-ospemifene | ||
| CYP1A2 | Melatonin 6-hydroxylation (4 μM) | >100 | >100 | >100 |
| CYP2A6 | Coumarin 7-hydroxylation (2 μM) | >100 | >100 | >100 |
| CYP2B6 | Bupropion hydroxylation (1 μM) | 7.8 | 26.5 | 15 |
| CYP2C8 | Amodiaquine desethylation (2 μM) | 36.4 | 27.7 | 7 |
| CYP2C9 | Tolbutamide methylhydroxylation (4 μM) | 10 | 1.1 | 12 |
| CYP2C19 | Omeprazole 5-hydroxylation (2 μM) | 22.5 | 15.3 | 50 |
| CYP2C19 | Omeprazole demethylation (2 μM) | 47.2 | 19.7 | 50 |
| CYP2D6 | Dextromethorphan O-demethylation (0.2 μM) | 48.7 | 25.5 | 86 |
| CYP2E1 | Chlorzoxazone 6-hydroxylation (6 μM) | >100 | >100 | >100 |
| CYP3A4 | Midazolam 1′-hydroxylation (0.4 μM) | >100 | >100 | 78 |
| CYP3A4 | Testosterone 6β-hydroxylation (1 μM) | >100 | >100 | >100 |
| CYP3A4 | Omeprazole sulfoxidation (2 μM) | >100 | 35.9 | 85 |
| CYP3A4 | Omeprazole 3-hydroxylation (2 μM) | 37.9 | 50.0 | 54 |
| Parameter | S-Warfarin (N = 16) | R-Warfarin (N = 16) | Bupropion (N = 16) | Hydroxybupropion (N = 16) | ||||
|---|---|---|---|---|---|---|---|---|
| OSP (−) | OSP (+) | OSP (−) | OSP (+) | OSP (−) | OSP (+) | OSP (−) | OSP (+) | |
| AUC∞ (μg h/mL) | 18.5 (29.9) | 19.2 (30.2) | 31.3 (21.7) | 32.8 (23.1) | 0.90 (32.3) | 0.75 (38.5) | 15.5 (29.4) | 15.2 (32.4) |
| AUCt (μg h/mL) | 17.2 (27.4) | 18.0 (28.3) | 26.2 (18.7) | 27.8 (19.6) | 0.85 (33.7) | 0.70 (38.5) | 14.3 (28.1) | 14.5 (31.7) |
| Cmax (ng/mL) | 497 (14.5) | 513 (13.3) | 518 (15.2) | 538 (15.4) | 74.9 (36.6) | 62.9 (40.4) | 398 (30.6) | 462 (29.0) |
| tmax (h) † | 4 (2–6) | 4 (2–8) | 4 (2–6) | 4 (2–8) | 3.5 (1–5) | 3.0 (1–5) | 6.0 (5–24) | 6.1 (5–8) |
| t1/2 (h) | 30.4 (13.2) | 29.2 (12.7) | 43.8 (17.1) | 42.7 (15.6) | 30.8 (29.4) | 29.5 (26.7) | 26.7 (18.8) | 21.2 (18.0) |
| CL/F (mL/h) | 587 (29.2) | 564 (27.5) | 334 (21.2) | 320 (23.0) | 185 (38.1) | 230 (38.1) | n.d. | n.d. |
| Metabolic ratio | Ospemifene (−) (N = 12) | Ospemifene (+) (N = 12) | Ratio * (90% CI) (N = 12) | |
|---|---|---|---|---|
| 3 h conc. | Omeprazole/5-hydroxyomeprazole | 0.67 (78.4) | 0.65 (98.7) | 0.97 (0.77–1.22) |
| Omeprazole/Omeprazole sulfone | 1.19 (52.7) | 1.15 (100.9) | 0.97 (0.67–1.41) | |
| AUCt | Omeprazole/5-hydroxyomeprazole | 0.78 (59.0) | 0.76 (61.5) | 0.97 (0.88–1.08) |
| Omeprazole/Omeprazole sulfone | 1.36 (20.4) | 1.37 (24.5) | 1.00 (0.88–1.15) |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Turpeinen, M.; Uusitalo, J.; Lehtinen, T.; Kailajärvi, M.; Pelkonen, O.; Vuorinen, J.; Tapanainen, P.; Stjernschantz, C.; Lammintausta, R.; Scheinin, M. Effects of Ospemifene on Drug Metabolism Mediated by Cytochrome P450 Enzymes in Humans in Vitro and in Vivo. Int. J. Mol. Sci. 2013, 14, 14064-14075. https://doi.org/10.3390/ijms140714064
Turpeinen M, Uusitalo J, Lehtinen T, Kailajärvi M, Pelkonen O, Vuorinen J, Tapanainen P, Stjernschantz C, Lammintausta R, Scheinin M. Effects of Ospemifene on Drug Metabolism Mediated by Cytochrome P450 Enzymes in Humans in Vitro and in Vivo. International Journal of Molecular Sciences. 2013; 14(7):14064-14075. https://doi.org/10.3390/ijms140714064
Chicago/Turabian StyleTurpeinen, Miia, Jouko Uusitalo, Terhi Lehtinen, Marita Kailajärvi, Olavi Pelkonen, Jouni Vuorinen, Pasi Tapanainen, Camilla Stjernschantz, Risto Lammintausta, and Mika Scheinin. 2013. "Effects of Ospemifene on Drug Metabolism Mediated by Cytochrome P450 Enzymes in Humans in Vitro and in Vivo" International Journal of Molecular Sciences 14, no. 7: 14064-14075. https://doi.org/10.3390/ijms140714064