Potential Targets for Colorectal Cancer Prevention



Abstract
:1. Introduction
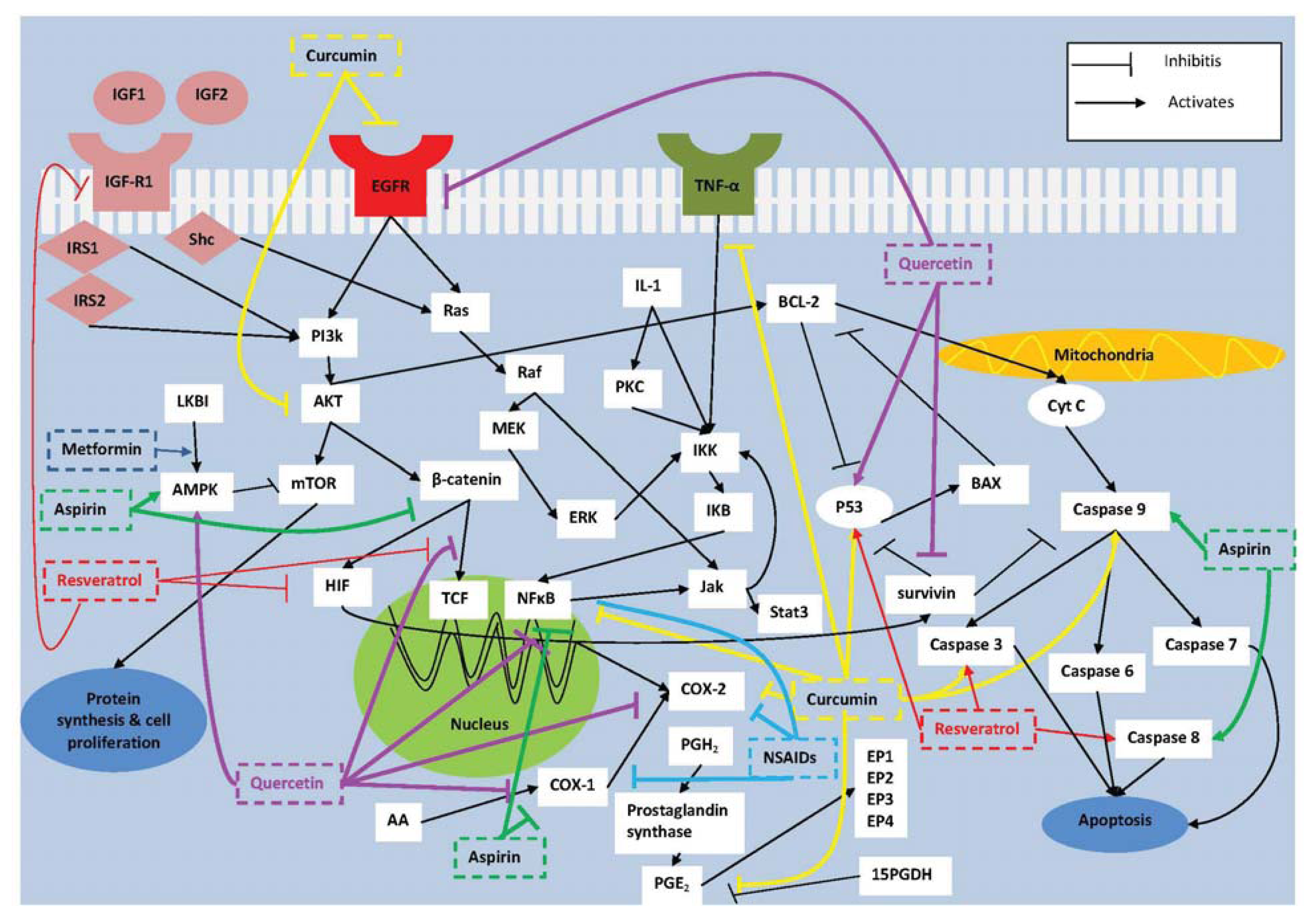
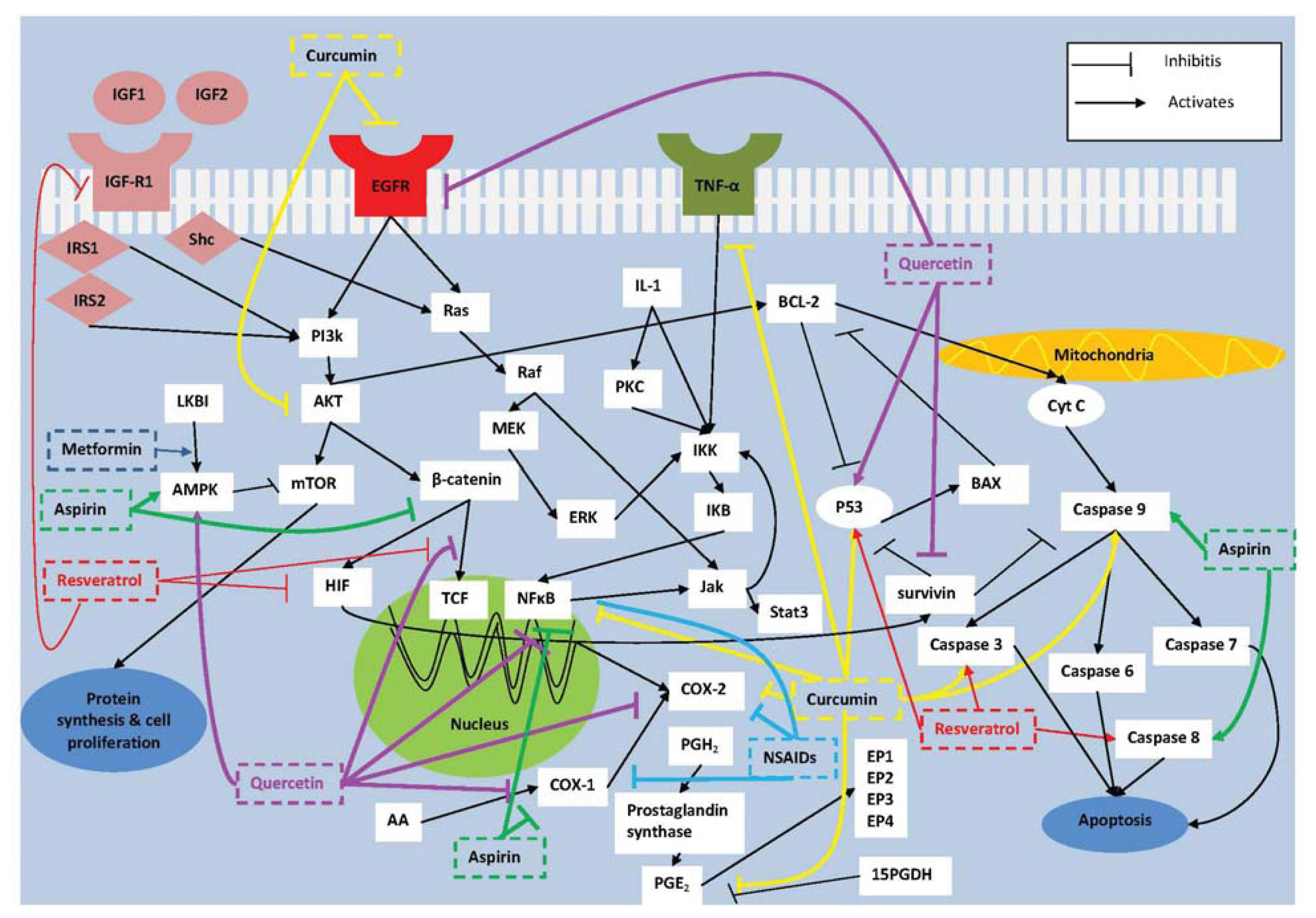
2. Molecular Targets for Prevention of Colorectal Tumorigenesis
2.1. COX-2
2.2. NF-κB
2.3. Survivin
2.4. IGF-I
3. Chemical Compounds with Chemopreventive Potential
3.1. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
Clinical Effects of NSAIDs on CRC
3.2. Aspirin
Clinical Effects of Aspirin on CRC
3.3. Metformin
Clinical Effects of Metformin on CRC
4. Natural Compounds with Chemopreventive Potential
4.1. Curcumin
4.2. Resveratrol
4.3. Quercetin
5. Combination Therapy
6. Conclusions
7. Future Directions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin 2011, 61, 69–90. [Google Scholar]
- Norat, T.; Bingham, S.; Ferrari, P.; Slimani, N.; Jenab, M.; Mazuir, M.; Overvad, K.; Olsen, A.; Tjønneland, A.; Clavel, F.; et al. Meat, fish, and colorectal cancer risk: The European Prospective Investigation into cancer and nutrition. J. Natl. Cancer Inst 2005, 97, 906–916. [Google Scholar]
- Robertson, D.J.; Sandler, R.S.; Haile, R.; Tosteson, T.D.; Greenberg, E.R.; Grau, M.; Baron, J.A. Fat, fiber, meat and the risk of colorectal adenomas. Am. J. Gastroenterol 2005, 100, 2789–2795. [Google Scholar]
- Wu, K.; Giovannucci, E.; Byrne, C.; Platz, E.A.; Fuchs, C.; Willett, W.C.; Sinha, R. Meat mutagens and risk of distal colon adenoma in a cohort of U.S. men. Cancer Epidemiol. Biomarkers Prev 2006, 15, 1120–1125. [Google Scholar]
- Liang, P.S.; Chen, T.Y.; Giovannucci, E. Cigarette smoking and colorectal cancer incidence and mortality: Systematic review and meta-analysis. Int. J. Cancer 2009, 124, 2406–2415. [Google Scholar]
- Cho, E.; Smith-Warner, S.A.; Ritz, J.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Freudenheim, J.L.; Giovannucci, E.; Goldbohm, R.A.; Graham, S.; et al. Alcohol intake and colorectal cancer: A pooled analysis of 8 cohort studies. Ann. Intern. Med 2004, 140, 603–613. [Google Scholar]
- Ferrari, P.; Jenab, M.; Norat, T.; Moskal, A.; Slimani, N.; Olsen, A.; Tjønneland, A.; Overvad, K.; Jensen, M.K.; Boutron-Ruault, M.C.; et al. Lifetime and baseline alcohol intake and risk of colon and rectal cancers in the European prospective investigation into cancer and nutrition (EPIC). Int. J. Cancer 2007, 121, 2065–2072. [Google Scholar]
- Ning, Y.; Wang, L.; Giovannucci, E.L. A quantitative analysis of body mass index and colorectal cancer: Findings from 56 observational studies. Obes. Rev 2010, 11, 19–30. [Google Scholar]
- Colbert, L.H.; Hartman, T.J.; Malila, N.; Limburg, P.J.; Pietinen, P.; Virtamo, J.; Taylor, P.R.; Albanes, D. Physical activity in relation to cancer of the colon and rectum in a cohort of male smokers. Cancer Epidemiol. Biomarkers Prev 2001, 10, 265–268. [Google Scholar]
- Colditz, G.A.; Cannuscio, C.C.; Frazier, A.L. Physical activity and reduced risk of colon cancer: Implications for prevention. Cancer Causes Control 1997, 8, 649–667. [Google Scholar]
- Hahn, E.; Kraus, S.; Arber, N. Role of cyclooxygenase-2 in pathogenesis and prevention of colorectal cancer. Dig. Dis 2010, 28, 585–589. [Google Scholar]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar]
- Smartt, H.J.; Greenhough, A.; Ordóñez-Morán, P.; Talero, E.; Cherry, C.A.; Wallam, C.A.; Parry, L.; Al Kharusi, M.; Roberts, H.R.; Mariadason, J.M.; et al. β-catenin represses expression of the tumour suppressor 15-prostaglandin dehydrogenase in the normal intestinal epithelium and colorectal tumour cells. Gut 2012, 61, 1306–1314. [Google Scholar]
- Shoji, Y.; Takahashi, M.; Kitamura, T.; Watanabe, K.; Kawamori, T.; Maruyama, T.; Sugimoto, Y.; Negishi, M.; Narumiya, S.; Sugimura, T.; et al. Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut 2004, 53, 1151–1158. [Google Scholar]
- O’Callaghan, G.; Ryan, A.; Neary, P.; O’Mahony, C.; Shanahan, F.; Houston, A. Targeting the EP1 receptor reduces Fas ligand expression and increases the antitumor immune response in an in vivo model of colon cancer. Int. J. Cancer 2013, 133, 825–834. [Google Scholar]
- Wang, D.; Wang, H.; Shi, Q.; Katkuri, S.; Walhi, W.; Desvergne, B.; Das, S.K.; Dey, S.K.; DuBois, R.N. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell 2004, 6, 285–295. [Google Scholar]
- Buchanan, F.G.; DuBois, R.N. Connecting COX-2 and Wnt in cancer. Cancer Cell 2006, 9, 6–8. [Google Scholar]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med 2002, 8, 289–293. [Google Scholar]
- Fukuda, R.; Kelly, B.; Semenza, G.L. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res 2003, 63, 2330–2334. [Google Scholar]
- Sheng, H.; Shao, J.; Morrow, J.D.; Beauchamp, R.D.; DuBois, R.N. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res 1998, 58, 362–366. [Google Scholar]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar]
- Kojima, M.; Morisaki, T.; Izuhara, K.; Uchiyama, A.; Matsunari, Y.; Katano, M.; Tanaka, M. Lipopolysaccharide increases cyclo-oxygenase-2 expression in a colon carcinoma cell line through nuclear factor-kappa B activation. Oncogene 2000, 19, 1225–1231. [Google Scholar]
- Schmedtje, J.F.; Ji, Y.S.; Liu, W.L.; DuBois, R.N.; Runge, M.S. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem 1997, 272, 601–608. [Google Scholar]
- Singer, I.I.; Kawka, D.W.; Schloemann, S.; Tessner, T.; Riehl, T.; Stenson, W.F. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 1998, 115, 297–306. [Google Scholar]
- Kim, Y.; Fischer, S.M. Transcriptional regulation of cyclooxygenase-2 in mouse skin carcinoma cells. Regulatory role of CCAAT/enhancer-binding proteins in the differential expression of cyclooxygenase-2 in normal and neoplastic tissues. J. Biol. Chem 1998, 273, 27686–27694. [Google Scholar]
- Shao, J.; Sheng, H.; Inoue, H.; Morrow, J.D.; DuBois, R.N. Regulation of constitutive cyclooxygenase-2 expression in colon carcinoma cells. J. Biol. Chem 2000, 275, 33951–33956. [Google Scholar]
- Araki, Y.; Okamura, S.; Hussain, S.P.; Nagashima, M.; He, P.; Shiseki, M.; Miura, K.; Harris, C.C. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res 2003, 63, 728–734. [Google Scholar]
- Howe, L.R.; Subbaramaiah, K.; Chung, W.J.; Dannenberg, A.J.; Brown, A.M. Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res 1999, 59, 1572–1577. [Google Scholar]
- Mei, J.M.; Hord, N.G.; Winterstein, D.F.; Donald, S.P.; Phang, J.M. Differential expression of prostaglandin endoperoxide H synthase-2 and formation of activated beta-catenin-LEF-1 transcription complex in mouse colonic epithelial cells contrasting in Apc. Carcinogenesis 1999, 20, 737–740. [Google Scholar]
- Subbaramaiah, K.; Cole, P.A.; Dannenberg, A.J. Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mechanisms. Cancer Res 2002, 62, 2522–2530. [Google Scholar]
- Hernández, G.L.; Volpert, O.V.; Iñiguez, M.A.; Lorenzo, E.; Martínez-Martínez, S.; Grau, R.; Fresno, M.; Redondo, J.M. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: Roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med 2001, 193, 607–620. [Google Scholar]
- Miller, C.; Zhang, M.; He, Y.; Zhao, J.; Pelletier, J.P.; Martel-Pelletier, J.; Di Battista, J.A. Transcriptional induction of cyclooxygenase-2 gene by okadaic acid inhibition of phosphatase activity in human chondrocytes: Co-stimulation of AP-1 and CRE nuclear binding proteins. J. Cell. Biochem 1998, 69, 392–413. [Google Scholar]
- Subbaramaiah, K.; Norton, L.; Gerald, W.; Dannenberg, A.J. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: Evidence for involvement of AP-1 and PEA3. J. Biol. Chem 2002, 277, 18649–18657. [Google Scholar]
- Meade, E.A.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J. Biol. Chem 1999, 274, 8328–8334. [Google Scholar]
- Bazan, N.G.; Lukiw, W.J. Cyclooxygenase-2 and presenilin-1 gene expression induced by interleukin-1beta and amyloid beta 42 peptide is potentiated by hypoxia in primary human neural cells. J. Biol. Chem 2002, 277, 30359–30367. [Google Scholar]
- Kaidi, A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res 2006, 66, 6683–6691. [Google Scholar]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest 2001, 108, 1657–1665. [Google Scholar]
- Young, L.E.; Sanduja, S.; Bemis-Standoli, K.; Pena, E.A.; Price, R.L.; Dixon, D.A. The mRNA binding proteins HuR and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis. Gastroenterology 2009, 136, 1669–1679. [Google Scholar]
- Dixon, D.A. Regulation of COX-2 expression in human cancers. Prog. Exp. Tumor Res 2003, 37, 52–71. [Google Scholar]
- Mukhopadhyay, D.; Houchen, C.W.; Kennedy, S.; Dieckgraefe, B.K.; Anant, S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol. Cell 2003, 11, 113–126. [Google Scholar]
- Anant, S.; Houchen, C.W. HuR and TTP: Two RNA binding proteins that deliver message from the 3′ end. Gastroenterology 2009, 136, 1495–1498. [Google Scholar]
- Dixon, D.A.; Balch, G.C.; Kedersha, N.; Anderson, P.; Zimmerman, G.A.; Beauchamp, R.D.; Prescott, S.M. Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J. Exp. Med 2003, 198, 475–481. [Google Scholar]
- Piecyk, M.; Wax, S.; Beck, A.R.; Kedersha, N.; Gupta, M.; Maritim, B.; Chen, S.; Gueydan, C.; Kruys, V.; Streuli, M.; et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J 2000, 19, 4154–4163. [Google Scholar]
- Sureban, S.M.; Ramalingam, S.; Natarajan, G.; May, R.; Subramaniam, D.; Bishnupuri, K.S.; Morrison, A.R.; Dieckgraefe, B.K.; Brackett, D.J.; Postier, R.G.; et al. Translation regulatory factor RBM3 is a proto-oncogene that prevents mitotic catastrophe. Oncogene 2008, 27, 4544–4556. [Google Scholar]
- Anant, S.; Houchen, C.W.; Pawar, V.; Ramalingam, S. Role of RNA-binding proteins in colorectal carcinogenesis. Curr. Colorectal Cancer Rep 2010, 6, 68–73. [Google Scholar]
- Poligone, B.; Baldwin, A.S. Positive and negative regulation of NF-kappaB by COX-2: Roles of different prostaglandins. J. Biol. Chem 2001, 276, 38658–38664. [Google Scholar]
- Yasui, H.; Adachi, M.; Imai, K. Combination of tumor necrosis factor-alpha with sulindac augments its apoptotic potential and suppresses tumor growth of human carcinoma cells in nude mice. Cancer 2003, 97, 1412–1420. [Google Scholar]
- Cornejo, M.G.; Boggon, T.J.; Mercher, T. JAK3: A two-faced player in hematological disorders. Int. J. Biochem. Cell Biol 2009, 41, 2376–2379. [Google Scholar]
- Lin, Q.; Lai, R.; Chirieac, L.R.; Li, C.; Thomazy, V.A.; Grammatikakis, I.; Rassidakis, G.Z.; Zhang, W.; Fujio, Y.; Kunisada, K.; et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: Inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am. J. Pathol 2005, 167, 969–980. [Google Scholar]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schütz, A.; Kovacic, B.; Friedrich, K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 2007, 9, 279–291. [Google Scholar]
- Qiao, L.; Wong, B.C. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist. Updat 2009, 12, 55–64. [Google Scholar]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res 2008, 14, 5000–5005. [Google Scholar]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res 2004, 64, 7183–7190. [Google Scholar]
- Yang, Y.L.; Li, X.M. The IAP family: Endogenous caspase inhibitors with multiple biological activities. Cell Res 2000, 10, 169–177. [Google Scholar]
- Cheung, C.H.; Chen, H.H.; Cheng, L.T.; Lyu, K.W.; Kanwar, J.R.; Chang, J.Y. Targeting Hsp90 with small molecule inhibitors induces the over-expression of the anti-apoptotic molecule, survivin, in human A549, HONE-1 and HT-29 cancer cells. Mol. Cancer 2010, 9, 77. [Google Scholar]
- Wu, X.Y.; Fu, Z.X.; Wang, X.H. Effect of hypoxia-inducible factor 1-α on Survivin in colorectal cancer. Mol. Med. Rep 2010, 3, 409–415. [Google Scholar]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995, 55, 5187–5190. [Google Scholar]
- Huerta, S.; Goulet, E.J.; Livingston, E.H. Colon cancer and apoptosis. Am. J. Surg 2006, 191, 517–526. [Google Scholar]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar]
- Zha, J.; Lackner, M.R. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin. Cancer Res 2010, 16, 2512–2517. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. An expanding role for mTOR in cancer. Trends Mol. Med 2005, 11, 353–361. [Google Scholar]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev 1999, 13, 2905–2927. [Google Scholar]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar]
- Tai, H.H.; Chi, X.; Tong, M. Regulation of 15-hydroxyprostaglandin dehydrogenase (15-PGDH) by non-steroidal anti-inflammatory drugs (NSAIDs). Prostaglandins Other Lipid Mediat 2011, 96, 37–40. [Google Scholar]
- Iguchi, G.; Chrysovergis, K.; Lee, S.H.; Baek, S.J.; Langenbach, R.; Eling, T.E. A reciprocal relationship exists between non-steroidal anti-inflammatory drug-activated gene-1 (NAG-1) and cyclooxygenase-2. Cancer Lett 2009, 282, 152–158. [Google Scholar]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar]
- Vaish, V.; Sanyal, S.N. Chemopreventive effects of NSAIDs on cytokines and transcription factors during the early stages of colorectal cancer. Pharmacol. Rep 2011, 63, 1210–1221. [Google Scholar]
- Baron, J.A.; Sandler, R.S.; Bresalier, R.S.; Quan, H.; Riddell, R.; Lanas, A.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Loftus, S.; et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology 2006, 131, 1674–1682. [Google Scholar]
- Arber, N.; Eagle, C.J.; Spicak, J.; Rácz, I.; Dite, P.; Hajer, J.; Zavoral, M.; Lechuga, M.J.; Gerletti, P.; Tang, J.; et al. Celecoxib for the prevention of colorectal adenomatous polyps. N. Engl. J. Med 2006, 355, 885–895. [Google Scholar]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Redston, M.; Solomon, S.D.; Kim, K.; Tang, J.; Rosenstein, R.B.; Wittes, J.; Corle, D.; et al. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med 2006, 355, 873–884. [Google Scholar]
- Patrono, C.; García Rodríguez, L.A.; Landolfi, R.; Baigent, C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med 2005, 353, 2373–2383. [Google Scholar]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nüsing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost 2011, 9, 790–798. [Google Scholar]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J 2009, 23, 405–414. [Google Scholar]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar]
- Clària, J.; Lee, M.H.; Serhan, C.N. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med 1996, 2, 583–596. [Google Scholar]
- Morris, T.; Stables, M.; Hobbs, A.; de Souza, P.; Colville-Nash, P.; Warner, T.; Newson, J.; Bellingan, G.; Gilroy, D.W. Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol 2009, 183, 2089–2096. [Google Scholar]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar]
- Gu, Q.; Wang, J.D.; Xia, H.H.; Lin, M.C.; He, H.; Zou, B.; Tu, S.P.; Yang, Y.; Liu, X.G.; Lam, S.K.; et al. Activation of the caspase-8/Bid and Bax pathways in aspirin-induced apoptosis in gastric cancer. Carcinogenesis 2005, 26, 541–546. [Google Scholar]
- Zimmermann, K.C.; Waterhouse, N.J.; Goldstein, J.C.; Schuler, M.; Green, D.R. Aspirin induces apoptosis through release of cytochrome c from mitochondria. Neoplasia 2000, 2, 505–513. [Google Scholar]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar]
- Burn, J.; Bishop, D.T.; Chapman, P.D.; Elliott, F.; Bertario, L.; Dunlop, M.G.; Eccles, D.; Ellis, A.; Evans, D.G.; Fodde, R.; et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev. Res. (Phila. ) 2011, 4, 655–665. [Google Scholar]
- Ishikawa, H.; Keiji, W.; Sadao, S.; Michihiro, M.; Keiji, H.; Tomiyo, N.; Ikuko, T.; Atsuko, K.; Nobuhisa, G.; Takashi, A.; et al. Preventive effects of low-dose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: Double-blind, randomized clinical trial. Cancer Med 2013, 2, 50–56. [Google Scholar]
- Burn, J.; Bishop, D.T.; Mecklin, J.P.; Macrae, F.; Möslein, G.; Olschwang, S.; Bisgaard, M.L.; Ramesar, R.; Eccles, D.; Maher, E.R.; et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med 2008, 359, 2567–2578. [Google Scholar]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med 2003, 348, 891–899. [Google Scholar]
- Benamouzig, R.; Deyra, J.; Martin, A.; Girard, B.; Jullian, E.; Piednoir, B.; Couturier, D.; Coste, T.; Little, J.; Chaussade, S. Daily soluble aspirin and prevention of colorectal adenoma recurrence: One-year results of the APACC trial. Gastroenterology 2003, 125, 328–336. [Google Scholar]
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med 2003, 348, 883–890. [Google Scholar]
- Logan, R.F.; Grainge, M.J.; Shepherd, V.C.; Armitage, N.C.; Muir, K.R. ukCAP Trial Group. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 2008, 134, 29–38. [Google Scholar]
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: Meta-analysis of the randomized trials. J. Natl. Cancer Inst 2009, 101, 256–266. [Google Scholar]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar]
- Chan, A.T.; Arber, N.; Burn, J.; Chia, W.K.; Elwood, P.; Hull, M.A.; Logan, R.F.; Rothwell, P.M.; Schrör, K.; Baron, J.A. Aspirin in the chemoprevention of colorectal neoplasia: An overview. Cancer Prev. Res 2012, 5, 164–178. [Google Scholar]
- Delage, B.; Rullier, A.; Capdepont, M.; Rullier, E.; Cassand, P. The effect of body weight on altered expression of nuclear receptors and cyclooxygenase-2 in human colorectal cancers. Nutr. J 2007, 6, 20. [Google Scholar]
- Giovannucci, E. Insulin and colon cancer. Cancer Causes Control 1995, 6, 164–179. [Google Scholar]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst 2004, 96, 546–553. [Google Scholar]
- Jenab, M.; Riboli, E.; Cleveland, R.J.; Norat, T.; Rinaldi, S.; Nieters, A.; Biessy, C.; Tjønneland, A.; Olsen, A.; Overvad, K.; et al. Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2007, 121, 368–376. [Google Scholar]
- Pawałowska, M.; Markowska, A. The influence of metformin in the etiology of selected cancers. Contemp. Oncol 2012, 16, 223–229. [Google Scholar]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Sakai, E.; Uchiyama, T.; Suzuki, K.; Iida, H.; Sakamoto, Y.; Yoneda, K.; et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev. Res. (Phila. ) 2010, 3, 1077–1083. [Google Scholar]
- Higurashi, T.; Takahashi, H.; Endo, H.; Hosono, K.; Yamada, E.; Ohkubo, H.; Sakai, E.; Uchiyama, T.; Hata, Y.; Fujisawa, N.; et al. Metformin efficacy and safety for colorectal polyps: A double-blind randomized controlled trial. BMC Cancer 2012, 12, 118. [Google Scholar]
- He, Z.Y.; Shi, C.B.; Wen, H.; Li, F.L.; Wang, B.L.; Wang, J. Upregulation of p53 expression in patients with colorectal cancer by administration of curcumin. Cancer Invest 2011, 29, 208–213. [Google Scholar]
- Guo, L.D.; Chen, X.J.; Hu, Y.H.; Yu, Z.J.; Wang, D.; Liu, J.Z. Curcumin inhibits proliferation and induces apoptosis of human colorectal cancer cells by activating the mitochondria apoptotic pathway. Phytother. Res 2013, 27, 422–430. [Google Scholar]
- Blakemore, L.M.; Boes, C.; Cordell, R.; Manson, M.M. Curcumin-induced mitotic arrest is characterized by spindle abnormalities, defects in chromosomal congression and DNA damage. Carcinogenesis 2013, 34, 351–360. [Google Scholar]
- Sandur, S.K.; Deorukhkar, A.; Pandey, M.K.; Pabón, A.M.; Shentu, S.; Guha, S.; Aggarwal, B.B.; Krishnan, S. Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-kappaB activity. Int. J. Radiat. Oncol. Biol. Phys 2009, 75, 534–542. [Google Scholar]
- Villegas, I.; Sánchez-Fidalgo, S.; de la Lastra, C.A. Chemopreventive effect of dietary curcumin on inflammation-induced colorectal carcinogenesis in mice. Mol. Nutr. Food Res 2011, 55, 259–267. [Google Scholar]
- Su, C.C.; Chen, G.W.; Lin, J.G.; Wu, L.T.; Chung, J.G. Curcumin inhibits cell migration of human colon cancer colo 205 cells through the inhibition of nuclear factor kappa B/p65 and down-regulates cyclooxygenase-2 and matrix metalloproteinase-2 expressions. Anticancer Res 2006, 26, 1281–1288. [Google Scholar]
- Lev-Ari, S.; Maimon, Y.; Strier, L.; Kazanov, D.; Arber, N. Down-regulation of prostaglandin E2 by curcumin is correlated with inhibition of cell growth and induction of apoptosis in human colon carcinoma cell lines. J. Soc. Integr. Oncol 2006, 4, 21–26. [Google Scholar]
- Chen, A.; Xu, J.; Johnson, A.C. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006, 25, 278–287. [Google Scholar]
- Johnson, S.M.; Gulhati, P.; Arrieta, I.; Wang, X.; Uchida, T.; Gao, T.; Evers, B.M. Curcumin inhibits proliferation of colorectal carcinoma by modulating Akt/mTOR signaling. Anticancer Res 2009, 29, 3185–3190. [Google Scholar]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res 2001, 7, 1894–1900. [Google Scholar]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res 2004, 10, 6847–6854. [Google Scholar]
- Garcea, G.; Berry, D.P.; Jones, D.J.; Singh, R.; Dennison, A.R.; Farmer, P.B.; Sharma, R.A.; Steward, W.P.; Gescher, A.J. Consumption of the putative chemopreventive agent curcumin by cancer patients: Assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol. Biomarkers Prev 2005, 14, 120–125. [Google Scholar]
- Carroll, R.E.; Benya, R.V.; Turgeon, D.K.; Vareed, S.; Neuman, M.; Rodriguez, L.; Kakarala, M.; Carpenter, P.M.; McLaren, C.; Meyskens, F.L.; et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev. Res 2011, 4, 354–364. [Google Scholar]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm 2007, 4, 807–818. [Google Scholar]
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med 2006, 6, 10. [Google Scholar]
- Irving, G.R.; Howells, L.M.; Sale, S.; Kralj-Hans, I.; Atkin, W.S.; Clark, S.K.; Britton, R.G.; Jones, D.J.; Scott, E.N.; Berry, D.P.; et al. Prolonged biologically active colonic tissue levels of curcumin achieved after oral administration—A clinical pilot study including assessment of patient acceptability. Cancer Prev. Res 2013, 6, 119–128. [Google Scholar]
- Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. Curcumin nanoformulations: A future nanomedicine for cancer. Drug Discov. Today 2012, 17, 71–80. [Google Scholar]
- Chen, H.J.; Hsu, L.S.; Shia, Y.T.; Lin, M.W.; Lin, C.M. The β-catenin/TCF complex as a novel target of resveratrol in the Wnt/β-catenin signaling pathway. Biochem. Pharmacol 2012, 84, 1143–1153. [Google Scholar]
- Wu, H.; Liang, X.; Fang, Y.; Qin, X.; Zhang, Y.; Liu, J. Resveratrol inhibits hypoxia-induced metastasis potential enhancement by restricting hypoxia-induced factor-1 alpha expression in colon carcinoma cells. Biomed. Pharmacother 2008, 62, 613–621. [Google Scholar]
- Vanamala, J.; Reddivari, L.; Radhakrishnan, S.; Tarver, C. Resveratrol suppresses IGF-1 induced human colon cancer cell proliferation and elevates apoptosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer 2010, 10, 238. [Google Scholar]
- Fouad, M.; Agha, A.; Merzabani, M.A.; Shouman, S. Resveratrol inhibits proliferation, angiogenesis and induces apoptosis in colon cancer cells: Calorie restriction is the force to the cytotoxicity. Hum. Exp. Toxicol. 2013. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol 2012, 40, 1020–1028. [Google Scholar]
- Panaro, M.A.; Carofiglio, V.; Acquafredda, A.; Cavallo, P.; Cianciulli, A. Anti-inflammatory effects of resveratrol occur via inhibition of lipopolysaccharide-induced NF-κB activation in Caco-2 and SW480 human colon cancer cells. Br. J. Nutr 2012, 108, 1623–1632. [Google Scholar]
- Schneider, Y.; Duranton, B.; Gossé, F.; Schleiffer, R.; Seiler, N.; Raul, F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr. Cancer 2001, 39, 102–107. [Google Scholar]
- Patel, K.R.; Brown, V.A.; Jones, D.J.; Britton, R.G.; Hemingway, D.; Miller, A.S.; West, K.P.; Booth, T.D.; Perloff, M.; Crowell, J.A.; et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res 2010, 70, 7392–7399. [Google Scholar]
- Boocock, D.J.; Faust, G.E.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomarkers Prev 2007, 16, 1246–1252. [Google Scholar]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov 2006, 5, 493–506. [Google Scholar]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. (Phila. ) 2011, 4, 1419–1425. [Google Scholar]
- Mutoh, M.; Takahashi, M.; Fukuda, K.; Komatsu, H.; Enya, T.; Matsushima-Hibiya, Y.; Mutoh, H.; Sugimura, T.; Wakabayashi, K. Suppression by flavonoids of cyclooxygenase-2 promoter-dependent transcriptional activity in colon cancer cells: Structure-activity relationship. Jpn. J. Cancer Res 2000, 91, 686–691. [Google Scholar]
- Priego, S.; Feddi, F.; Ferrer, P.; Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Estrela, J.M. Natural polyphenols facilitate elimination of HT-29 colorectal cancer xenografts by chemoradiotherapy: A Bcl-2- and superoxide dismutase 2-dependent mechanism. Mol. Cancer Ther 2008, 7, 3330–3342. [Google Scholar]
- Kim, H.J.; Kim, S.K.; Kim, B.S.; Lee, S.H.; Park, Y.S.; Park, B.K.; Kim, S.J.; Kim, J.; Choi, C.; Kim, J.S.; et al. Apoptotic effect of quercetin on HT-29 colon cancer cells via the AMPK signaling pathway. J. Agric. Food Chem 2010, 58, 8643–8650. [Google Scholar]
- Fridrich, D.; Teller, N.; Esselen, M.; Pahlke, G.; Marko, D. Comparison of delphinidin, quercetin and (−)-epigallocatechin-3-gallate as inhibitors of the EGFR and the ErbB2 receptor phosphorylation. Mol. Nutr. Food Res 2008, 52, 815–822. [Google Scholar]
- Park, C.H.; Chang, J.Y.; Hahm, E.R.; Park, S.; Kim, H.K.; Yang, C.H. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem. Biophys. Res. Commun 2005, 328, 227–234. [Google Scholar]
- Shan, B.E.; Wang, M.X.; Li, R.Q. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest 2009, 27, 604–612. [Google Scholar]
- Chirumbolo, S. Quercetin in cancer prevention and therapy. Integr. Cancer Ther 2013, 12, 97–102. [Google Scholar]
- Cai, X.; Fang, Z.; Dou, J.; Yu, A.; Zhai, G. Bioavailability of quercetin: Problems and promises. Curr. Med. Chem 2013, 20, 2572–2582. [Google Scholar]
- Li, H.; Zhao, X.; Ma, Y.; Zhai, G.; Li, L.; Lou, H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J. Control Release 2009, 133, 238–244. [Google Scholar]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515, e3.. [Google Scholar]
- Shpitz, B.; Giladi, N.; Sagiv, E.; Lev-Ari, S.; Liberman, E.; Kazanov, D.; Arber, N. Celecoxib and curcumin additively inhibit the growth of colorectal cancer in a rat model. Digestion 2006, 74, 140–144. [Google Scholar]
- Lev-Ari, S.; Strier, L.; Kazanov, D.; Madar-Shapiro, L.; Dvory-Sobol, H.; Pinchuk, I.; Marian, B.; Lichtenberg, D.; Arber, N. Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin. Cancer Res 2005, 11, 6738–6744. [Google Scholar]
- Cruz-Correa, M.; Shoskes, D.A.; Sanchez, P.; Zhao, R.; Hylind, L.M.; Wexner, S.D.; Giardiello, F.M. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin. Gastroenterol. Hepatol 2006, 4, 1035–1038. [Google Scholar]
- Aftab, N.; Vieira, A. Antioxidant activities of curcumin and combinations of this curcuminoid with other phytochemicals. Phytother. Res 2010, 24, 500–502. [Google Scholar]
- Majumdar, A.P.; Banerjee, S.; Nautiyal, J.; Patel, B.B.; Patel, V.; Du, J.; Yu, Y.; Elliott, A.A.; Levi, E.; Sarkar, F.H. Curcumin synergizes with resveratrol to inhibit colon cancer. Nutr. Cancer 2009, 61, 544–553. [Google Scholar]

{kind=link}
| Title | Design/phase | Intervention | Primary endpoint | Identifier |
|---|---|---|---|---|
| Study Investigating the Ability of Plant Exosomes to Deliver Curcumin to Normal and Colon Cancer Tissue | Phase I | Arm 1: curcumin alone Arm 2: curcumin with plant exosomes Arm 3: no treatment | To estimate the effect of a fixed concentration of curcumin when delivered by plant exosomes compared to oral tablets of curcumin alone | NCT01294072 |
| Phase II A Trial of Curcumin Among Patients With Prevalent Subclinical Neoplastic Lesions (Aberrant Crypt Foci) | Phase II | Patients receive 1 of 2 doses of oral curcumin once daily | To determine mean percentage change from baseline in prostaglandin E2 (PGE2) within ACF pre and post 30 days of curcumin administration at a specified dose | NCT00365209 |
| Curcumin for the Chemoprevention of Colorectal Cancer | Phase II | 4 g curcumin daily for 4 months or placebo | Cellular proliferation and apoptosis in the colonic mucosa and COX-2 expression and activity | NCT00118989 |
| Use of Curcumin for Treatment of Intestinal Adenomas in Familial Adenomatous Polyposis (FAP) | Randomized double blind placebo controlled trial | Curcumin 2 pills twice per day or placebo 2 pills twice per day for 12 months | To determine the tolerability and efficacy of curcumin to regress intestinal adenomas by measuring duodenal and colorectal/ileal polyp number, and polyp size in patients with FAP | NCT00927485 |
| Curcumin in Treating Patients With Familial Adenomatous Polyposis | Randomized double blind placebo controlled trial | Curcumin 2 pills per day or placebo 2 pills per day for 12 months | To determine the tolerability and efficacy of curcumin to regress intestinal adenomas by measuring duodenal and colorectal/ileal polyp number, and polyp size in FAP patients | NCT00641147 |
| Curcumin Biomarkers | Phase I | 4 g curcumin C3 tablets daily for 30 days | To identify genes that are modified by curcumin that could be used as biomarkers in future chemoprevention studies. The study will also evaluate tolerability and toxicity | NCT01333917 |
| Resveratrol in Treating Patients With Colorectal Cancer That Can Be Removed By Surgery | Phase I | Patients with colorectal adenocarcinomas receive resveratrol for days 1 to 8 and on day 9 undergo colorectomy. Tumor biopsy will be retrieved | To study the side effects and best dose of resveratrol in treating patients with colorectal cancer that can be removed by surgery | NCT00433576 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Temraz, S.; Mukherji, D.; Shamseddine, A. Potential Targets for Colorectal Cancer Prevention. Int. J. Mol. Sci. 2013, 14, 17279-17303. https://doi.org/10.3390/ijms140917279
Temraz S, Mukherji D, Shamseddine A. Potential Targets for Colorectal Cancer Prevention. International Journal of Molecular Sciences. 2013; 14(9):17279-17303. https://doi.org/10.3390/ijms140917279
Chicago/Turabian StyleTemraz, Sally, Deborah Mukherji, and Ali Shamseddine. 2013. "Potential Targets for Colorectal Cancer Prevention" International Journal of Molecular Sciences 14, no. 9: 17279-17303. https://doi.org/10.3390/ijms140917279