The DNA-Damage Response to γ-Radiation Is Affected by miR-27a in A549 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
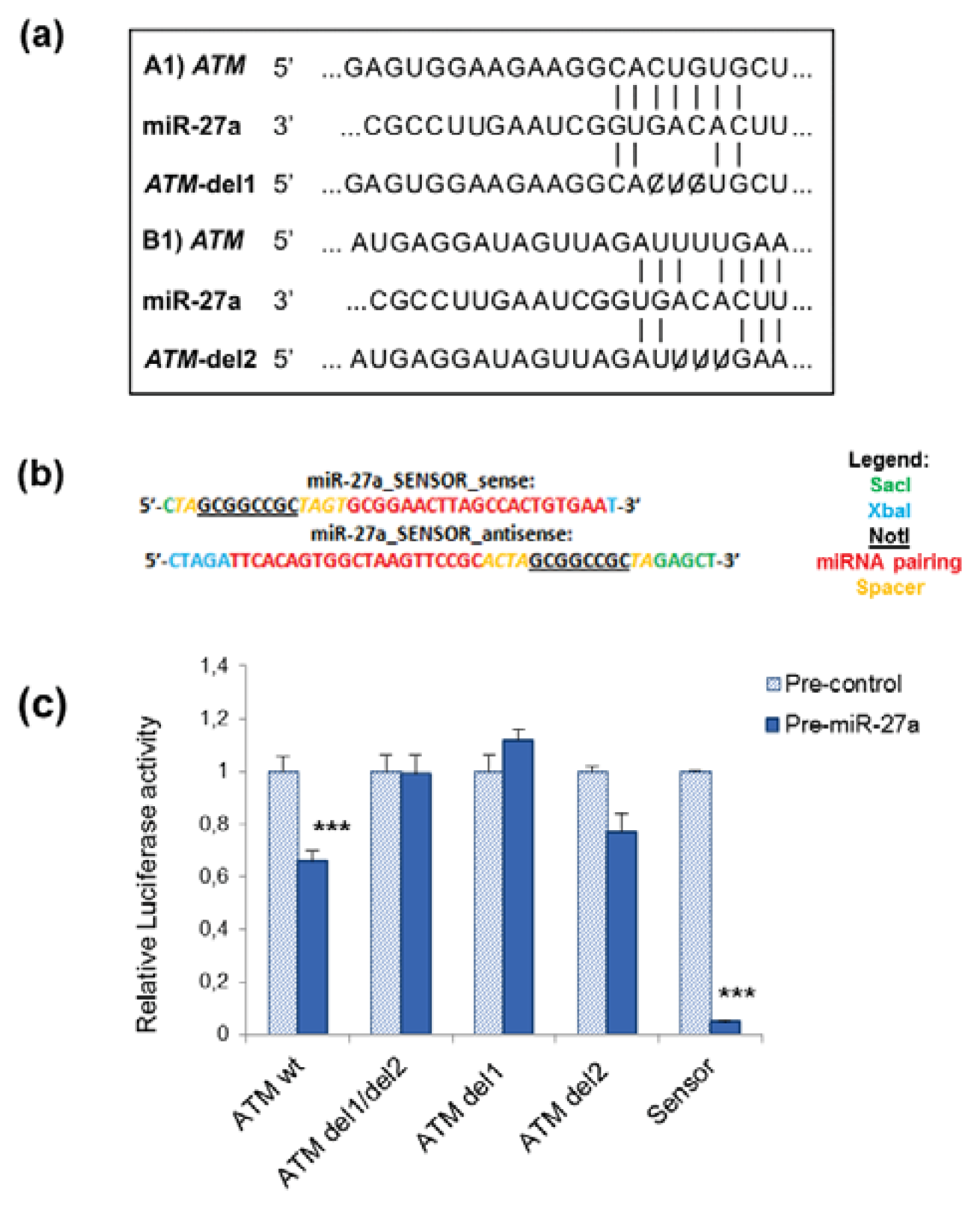
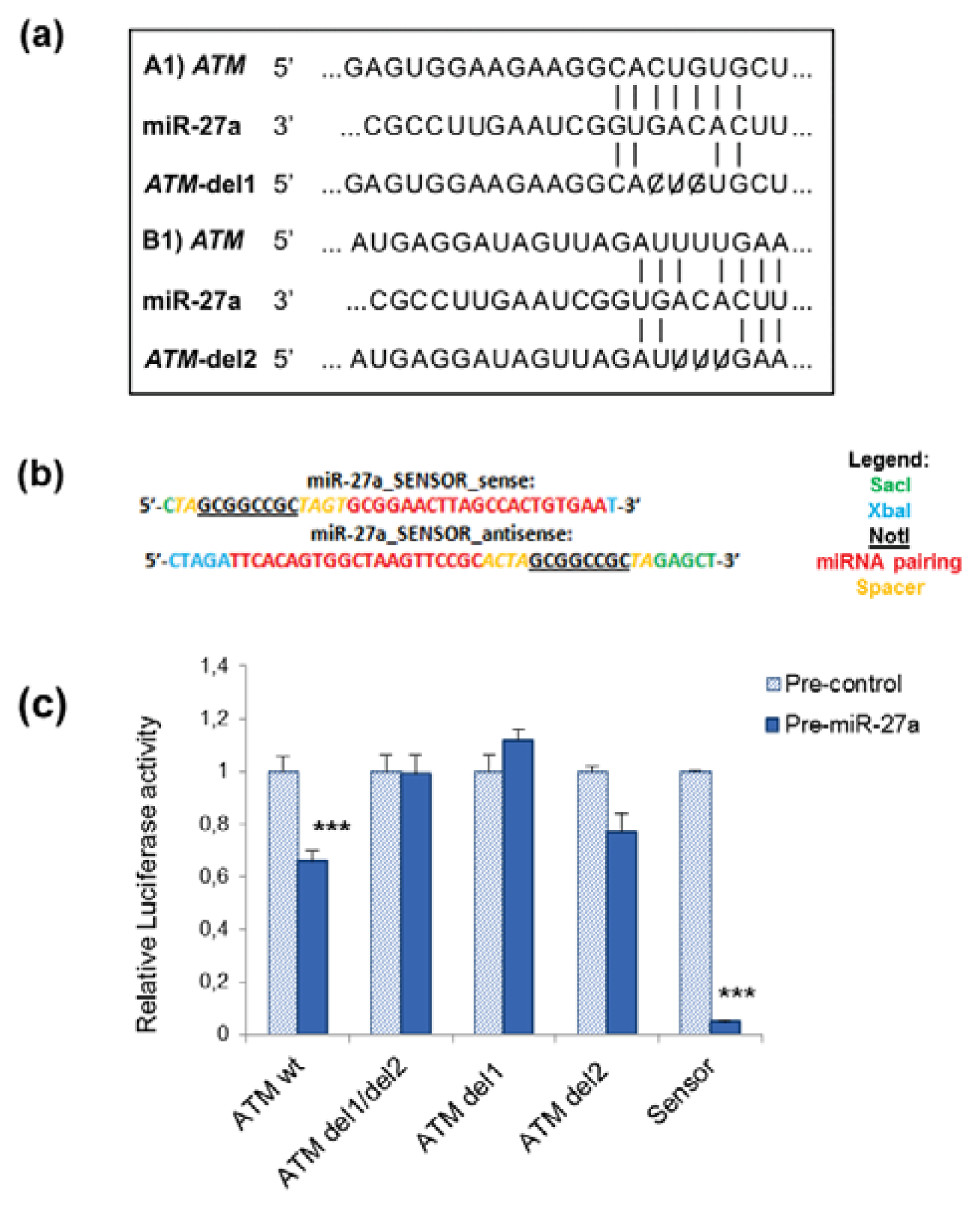
2.1. miR-27a Interacts with 3′UTR of ATM Gene
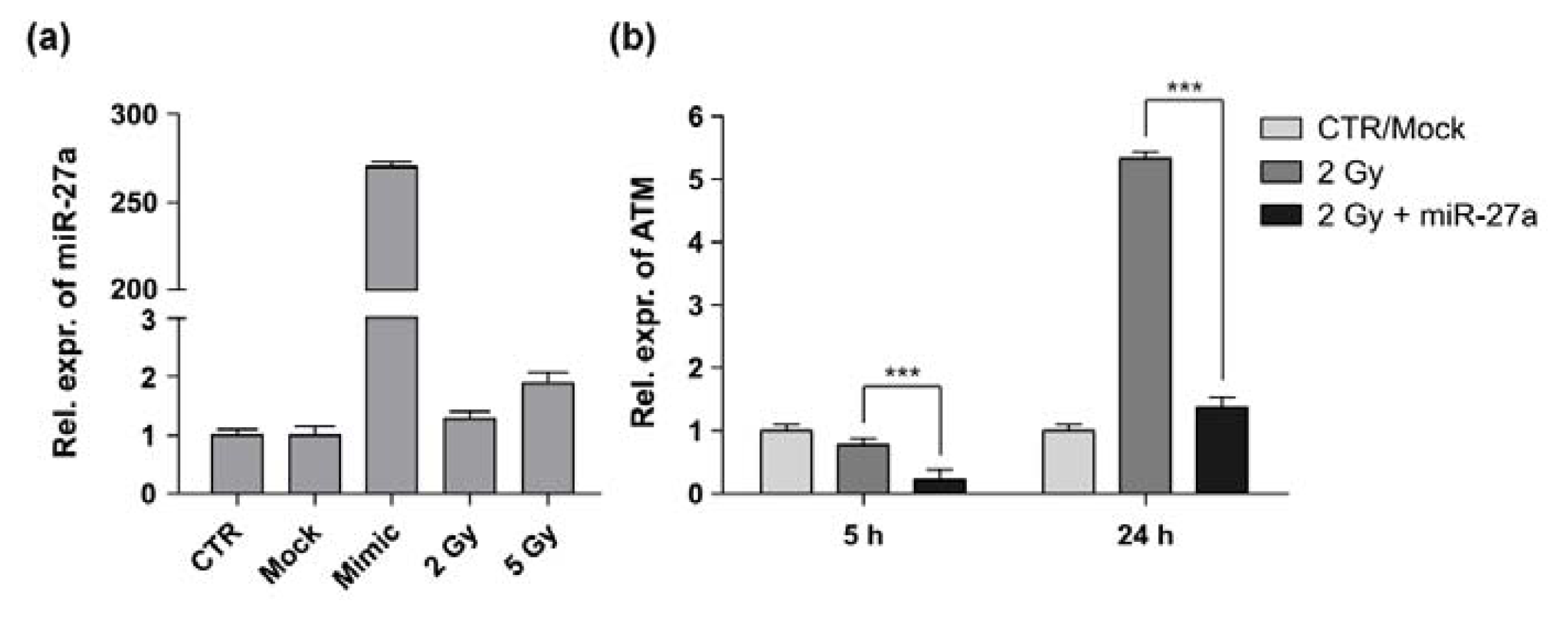
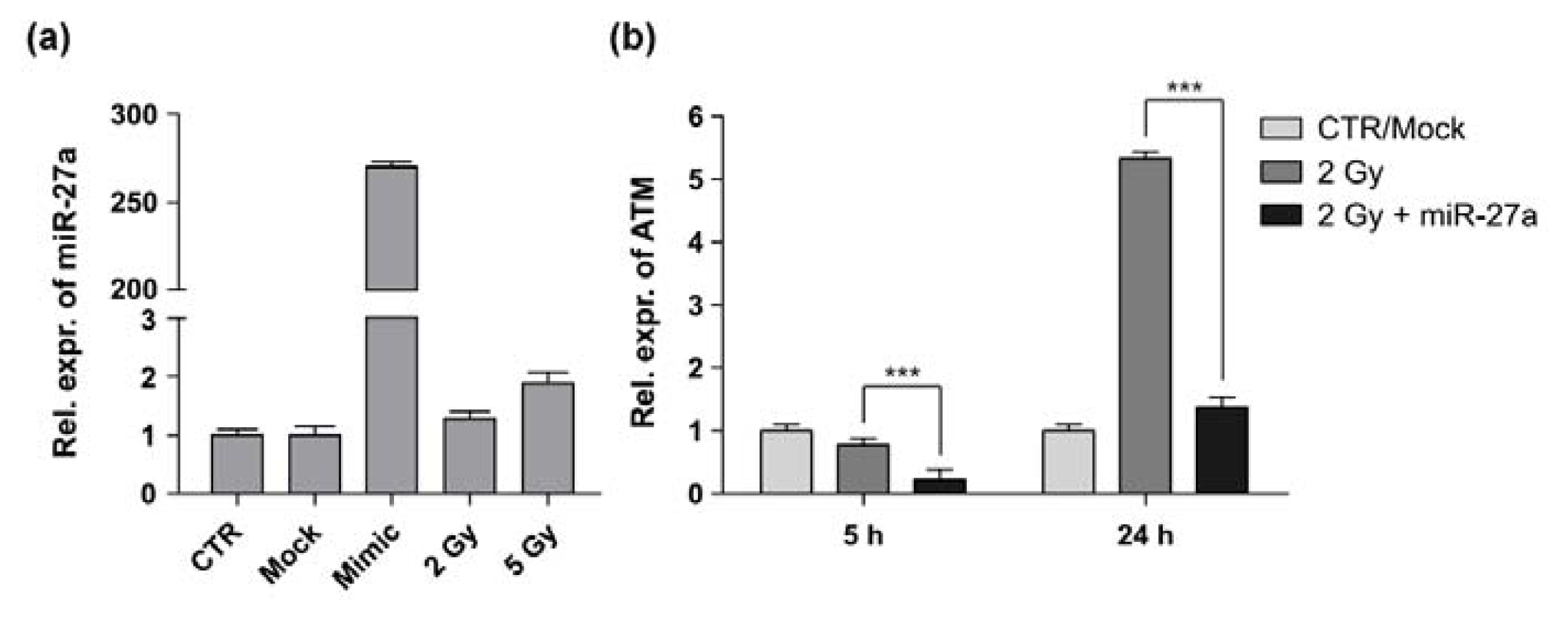
2.2. miR-27a Affects ATM Expression Levels in A549 Cells
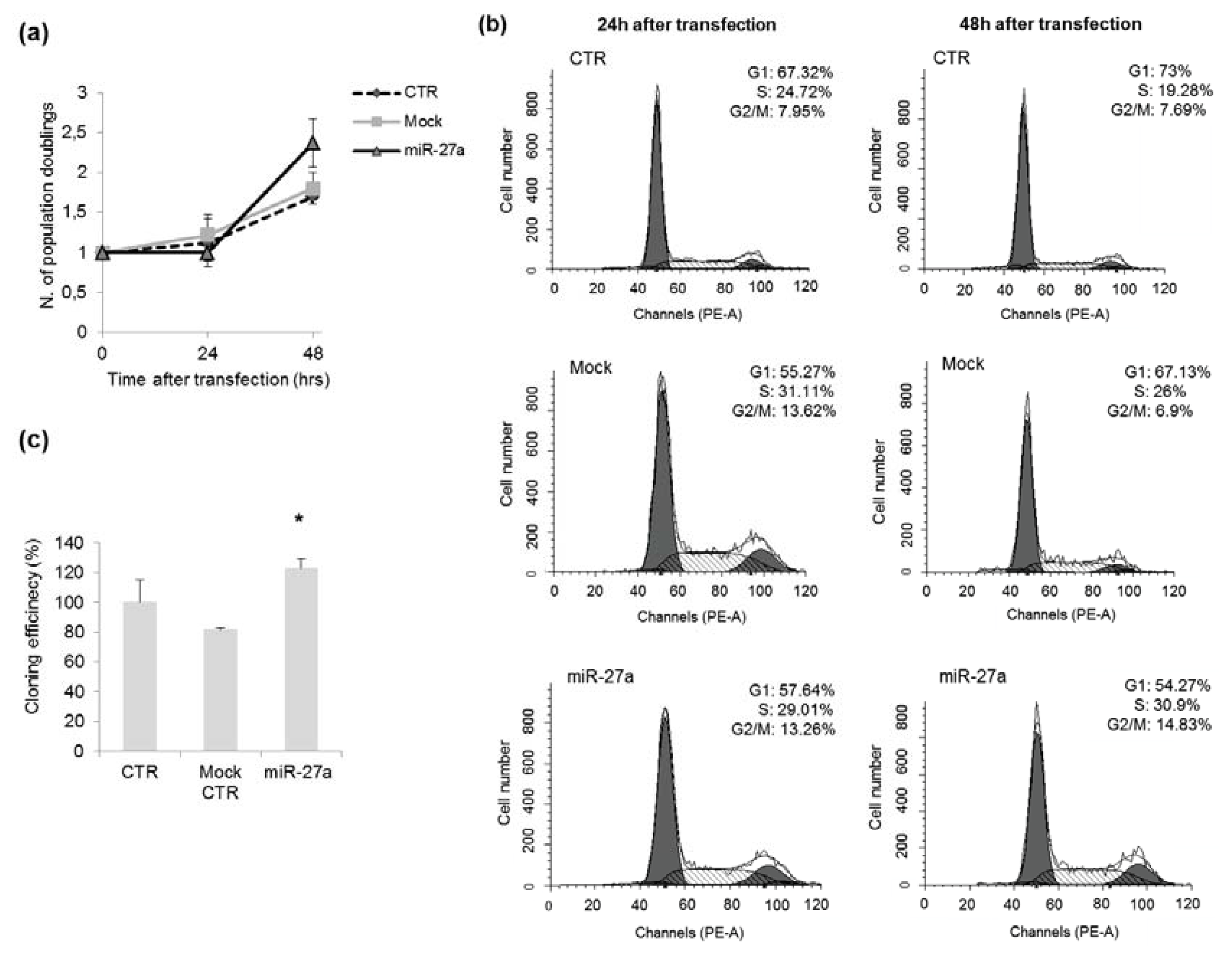
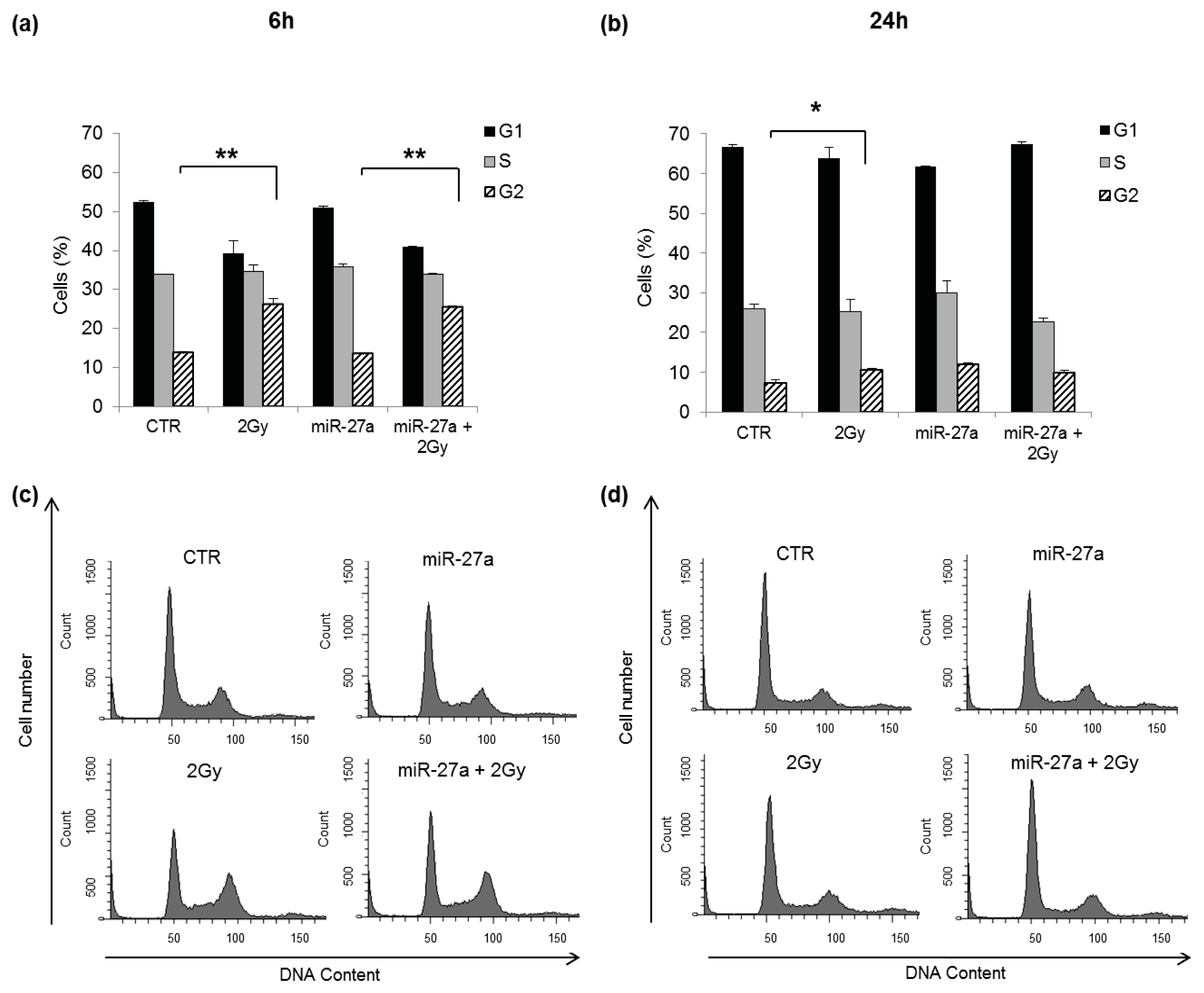
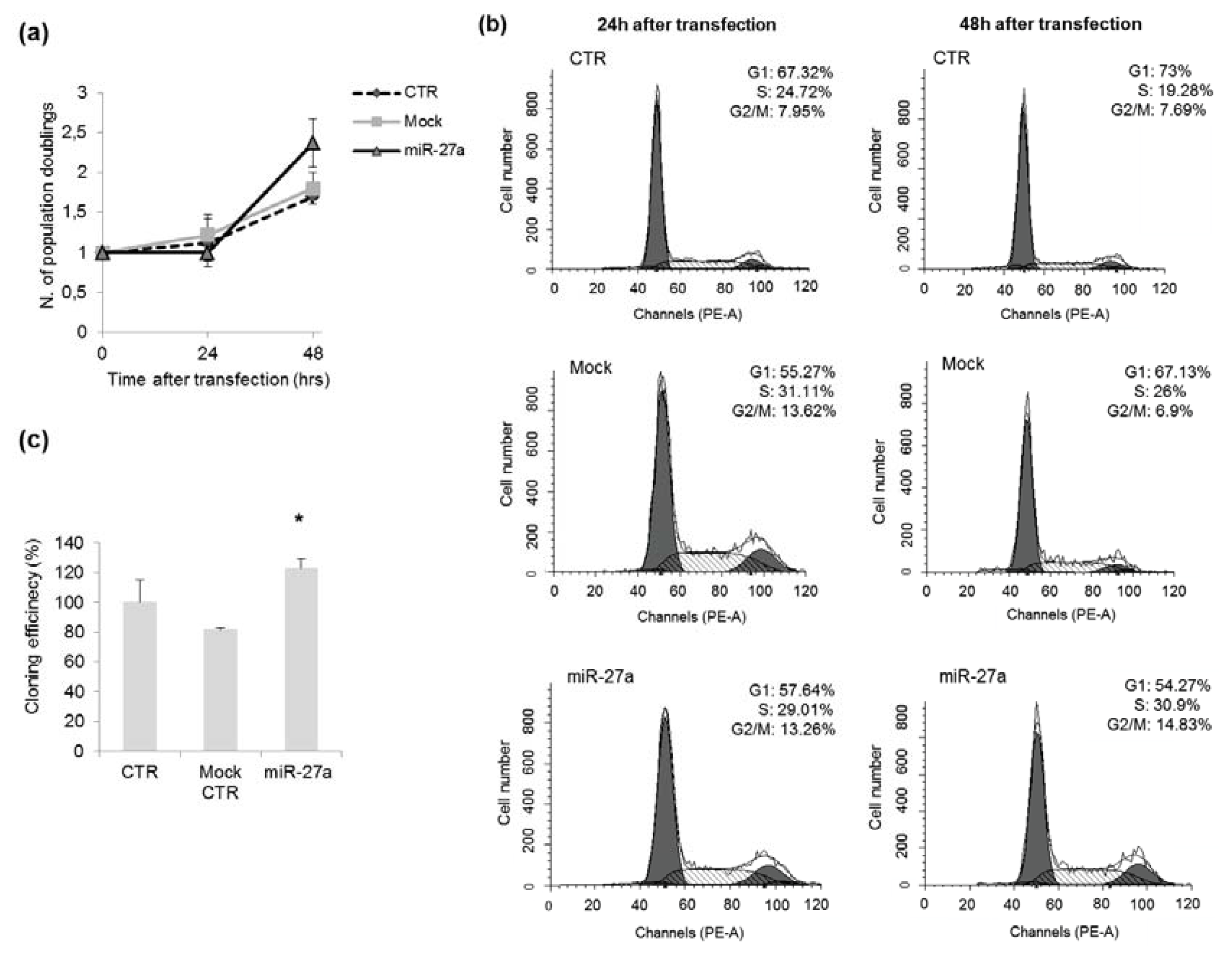
2.3. Effects of miR-27a on Proliferation of A549 Cells
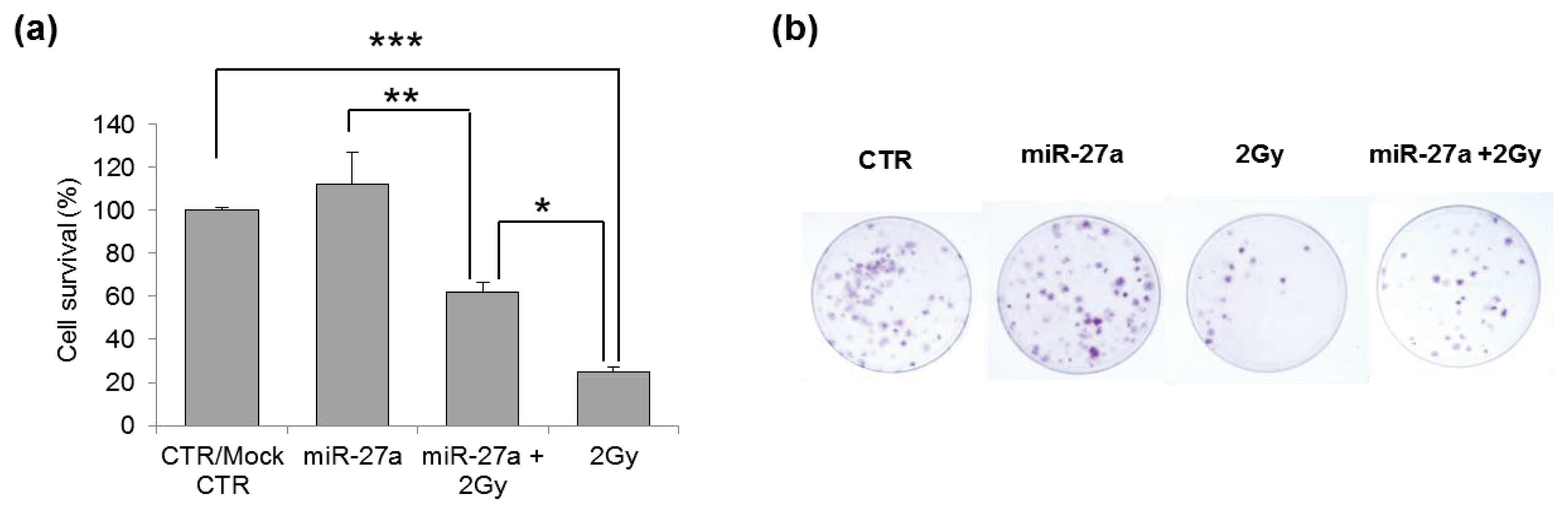
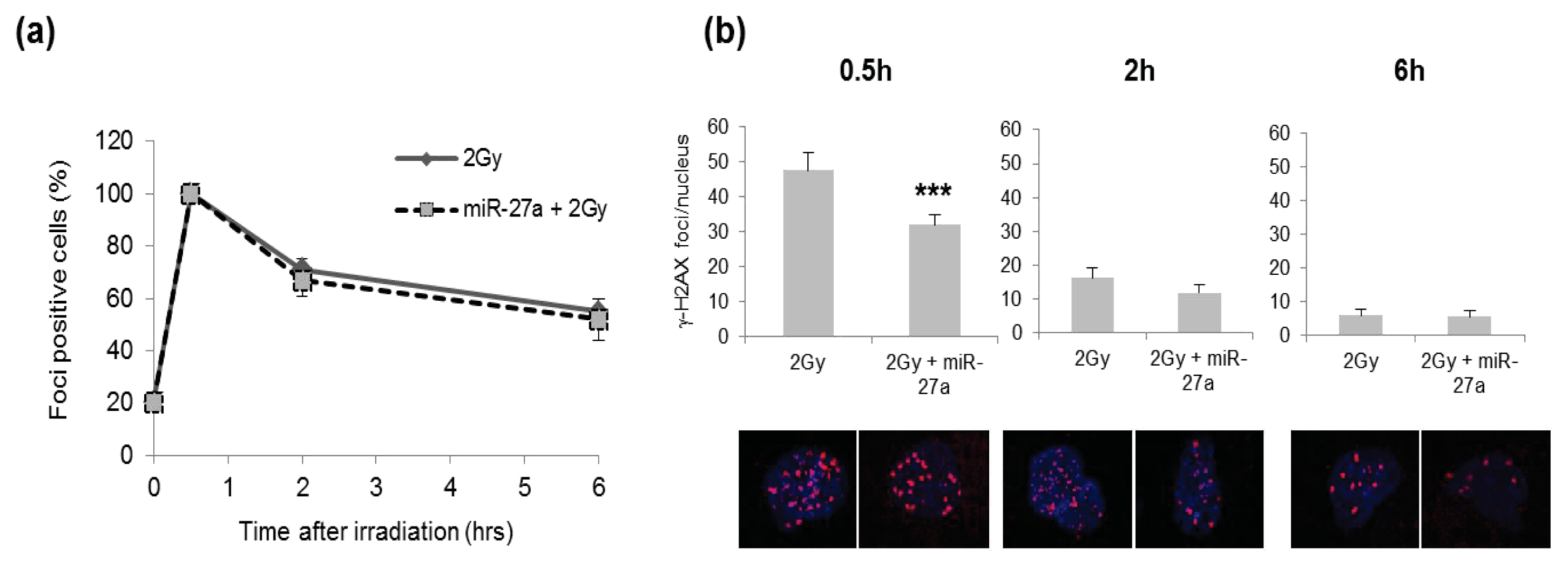
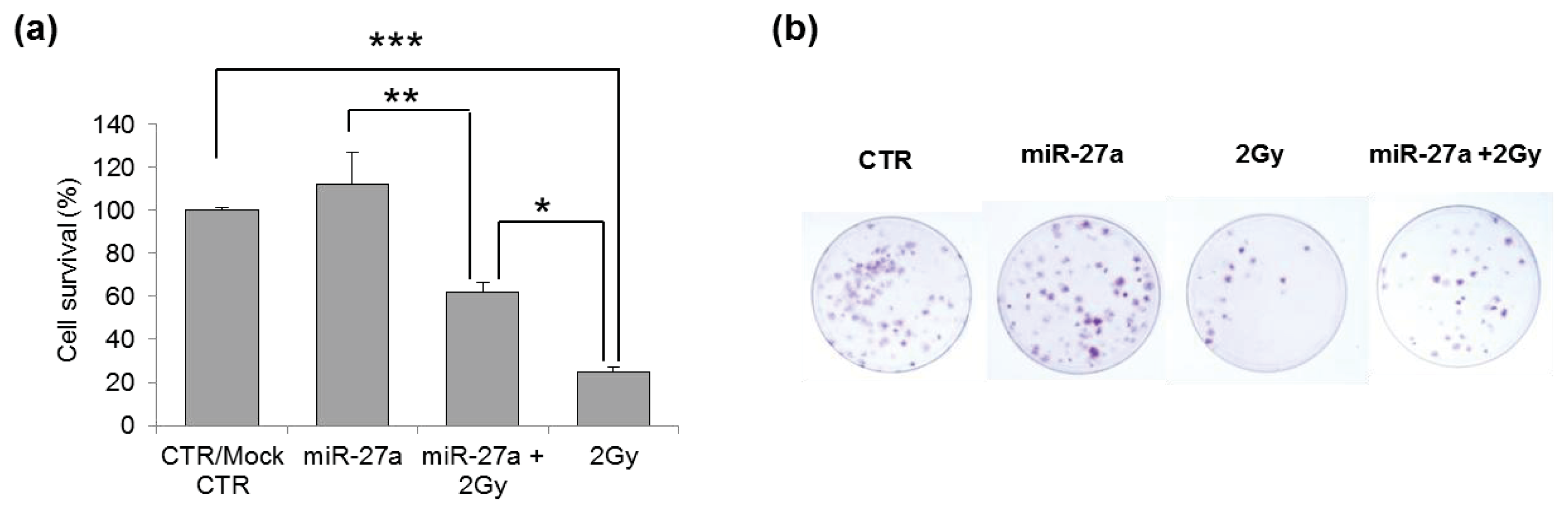
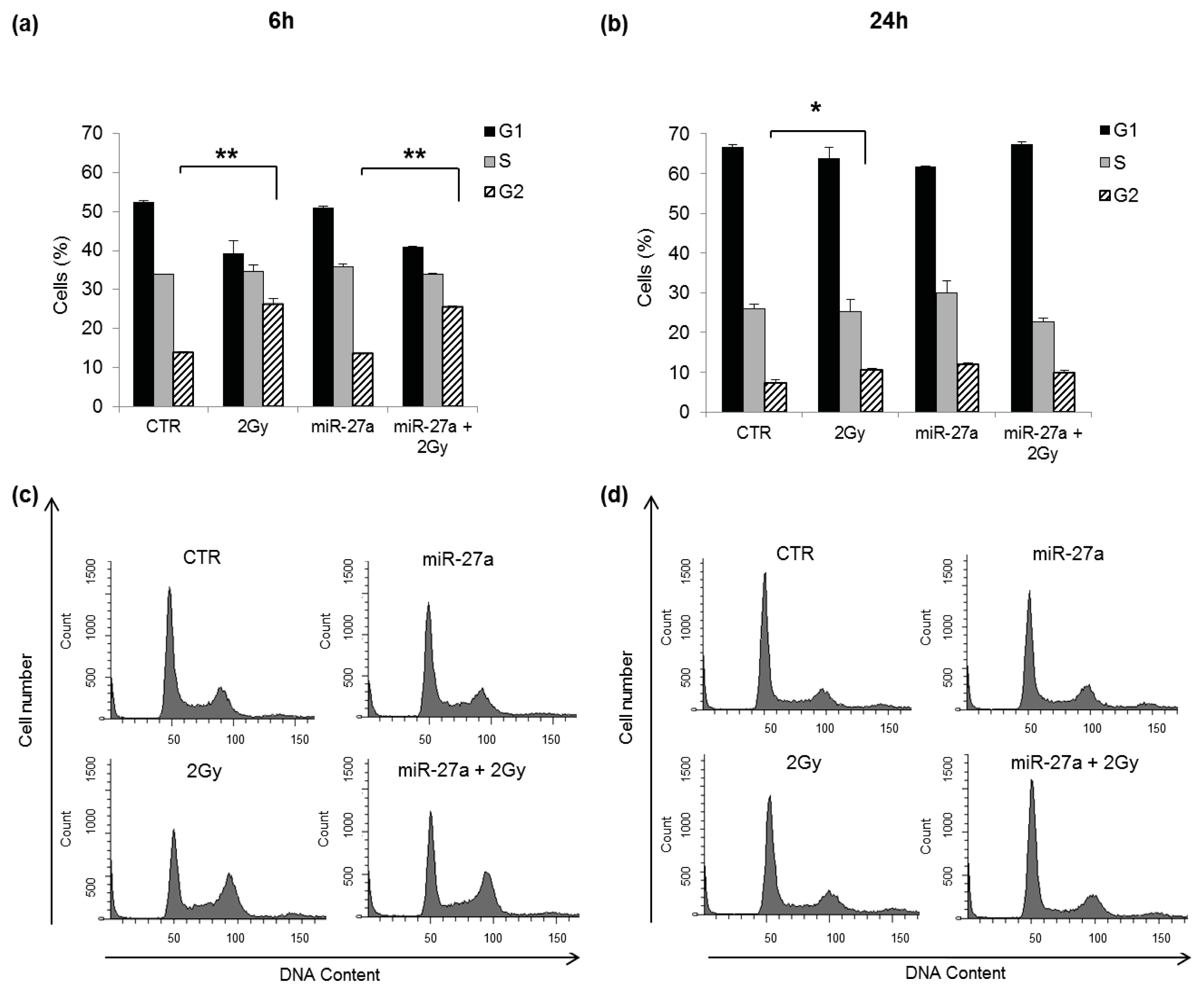
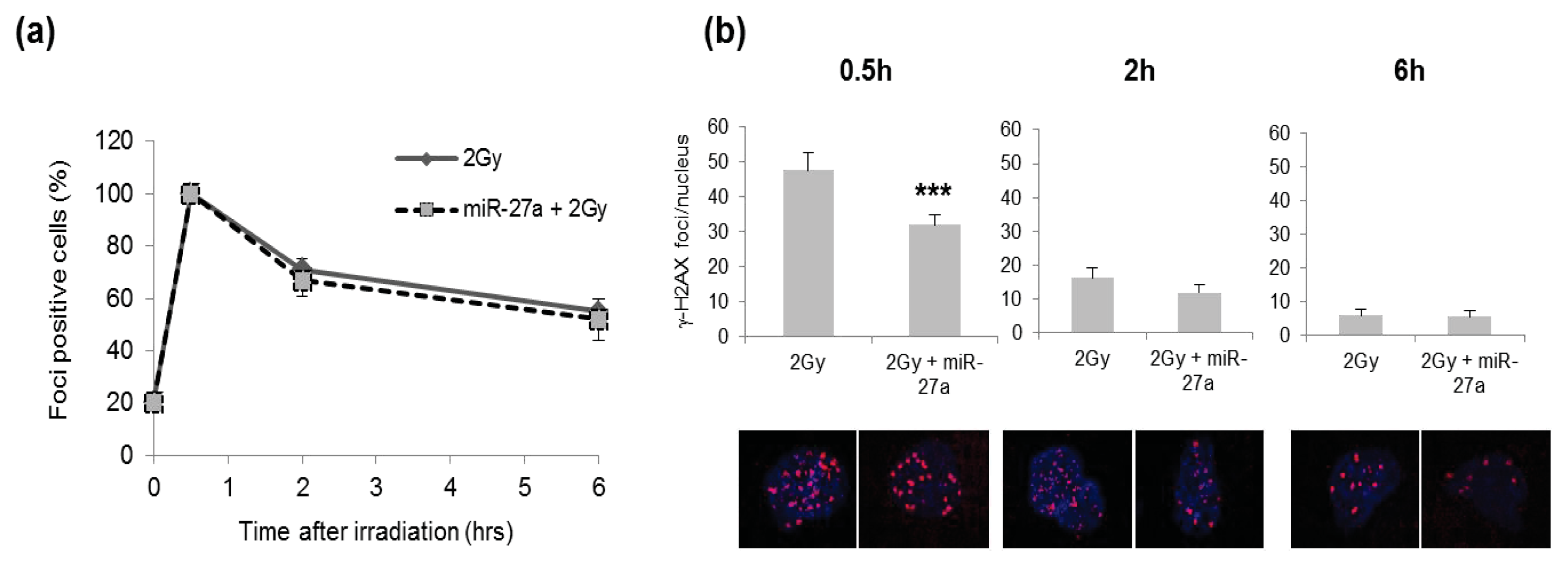
2.4. Effects of miR-27a Up-Regulation on Radiosensitivity of A549 Cells
3. Experimental Section
3.1. Cell Culture
3.2. Construction of Recombinant Vectors and Site-Directed Mutagenesis
- ATM Fwd-5′-ATCTAGGAGCTCAGGAGTGGAAGAAGGCACTG-3′
- ATM Rev-5′-ATCTAGTCTAGAACGCTGTCCAAAGTTTTTCC-3′
- ATMdel1 Fwd-5′-AGTGGAAGAAGGCACTCTCAGTGTTGGTGGAC-3′
- ATMdel1 Rev-5′-GTCCACCAACACTGAGAGTGCCTTCTTCCACT-3′
- ATMdel2 Fwd-5′-CAAGGACAAATGAGGAGTAGTTAGATGAAAATATTAATCATAGAATAGTTGTT-3′
- ATMdel2 Rev-5′-AACAACTATTCTATGATTAATATTTTCATCTAACTACTCCTCATTTGTCCTTG-3′
3.3. Transient Transfection and Cell Irradiation
3.4. Total RNA Isolation and qRT-PCR
3.5. Luciferase Reporter Assays
3.6. Clonogenic Survival Assay
3.7. Cell Cycle Analysis
3.8. Immunofluorescence Staining
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanović, M. The ATM signaling network in development and disease. Front. Genet 2013, 4, 37. [Google Scholar]
- Boucas, J.; Riabinska, A.; Jokic, M.; Herter-Sprie, G.S.; Chen, S.; Höpker, K.; Reinhardt, H.C. Posttranscriptional regulation of gene expression-adding another layer of complexity to the DNA damage response. Front. Genet 2012, 3, 159. [Google Scholar]
- Bartel, D.P. microRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Lee, H.-J. Exceptional stories of microRNAs. Exp. Biol. Med 2013, 238, 339–343. [Google Scholar]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet 2005, 37, 766–770. [Google Scholar]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; Song, E. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar]
- Bae, H.J.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Jung, K.H.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Kim, S.J.; Park, W.S.; et al. microRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene 2013. [Google Scholar] [CrossRef]
- Bisso, A.; Faleschini, M.; Zampa, F.; Capaci, V.; de Santa, J.; Santarpia, L.; Piazza, S.; Cappelletti, V.; Daidone, M.; Agami, R.; et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle 2013, 12, 1679–1687. [Google Scholar]
- Wang, H.F.; Chen, H.; Ma, M.W.; Wang, J.A.; Tang, T.T.; Ni, L.S.; Yu, J.L.; Li, Y.Z.; Bai, B.X. miR-573 regulates melanoma progression by targeting the melanoma cell adhesion molecule. Oncol. Rep 2013, 30, 520–526. [Google Scholar]
- He, L.; He, X.; Lowe, S.W.; Hannon, G.J. microRNAs join the p53 network—Another piece in the tumour-suppression puzzle. Nat. Rev. Cancer 2007, 7, 819–822. [Google Scholar]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar]
- Wouters, M.D.; van Gent, D.C.; Hoeijmakers, J.H.; Pothof, J. microRNAs, the DNA damage response and cancer. Mutat. Res 2011, 717, 54–66. [Google Scholar]
- Girardi, C.; de Pittà, C.; Casara, S.; Sales, G.; Lanfranchi, G.; Celotti, L.; Mognato, M. Analysis of miRNA and mRNA expression profiles highlights alterations in ionizing radiation response of human lymphocytes under modeled microgravity. PLoS One 2012, 7, e31293. [Google Scholar]
- Guttilla, I.K.; White, B.A. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J. Biol. Chem 2009, 284, 23204–23216. [Google Scholar]
- Gottardo, F.; Liu, C.G.; Ferracin, M.; Calin, G.A.; Fassan, M.; Bassi, P.; Sevignani, C.; Byrne, D.; Negrini, M.; Pagano, F.; et al. micro-RNA profiling in kidney and bladder cancers. Urol. Oncol 2007, 25, 387–392. [Google Scholar]
- Liu, T.; Tang, H.; Lang, Y.; Liu, M.; Li, X. microRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett 2009, 273, 233–242. [Google Scholar]
- Wu, X.J.; Li, Y.; Liu, D.; Zhao, L.D.; Bai, B.; Xue, M.H. miR-27a as an oncogenic microRNA of hepatitis B virus-related hepatocellular carcinoma. Asian Pac. J. Cancer Prev 2013, 14, 885–889. [Google Scholar]
- Lerner, M.; Lundgren, J.; Akhoondi, S.; Jahn, A.; Ng, H.F.; Akbari Moqadam, F.; Oude Vrielink, J.A.; Agami, R.; den Boer, M.L.; Grandér, D.; et al. miRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle 2011, 10, 2172–2183. [Google Scholar]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar]
- Kozaki, K.; Imoto, I.; Mogi, S.; Omura, K.; Inazawa, J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 2008, 68, 2094–2105. [Google Scholar]
- Heegaard, N.H.; Schetter, A.J.; Welsh, J.A.; Yoneda, M.; Bowman, E.D.; Harris, C.C. Circulating micro-RNA expression profiles in early stage nonsmall cell lung cancer. Int. J. Cancer 2012, 130, 1378–1386. [Google Scholar]
- Chhabra, R.; Dubey, R.; Saini, N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24-2 cluster and its implication in human diseases. Mol. Cancer 2010, 9, 232. [Google Scholar]
- Li, X.; Liu, X.; Xu, W.; Zhou, P.; Gao, P.; Jiang, S.; Lobie, P.E.; Zhu, T. C-MYC regulated miR-23a~24-2~27a cluster promotes mammary carcinoma cell invasion and hepatic metastasis by targeting Sprouty2. J. Biol. Chem 2013, 288, 18121–18133. [Google Scholar]
- Zhou, Q.; Gallagher, R.; Ufret-Vincenty, R.; Li, X.; Olson, E.N.; Wang, S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc. Natl. Acad. Sci. USA 2011, 108, 8287–8292. [Google Scholar]
- Mansour, W.Y.; Bogdanova, N.V.; Kasten-Pisula, U.; Rieckmann, T.; Köcher, S.; Borgmann, K.; Baumann, M.; Krause, M.; Petersen, C.; Hu, H.; et al. Aberrant overlexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother. Oncol 2013, 106, 147–154. [Google Scholar]
- Ghosh, S.; Bhat, N.N.; Santra, S.; Thomas, R.G.; Gupta, S.K.; Choudhury, R.K.; Krishna, M. Low energy proton beam induces efficient cell killing in A549 lung adenocarcinoma cells. Cancer Invest 2010, 28, 615–622. [Google Scholar]
- Clyde, R.G.; Craig, A.L.; de Breed, L.; Bown, J.L.; Forrester, L.; Vojtesek, B.; Smith, G.; Hupp, T.; Crawford, J. A novel ataxia-telengiectasia mutated autoregolatory feedback mechanism in murine embryonic stem cells. J. R. Soc. Interface 2009, 6, 1167–1177. [Google Scholar]
- Mertens-Talcott, S.U.; Chintharlapalli, S.; Li, X.; Safe, S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res 2007, 67, 11001–11011. [Google Scholar]
- Xu, W.; Liu, M.; Peng, X.; Zhou, P.; Zhou, J.; Xu, K.; Xu, H.; Jiang, S. miR-24-3p and miR-27a-3p promote cell proliferation in glioma cells via cooperative regulation of MXI1. Int. J. Oncol 2013, 42, 757–766. [Google Scholar]
- Huang, Z.; Chen, X.; Yu, B.; He, J.; Chen, D. microRNA-27a promotes myoblast proliferation by targeting myostatin. Biochem. Biophys. Res. Commun 2012, 423, 265–269. [Google Scholar]
- Derheimer, F.A.; Kastan, M.B. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett 2010, 584, 3675–3681. [Google Scholar]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; Redon, C.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol 2003, 5, 675–679. [Google Scholar]
- Song, L.; Lin, C.; Wu, Z.; Gong, H.; Zeng, Y.; Wu, J.; Li, M.; Li, J. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One 2011, 6, e25454. [Google Scholar]
- Beamish, H.; Lavin, M.F. Radiosensitivity in ataxia-telangiectasia: Anomalies in radiation-induced cell cycle delay. Int. J. Radiat. Biol 1994, 65, 175–184. [Google Scholar]
- Rotman, G.; Shiloh, Y. The ATM gene and protein: Possible roles in genome surveillance, checkpoint controls and cellular defense against oxidative stress. Cancer Surv 1997, 29, 285–304. [Google Scholar]
- Rotman, G.; Shiloh, Y. ATM: From gene to function. Hum. Mol. Genet 1998, 7, 1555–1563. [Google Scholar]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol 2013, 14, 197–210. [Google Scholar]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 2005, 123, 1065–1078. [Google Scholar]
- Moumen, A.; Magill, C.; Dry, K.L.; Jackson, S.P. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 2013, 12, 698–704. [Google Scholar]
- Shin, D.Y.; Sung Kang, H.; Kim, G.Y.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed. Pharmacother 2013, 67, 305–311. [Google Scholar]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004, 64, 2390–2396. [Google Scholar]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci 2006, 31, 402–410. [Google Scholar]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet 2007, 39, 1278–1284. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar]
- Huber, S.M.; Misovic, M.; Mayer, C.; Rodemann, H.P.; Dittmann, K. EGFR-mediated stimulation of sodium/glucose cotransport promotes survival of irradiated human A549 lung adenocarcinoma cells. Radiother. Oncol 2012, 103, 373–379. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Di Francesco, A.; De Pittà, C.; Moret, F.; Barbieri, V.; Celotti, L.; Mognato, M. The DNA-Damage Response to γ-Radiation Is Affected by miR-27a in A549 Cells. Int. J. Mol. Sci. 2013, 14, 17881-17896. https://doi.org/10.3390/ijms140917881
Di Francesco A, De Pittà C, Moret F, Barbieri V, Celotti L, Mognato M. The DNA-Damage Response to γ-Radiation Is Affected by miR-27a in A549 Cells. International Journal of Molecular Sciences. 2013; 14(9):17881-17896. https://doi.org/10.3390/ijms140917881
Chicago/Turabian StyleDi Francesco, Andrea, Cristiano De Pittà, Francesca Moret, Vito Barbieri, Lucia Celotti, and Maddalena Mognato. 2013. "The DNA-Damage Response to γ-Radiation Is Affected by miR-27a in A549 Cells" International Journal of Molecular Sciences 14, no. 9: 17881-17896. https://doi.org/10.3390/ijms140917881