The Role of Initial Oligomers in Amyloid Fibril Formation by Human Stefin B

Abstract
:1. Introduction
2. Results and Discussion
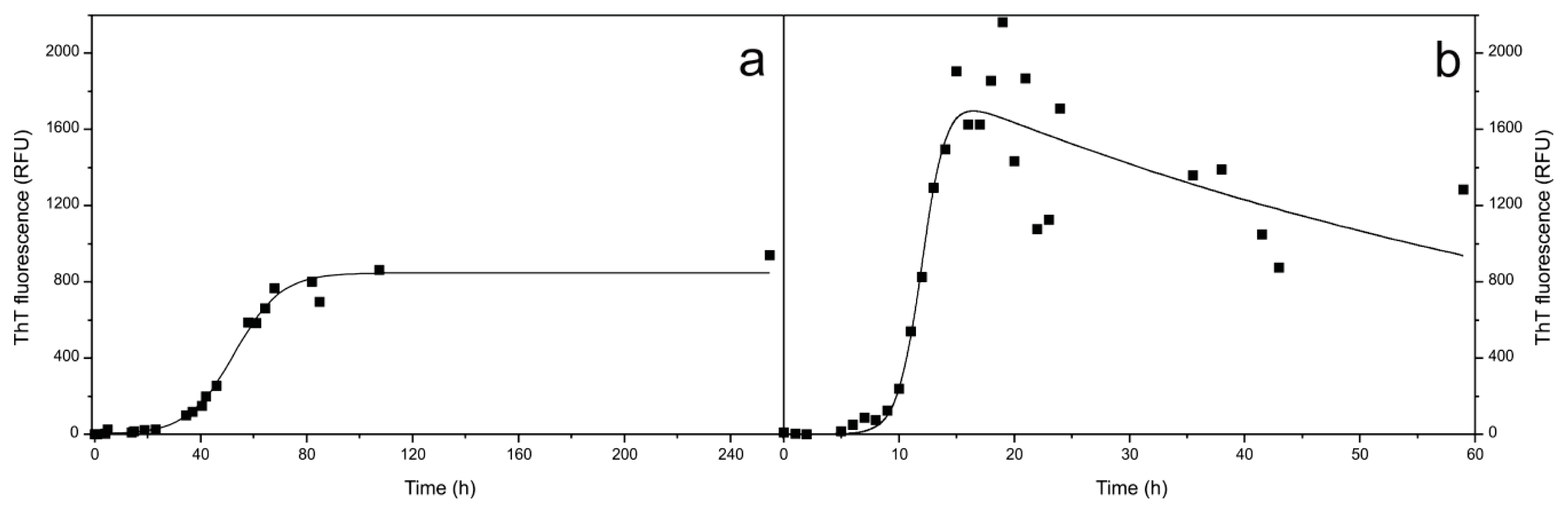
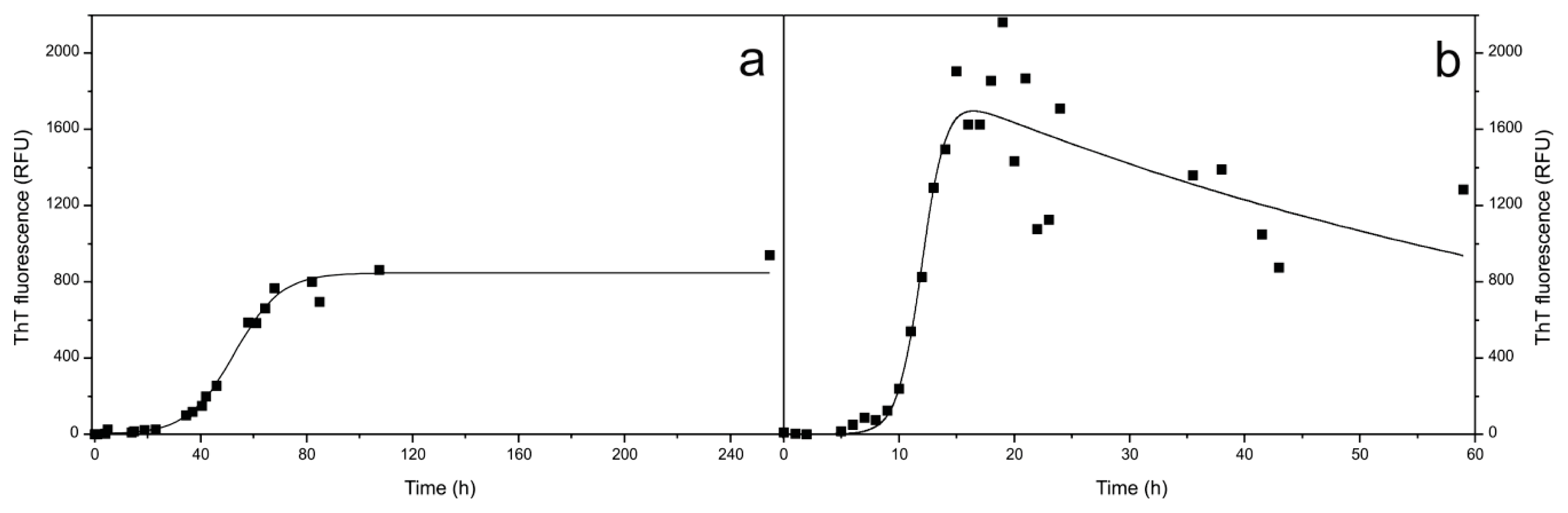
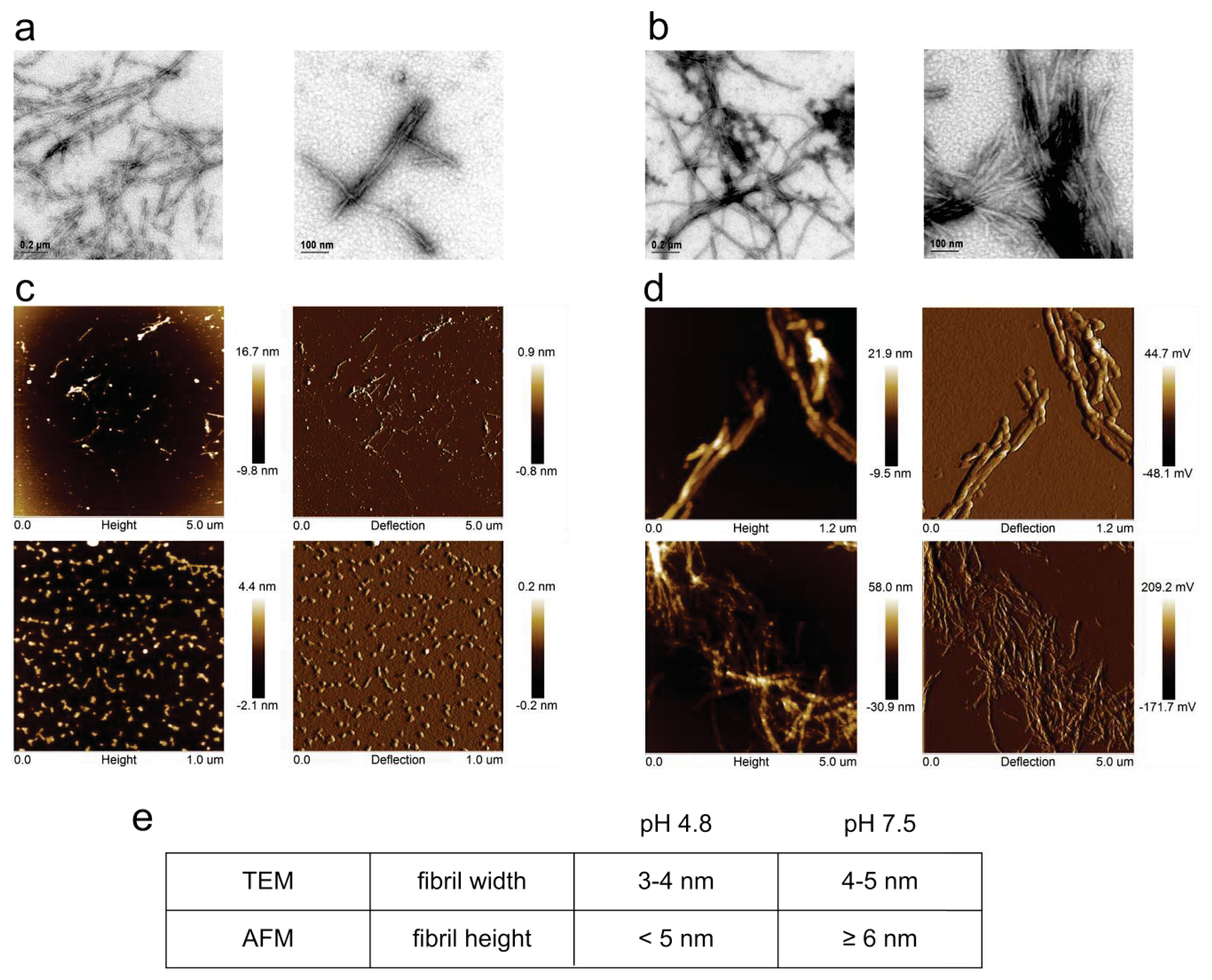
2.1. Fibril Formation by Stefin B; ThT Fluorescence, TEM and AFM Observations
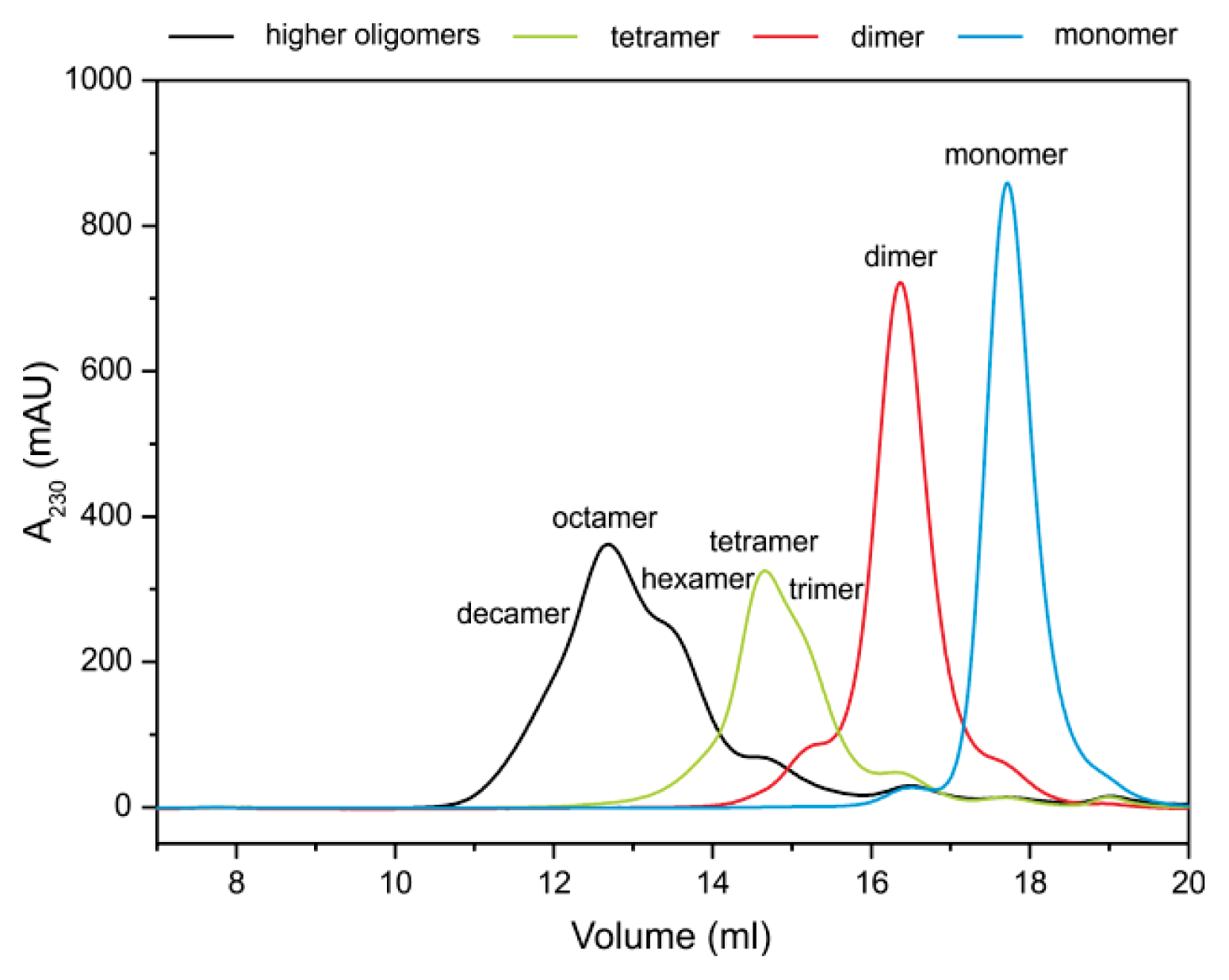
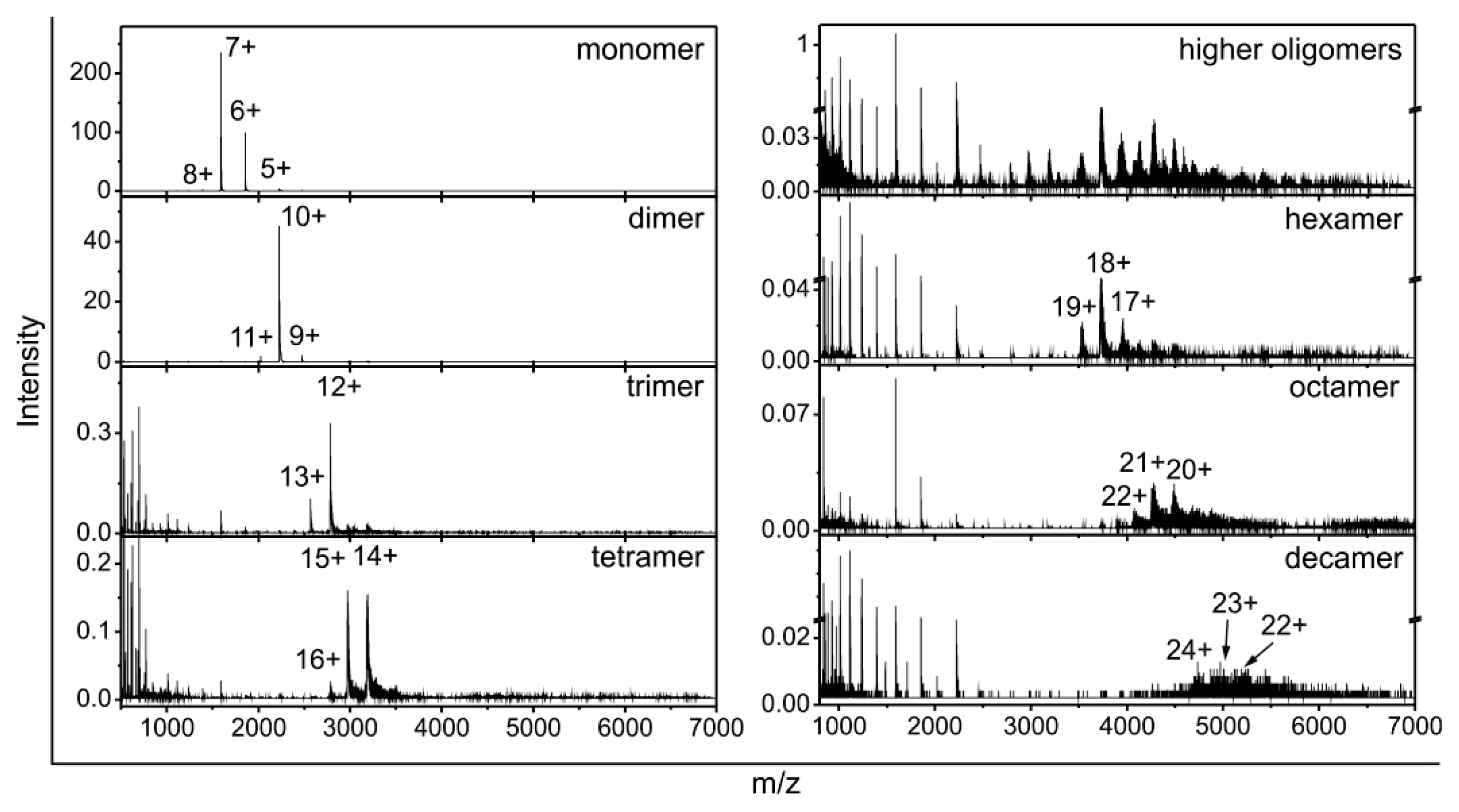
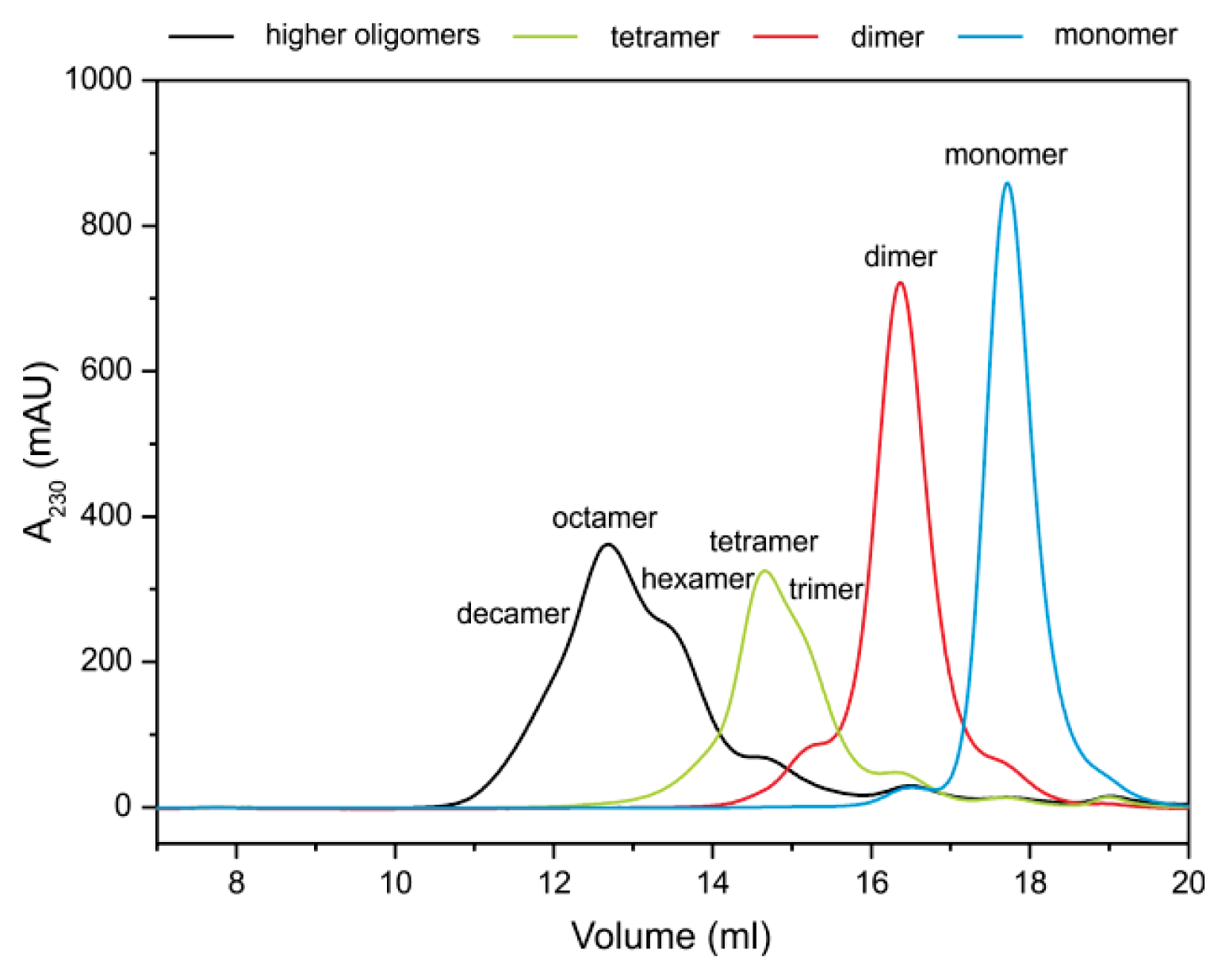
2.2. Stefin B Oligomers
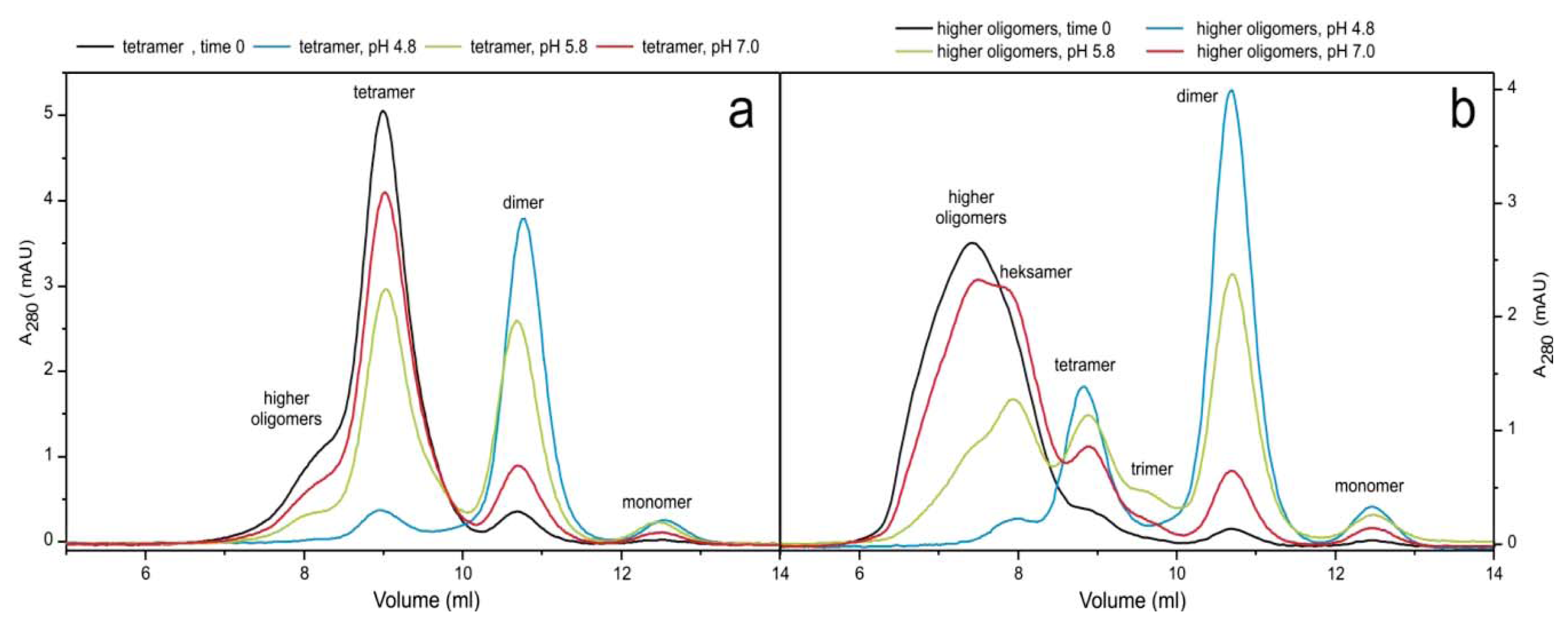
2.3. pH Dependent Transitions Between Oligomers
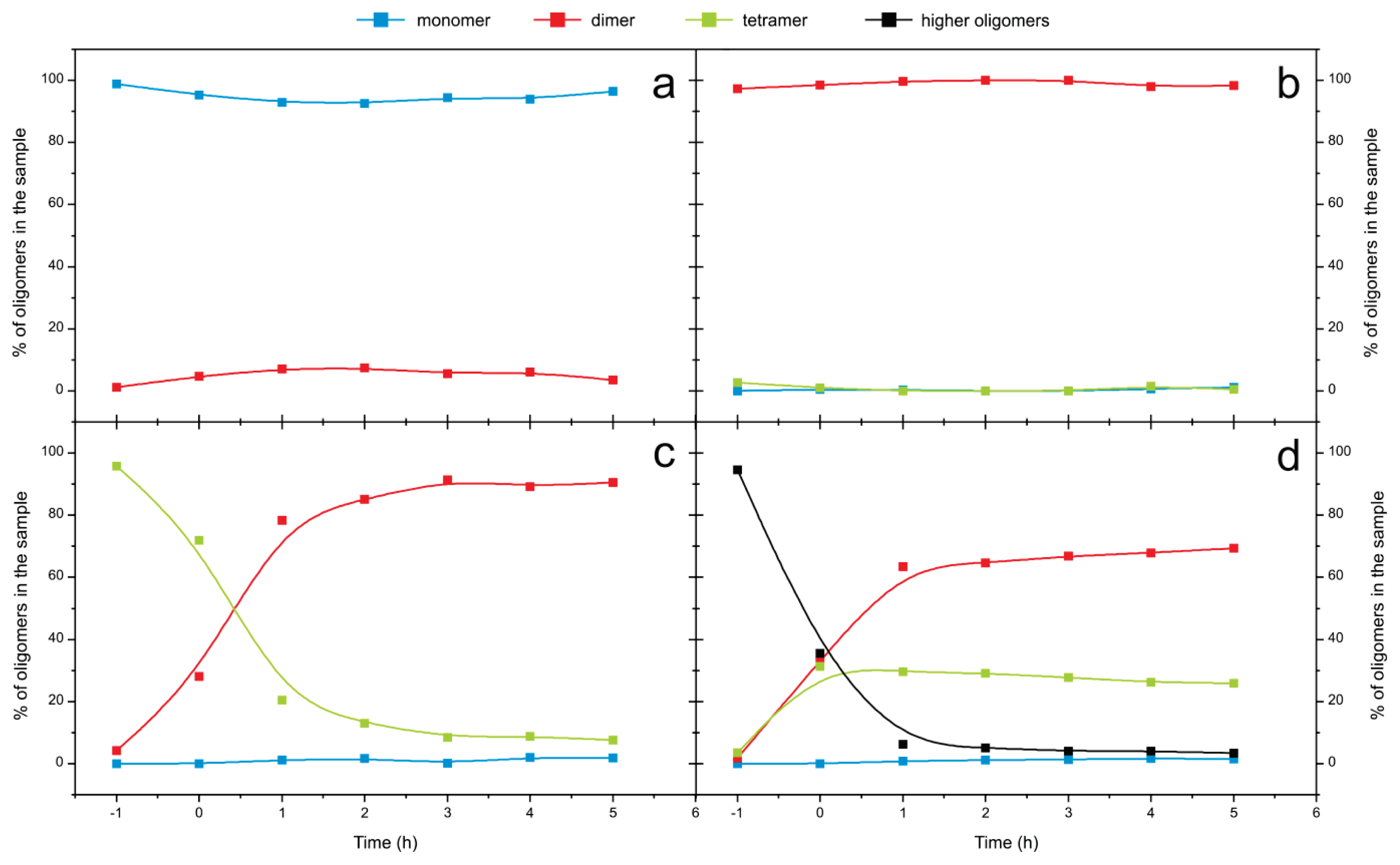
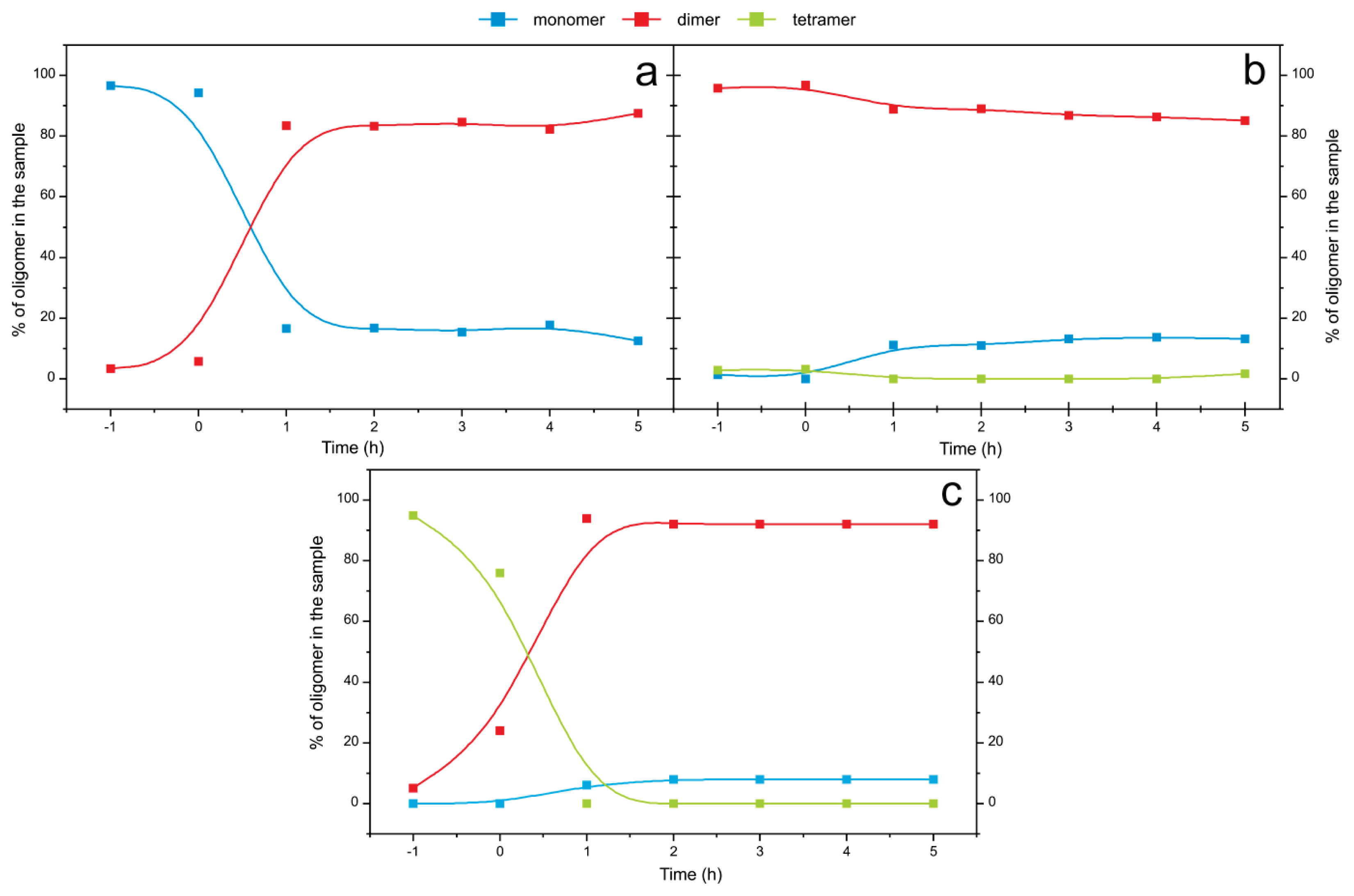
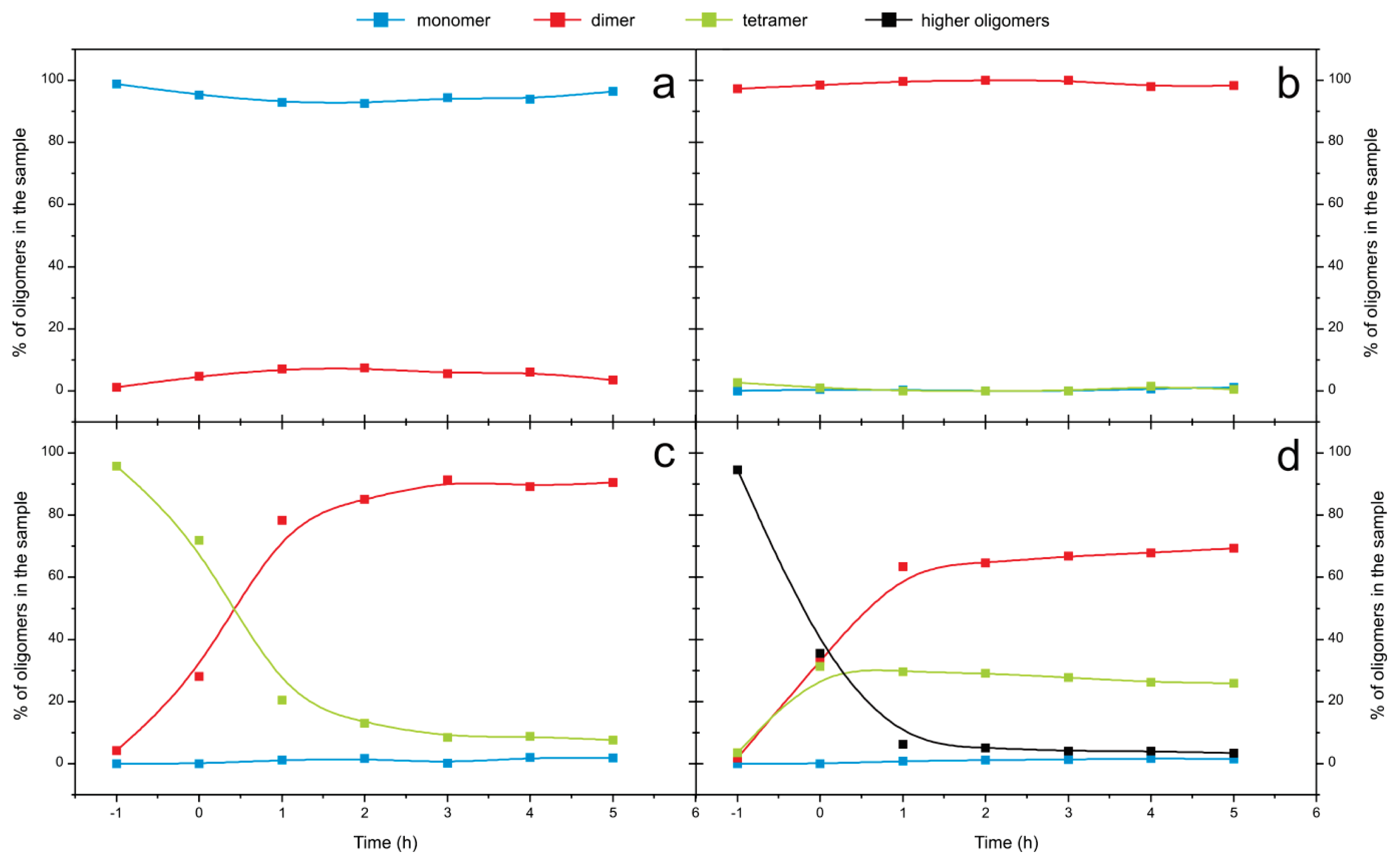
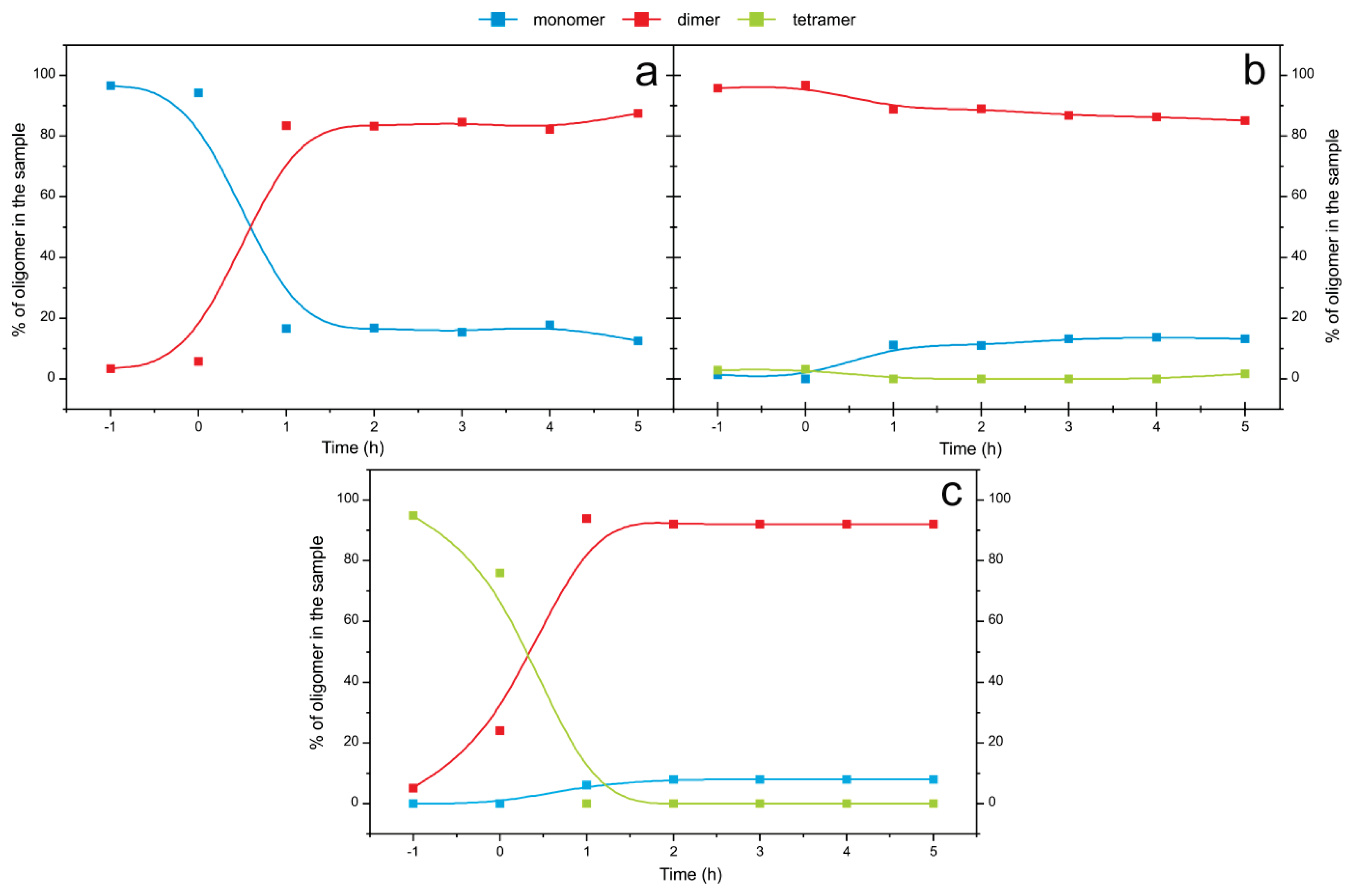
2.4. Time Course of the Transitions between Oligomers at pH 5.8 at Room Temperature Monitored by SEC and ESI MS—the Beginning of the Lag Phase
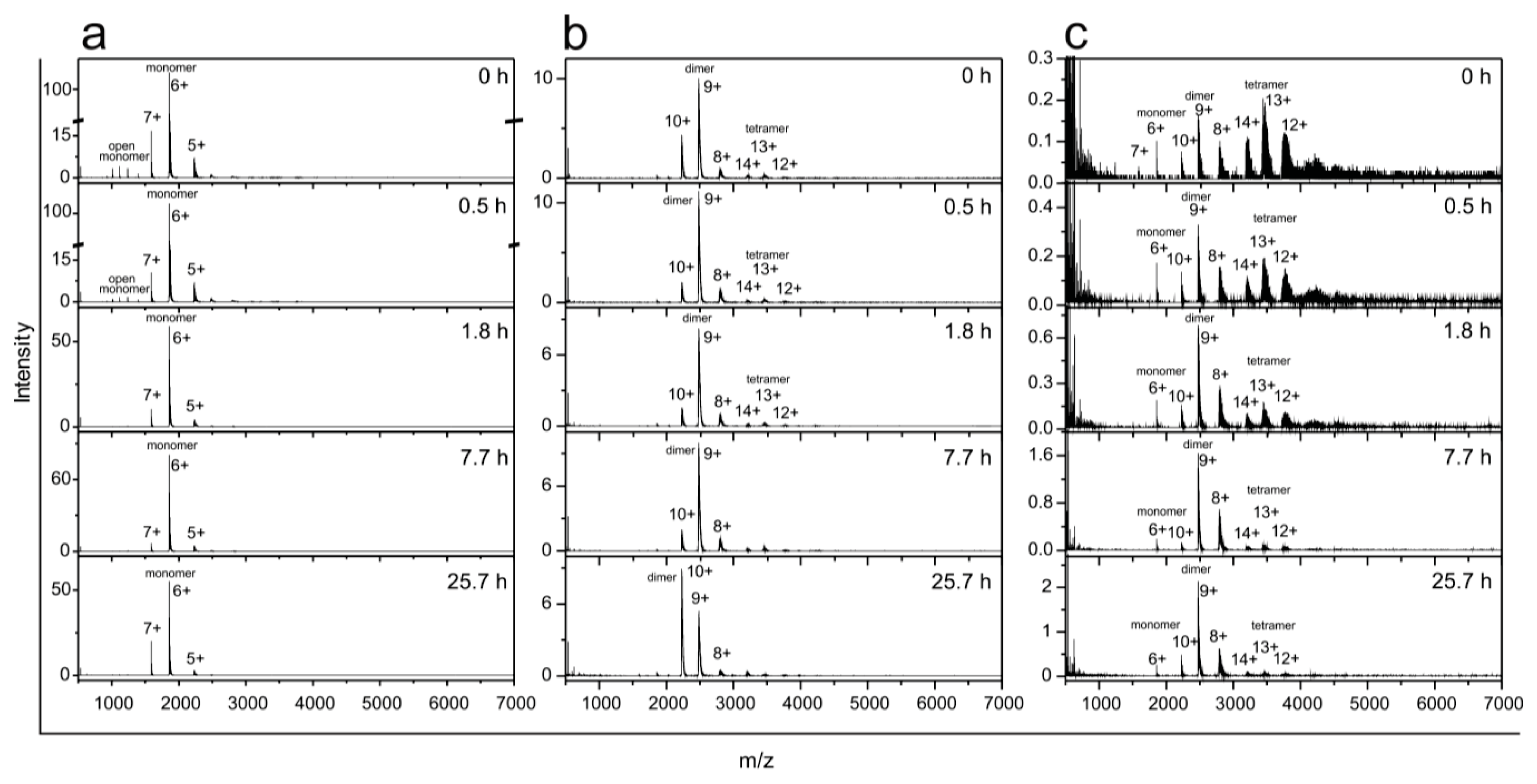
2.5. Time Course of the Transitions between Oligomers at pH 7.5 and 50 °C Monitored by SEC and ESI MS—the Whole Lag Phase
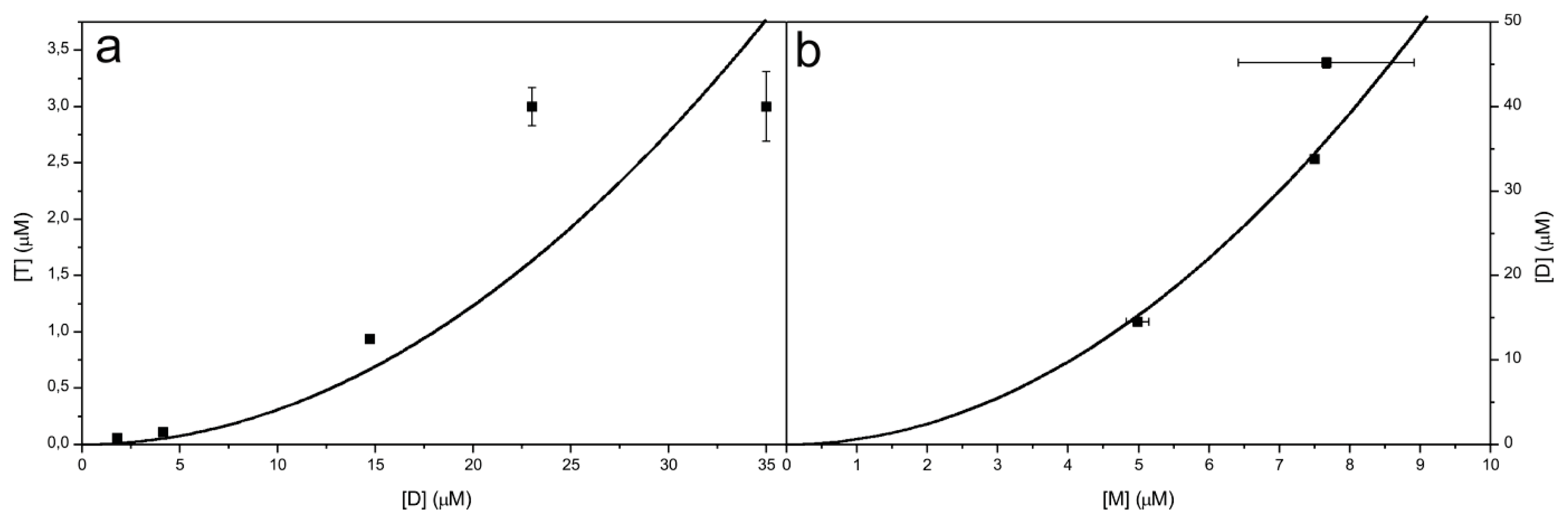
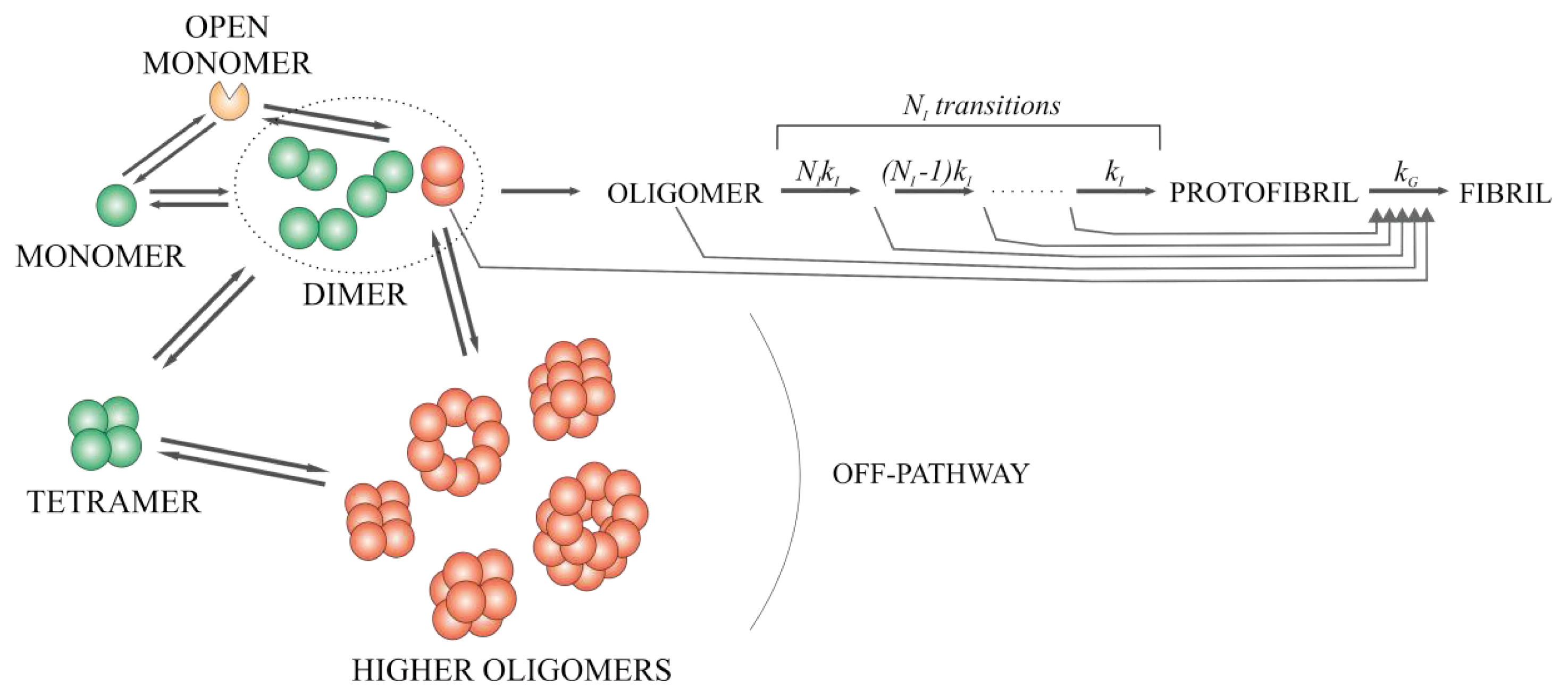
2.6. Model for the Mechanism of Amyloid Fibril Formation by Stefin B
3. Experimental Section
3.1. Expression and Purification of Recombinant Stefin B
3.2. Size Exclusion Chromatography (SEC on FPLC)
3.3. Electrospray Ionization Mass Spectrometry (ESI MS) Studies of Stefin B
3.4. Monitoring Fibrillation of Stefin B by Thioflavin T (ThT) Fluorescence
3.5. Transmission Electron Microscopy (TEM)
3.6. Atomic Force Microscopy (AFM)
3.7. Monitoring Oligomer Transitions by SEC
3.8. Monitoring Oligomer Transitions by ESI MS
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ii, K.; Ito, H.; Kominami, E.; Hirano, A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on guam, and senile dementia and in the aged. Virchows Arch. A 1993, 423, 185–194. [Google Scholar]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des 2002, 8, 1623–1637. [Google Scholar]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett 1991, 285, 213–219. [Google Scholar]
- Jerala, R.; Trstenjak, M.; Lenarcic, B.; Turk, V. Cloning a synthetic gene for human stefin B and its expression in E. coli. FEBS Lett 1988, 239, 41–44. [Google Scholar]
- Brzin, J.; Kopitar, M.; Turk, V.; Machleidt, W. Protein inhibitors of cysteine proteinases. I. Isolation and characterization of stefin, a cytosolic protein inhibitor of cysteine proteinases from human polymorphonuclear granulocytes. Hoppe-Seyler’s Z. Physiol. Chem 1983, 364, 1475–1480. [Google Scholar]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci 2008, 13, 5406–5420. [Google Scholar]
- Rawlings, N.D.; Tolle, D.P.; Barrett, A.J. Merops: The peptidase database. Nucleic Acids Res 2004, 32, D160–D164. [Google Scholar]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The refined 2.4 a X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: A novel type of proteinase inhibitor interaction. EMBO J 1990, 9, 1939–1947. [Google Scholar]
- Staniforth, R.A.; Giannini, S.; Higgins, L.D.; Conroy, M.J.; Hounslow, A.M.; Jerala, R.; Craven, C.J.; Waltho, J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J 2001, 20, 4774–4781. [Google Scholar]
- Jenko Kokalj, S.; Guncar, G.; Stern, I.; Morgan, G.; Rabzelj, S.; Kenig, M.; Staniforth, R.A.; Waltho, J.P.; Zerovnik, E.; Turk, D. Essential role of proline isomerization in stefin B tetramer formation. J. Mol. Biol 2007, 366, 1569–1579. [Google Scholar]
- Morgan, G.J.; Giannini, S.; Hounslow, A.M.; Craven, C.J.; Zerovnik, E.; Turk, V.; Waltho, J.P.; Staniforth, R.A. Exclusion of the native alpha-helix from the amyloid fibrils of a mixed alpha/beta protein. J. Mol. Biol 2008, 375, 487–498. [Google Scholar]
- Kos, J.; Krasovec, M.; Cimerman, N.; Nielsen, H.J.; Christensen, I.J.; Brunner, N. Cysteine proteinase inhibitors stefin A, stefin B, and cystatin C in sera from patients with colorectal cancer: Relation to prognosis. Clin. Cancer Res 2000, 6, 505–511. [Google Scholar]
- Riccio, M.; di Giaimo, R.; Pianetti, S.; Palmieri, P.P.; Melli, M.; Santi, S. Nuclear localization of cystatin b, the cathepsin inhibitor implicated in myoclonus epilepsy (epm1). Exp. Cell Res 2001, 262, 84–94. [Google Scholar]
- Bespalova, I.N.; Adkins, S.; Pranzatelli, M.; Burmeister, M. Novel cystatin b mutation and diagnostic pcr assay in an unverricht-lundborg progressive myoclonus epilepsy patient. Am. J. Med. Genet 1997, 74, 467–471. [Google Scholar]
- Kagitani-Shimono, K.; Imai, K.; Okamoto, N.; Ono, J.; Okada, S. Unverricht-lundborg disease with cystatin B gene abnormalities. Pediatr. Neurol 2002, 26, 55–60. [Google Scholar]
- Pennacchio, L.A.; Lehesjoki, A.E.; Stone, N.E.; Willour, V.L.; Virtaneva, K.; Miao, J.; D’Amato, E.; Ramirez, L.; Faham, M.; Koskiniemi, M.; et al. Mutations in the gene encoding cystatin b in progressive myoclonus epilepsy (epm1). Science 1996, 271, 1731–1734. [Google Scholar]
- Ceru, S.; Rabzelj, S.; Kopitar-Jerala, N.; Turk, V.; Zerovnik, E. Protein aggregation as a possible cause for pathology in a subset of familial unverricht-lundborg disease. Med. Hypotheses 2005, 64, 955–959. [Google Scholar]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; et al. Cystatin b deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, epm1. J. Neurosci 2009, 29, 5910–5915. [Google Scholar]
- Kenig, M.; Berbic, S.; Krijestorac, A.; Kroon-Zitko, L.; Tusek, M.; Pompe-Novak, M.; Zerovnik, E. Differences in aggregation properties of three site-specific mutants of recombinant human stefin B. Protein Sci 2004, 13, 63–70. [Google Scholar]
- Polajnar, M.; Vidmar, R.; Vizovisek, M.; Fonovic, M.; Kopitar-Jerala, N.; Zerovnik, E. Influence of partial unfolding and aggregation of human stefin B (cystatin B) epm1 mutants g50e and q71p on selective cleavages by cathepsins b and s. Biol. Chem 2013, 394, 783–790. [Google Scholar]
- Rabzelj, S.; Turk, V.; Zerovnik, E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with epm1. Protein Sci 2005, 14, 2713–2722. [Google Scholar]
- Zerovnik, E.; Jerala, R.; Virden, R.; Kroon Zitko, L.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: II. Folding from guhcl unfolded, tfe denatured, acid denatured, and acid intermediate states. Proteins 1998, 32, 304–313. [Google Scholar]
- Zerovnik, E.; Lohner, K.; Jerala, R.; Laggner, P.; Turk, V. Calorimetric measurements of thermal denaturation of stefins A and B. Comparison to predicted thermodynamics of stefin B unfolding. Eur. J. Biochem 1992, 210, 217–221. [Google Scholar]
- Zerovnik, E.; Virden, R.; Jerala, R.; Kroon-Zitko, L.; Turk, V.; Waltho, J.P. Differences in the effects of tfe on the folding pathways of human stefins A and B. Proteins 1999, 36, 205–216. [Google Scholar]
- Zerovnik, E.; Virden, R.; Jerala, R.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: I. Comparison to homologous stefin A. Influence of ph and trifluoroethanol on the fast and slow folding phases. Proteins 1998, 32, 296–303. [Google Scholar]
- Jenko, S.; Skarabot, M.; Kenig, M.; Guncar, G.; Musevic, I.; Turk, D.; Zerovnik, E. Different propensity to form amyloid fibrils by two homologous proteins-human stefins A and B: Searching for an explanation. Proteins 2004, 55, 417–425. [Google Scholar]
- Zerovnik, E.; Pompe-Novak, M.; Skarabot, M.; Ravnikar, M.; Musevic, I.; Turk, V. Human stefin B readily forms amyloid fibrils in vitro. Biochim. Biophys. Acta 2002, 1594, 1–5. [Google Scholar]
- Zerovnik, E.; Zavasnik-Bergant, V.; Kopitar-Jerala, N.; Pompe-Novak, M.; Skarabot, M.; Goldie, K.; Ravnikar, M.; Musevic, I.; Turk, V. Amyloid fibril formation by human stefin B in vitro: Immunogold labelling and comparison to stefin A. Biol. Chem 2002, 383, 859–863. [Google Scholar]
- Anderluh, G.; Gutierrez-Aguirre, I.; Rabzelj, S.; Ceru, S.; Kopitar-Jerala, N.; Macek, P.; Turk, V.; Zerovnik, E. Interaction of human stefin B in the prefibrillar oligomeric form with membranes. Correlation with cellular toxicity. FEBS J 2005, 272, 3042–3051. [Google Scholar]
- Anderluh, G.; Zerovnik, E. Pore formation by human stefin B in its native and oligomeric states and the consequent amyloid induced toxicity. Front. Mol. Neurosci 2012, 5, 85. [Google Scholar]
- Rabzelj, S.; Viero, G.; Gutierrez-Aguirre, I.; Turk, V.; Dalla Serra, M.; Anderluh, G.; Zerovnik, E. Interaction with model membranes and pore formation by human stefin B: Studying the native and prefibrillar states. FEBS J 2008, 275, 2455–2466. [Google Scholar]
- Ceru, S.; Kokalj, S.J.; Rabzelj, S.; Skarabot, M.; Gutierrez-Aguirre, I.; Kopitar-Jerala, N.; Anderluh, G.; Turk, D.; Turk, V.; Zerovnik, E. Size and morphology of toxic oligomers of amyloidogenic proteins: A case study of human stefin B. Amyloid 2008, 15, 147–159. [Google Scholar]
- Ceru, S.; Zerovnik, E. Similar toxicity of the oligomeric molten globule state and the prefibrillar oligomers. FEBS Lett 2008, 582, 203–209. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett 2004, 11, 213–228. [Google Scholar]
- Skerget, K.; Vilfan, A.; Pompe-Novak, M.; Turk, V.; Waltho, J.P.; Turk, D.; Zerovnik, E. The mechanism of amyloid-fibril formation by stefin B: Temperature and protein concentration dependence of the rates. Proteins 2009, 74, 425–436. [Google Scholar]
- He, X.; Giurleo, J.T.; Talaga, D.S. Role of small oligomers on the amyloidogenic aggregation free-energy landscape. J. Mol. Biol 2010, 395, 134–154. [Google Scholar]
- Singh, J.; Sabareesan, A.T.; Mathew, M.K.; Udgaonkar, J.B. Development of the structural core and of conformational heterogeneity during the conversion of oligomers of the mouse prion protein to worm-like amyloid fibrils. J. Mol. Biol 2012, 423, 217–231. [Google Scholar]
- Tay, W.M.; Huang, D.; Rosenberry, T.L.; Paravastu, A.K. The Alzheimer’s amyloid-beta(1–42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. J. Mol. Biol 2013, 425, 2494–2508. [Google Scholar]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar]
- Breuker, K.; McLafferty, F.W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc. Natl. Acad. Sci. USA 2008, 105, 18145–18152. [Google Scholar]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev 1997, 16, 1–23. [Google Scholar]
- Skerget, K.; Taler-Vercic, A.; Bavdek, A.; Hodnik, V.; Ceru, S.; Tusek-Znidaric, M.; Kumm, T.; Pitsi, D.; Pompe-Novak, M.; Palumaa, P.; et al. Interaction between oligomers of stefin B and amyloid-beta in vitro and in cells. J. Biol. Chem 2010, 285, 3201–3210. [Google Scholar]
- Kaddis, C.S.; Loo, J.A. Native protein ms and ion mobility: Large flying proteins with esi. Anal. Chem 2007, 79, 1778–1784. [Google Scholar]
- Kaltashov, I.; Eyles, S.J. Mass Spectrometry-based Approaches to Study Biomolecular Dynamics: Equilibrium Intermediates. In Mass Spectrometry in Biophysics: Conformation and Dynamics of Biomolecules; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2005; pp. 184–186. [Google Scholar]
- Frimpong, A.K.; Abzalimov, R.R.; Uversky, V.N.; Kaltashov, I.A. Characterization of intrinsically disordered proteins with electrospray ionization mass spectrometry: Conformational heterogeneity of alpha-synuclein. Proteins 2010, 78, 714–722. [Google Scholar]
- Grandori, R. Origin of the conformation dependence of protein charge-state distributions in electrospray ionization mass spectrometry. J. Mass Spectrom 2003, 38, 11–15. [Google Scholar]
- Konermann, L.; Douglas, D.J. Equilibrium unfolding of proteins monitored by electrospray ionization mass spectrometry: Distinguishing two-state from multi-state transitions. Rapid Commun. Mass Spectrom 1998, 12, 435–442. [Google Scholar]
- Konermann, L.; Pan, J.; Wilson, D.J. Protein folding mechanisms studied by time-resolved electrospray mass spectrometry. Biotechniques 2006, 40, 135–141. [Google Scholar]
- Palumaa, P.; Njunkova, O.; Pokras, L.; Eriste, E.; Jornvall, H.; Sillard, R. Evidence for non-isostructural replacement of Zn(2+) with Cd(2+) in the beta-domain of brain-specific metallothionein-3. FEBS Lett 2002, 527, 76–80. [Google Scholar]
- Smith, D.P.; Giles, K.; Bateman, R.H.; Radford, S.E.; Ashcroft, A.E. Monitoring copopulated conformational states during protein folding events using electrospray ionization-ion mobility spectrometry-mass spectrometry. J. Am. Soc.Mass Spectrom 2007, 18, 2180–2190. [Google Scholar]
- Woods, L.A.; Radford, S.E.; Ashcroft, A.E. Advances in ion mobility spectrometry-mass spectrometry reveal key insights into amyloid assembly. Biochim. Biophys. Acta 2013, 1834, 1257–1268. [Google Scholar]
- Jablonowska, A.; Bakun, M.; Kupniewska-Kozak, A.; Dadlez, M. Alzheimer’s disease Aβ peptide fragment 10–30 forms a spectrum of metastable oligomers with marked preference for n to n and c to c monomer termini proximity. J. Mol. Biol 2004, 344, 1037–1049. [Google Scholar]
- Diezemann, N.; Weigang, L.M.M.; Chakraborty, P.; Frauendorf, H.; Letzel, T.; Diederichsen, U. Organization of nucleobase-functionalized beta-peptides investigated by soft electrospray ionization mass spectrometry. J. Mass Spectrom. 2009, 44, 794–802. [Google Scholar]
- Nettleton, E.J.; Tito, P.; Sunde, M.; Bouchard, M.; Dobson, C.M.; Robinson, C.V. Characterization of the oligomeric states of insulin in self-assembly and amyloid fibril formation by mass spectrometry. Biophys. J 2000, 79, 1053–1065. [Google Scholar]
- McCammon, M.G.; Scott, D.J.; Keetch, C.A.; Greene, L.H.; Purkey, H.E.; Petrassi, H.M.; Kelly, J.W.; Robinson, C.V. Screening transthyretin amyloid fibril inhibitors: Characterization of novel multiprotein, multiligand complexes by mass spectrometry. Structure 2002, 10, 851–863. [Google Scholar]
- Nettleton, E.J.; Sunde, M.; Lai, Z.; Kelly, J.W.; Dobson, C.M.; Robinson, C.V. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: Evidence from electrospray mass spectrometry. J. Mol. Biol 1998, 281, 553–564. [Google Scholar]
- Smith, A.M.; Jahn, T.R.; Ashcroft, A.E.; Radford, S.E. Direct observation of oligomeric species formed in the early stages of amyloid fibril formation using electrospray ionisation mass spectrometry. J. Mol. Biol 2006, 364, 9–19. [Google Scholar]
- Sanglier, S.; Ramström, H.; Haiech, J.; Leize, E.; Dorsselaer, A.V. Electrospray ionization mass spectrometry analysis revealed a ~310 kDa noncovalent hexamer of hpr kinase/phosphatase from bacillus subtilis. Inter. J. Mass Spectrom 2002, 219, 681–696. [Google Scholar]
- Zal, F.; Chausson, F.; Leize, E.; van Dorsselaer, A.; Lallier, F.H.; Green, B.N. Quadrupole time-of-flight mass spectrometry of the native hemocyanin of the deep-sea crab bythograea thermydron. Biomacromolecules 2002, 3, 229–231. [Google Scholar]
- Tahallah, N.; Pinkse, M.; Maier, C.S.; Heck, A.J.R. The effect of the source pressure on the abundance of ions of noncovalent protein assemblies in an electrospray ionization orthogonal time-of-flight instrument. Rapid Comm. Mass Spectrom 2001, 15, 596–601. [Google Scholar]
- Sanglier, S.; Leize, E.; Dorsselaer, A.; Zal, F. Comparative esi-ms study of ~2.2 mda native hemocyanins from deep-sea and shore crabs: From protein oligomeric state to biotope. J. Am. Soc. Mass Spectrom 2003, 14, 419–429. [Google Scholar]
- Smith, D.P.; Woods, L.A.; Radford, S.E.; Ashcroft, A.E. Structure and dynamics of oligomeric intermediates in beta2-microglobulin self-assembly. Biophys. J 2011, 101, 1238–1247. [Google Scholar]
- Kloniecki, M.; Jablonowska, A.; Poznanski, J.; Langridge, J.; Hughes, C.; Campuzano, I.; Giles, K.; Dadlez, M. Ion mobility separation coupled with ms detects two structural states of Alzheimer’s disease abeta1–40 peptide oligomers. J. Mol. Biol 2011, 407, 110–124. [Google Scholar]
- Zerovnik, E.; Jerala, R.; Kroon-Zitko, L.; Pain, R.H.; Turk, V. Intermediates in denaturation of a small globular protein, recombinant human stefin B. J. Biol. Chem 1992, 267, 9041–9046. [Google Scholar]
- Zerovnik, E.; Jerala, R.; Poklar, N.; Kroon-Zitko, L.; Turk, V. Compactness of the molten globule in comparison to unfolded states as observed by size-exclusion chromatography. Biochim. Biophys. Acta 1994, 1209, 140–143. [Google Scholar]
- Jerala, R.; Zerovnik, E. Accessing the global minimum conformation of stefin A dimer by annealing under partially denaturing conditions. J. Mol. Biol 1999, 291, 1079–1089. [Google Scholar]
- Smajlovic, A.; Berbic, S.; Schiene-Fischer, C.; Tusek-Znidaric, M.; Taler, A.; Jenko-Kokalj, S.; Turk, D.; Zerovnik, E. Essential role of pro 74 in stefin B amyloid-fibril formation: Dual action of cyclophilin a on the process. FEBS Lett 2009, 583, 1114–1120. [Google Scholar]
- Zerovnik, E.; Skarabot, M.; Skerget, K.; Giannini, S.; Stoka, V.; Jenko-Kokalj, S.; Staniforth, R.A. Amyloid fibril formation by human stefin B: Influence of ph and tfe on fibril growth and morphology. Amyloid 2007, 14, 237–247. [Google Scholar]
- Cipollini, E.; Riccio, M.; di Giaimo, R.; dal Piaz, F.; Pulice, G.; Catania, S.; Caldarelli, I.; Dembic, M.; Santi, S.; Melli, M. Cystatin b and its epm1 mutants are polymeric and aggregate prone in vivo. Biochim. Biophys. Acta 2008, 1783, 312–322. [Google Scholar]
- Paramore, R.; Morgan, G.J.; Davis, P.J.; Sharma, C.A.; Hounslow, A.; Taler-Vercic, A.; Zerovnik, E.; Waltho, J.P.; Cliff, M.J.; Staniforth, R.A. Mapping local structural perturbations in the native state of stefin B (cystatin B) under amyloid forming conditions. Front. Mol. Neurosci 2012, 5, 94. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein concentration (μM) | Amount of oligomers in the sample (%) (time –1—beginning of the experiment, before changing the conditions) | Amount of oligomers in the sample (%) (time 5 h—end of the experiment) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| M | D | T | H | M | D | T | H | ||
| pH 5.8 | 4 | 5 | 56 | 28 | 11 | 4 | 89 | 7 | – |
| 9 | 5 | 56 | 28 | 11 | 4 | 91 | 5 | – | |
| 34 | 5 | 56 | 28 | 11 | 3 | 86 | 11 | – | |
| 75 | 20 | 53 | 21 | 6 | 23 | 58 | 16 | 3 | |
| 100 | 20 | 53 | 21 | 6 | 20 | 66 | 12 | 2 | |
| pH 7.5 | 34 | 20 | 53 | 21 | 6 | 15 | 85 | – | – |
| 75 | 20 | 53 | 21 | 6 | 10 | 90 | – | – | |
| 100 | 8 | 66 | 21 | 5 | 9 | 89 | 2 | – | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taler-Verčič, A.; Kirsipuu, T.; Friedemann, M.; Noormägi, A.; Polajnar, M.; Smirnova, J.; Žnidarič, M.T.; Žganec, M.; Škarabot, M.; Vilfan, A.; et al. The Role of Initial Oligomers in Amyloid Fibril Formation by Human Stefin B. Int. J. Mol. Sci. 2013, 14, 18362-18384. https://doi.org/10.3390/ijms140918362
Taler-Verčič A, Kirsipuu T, Friedemann M, Noormägi A, Polajnar M, Smirnova J, Žnidarič MT, Žganec M, Škarabot M, Vilfan A, et al. The Role of Initial Oligomers in Amyloid Fibril Formation by Human Stefin B. International Journal of Molecular Sciences. 2013; 14(9):18362-18384. https://doi.org/10.3390/ijms140918362
Chicago/Turabian StyleTaler-Verčič, Ajda, Tiina Kirsipuu, Merlin Friedemann, Andra Noormägi, Mira Polajnar, Julia Smirnova, Magda Tušek Žnidarič, Matjaž Žganec, Miha Škarabot, Andrej Vilfan, and et al. 2013. "The Role of Initial Oligomers in Amyloid Fibril Formation by Human Stefin B" International Journal of Molecular Sciences 14, no. 9: 18362-18384. https://doi.org/10.3390/ijms140918362