Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Brief Overview of the Line-Scan Approach
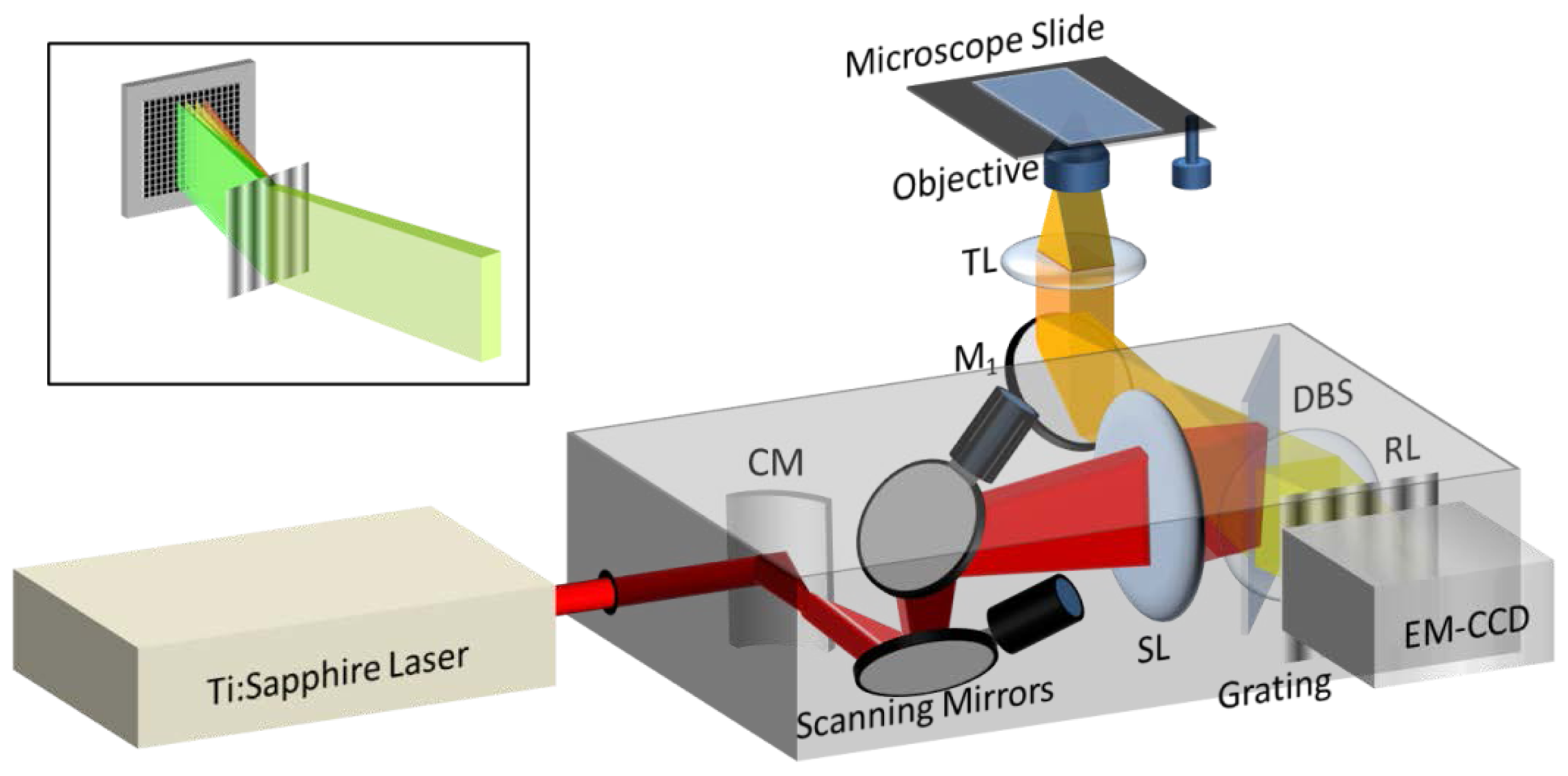
2.2. Design and Realization of the Optical Setups
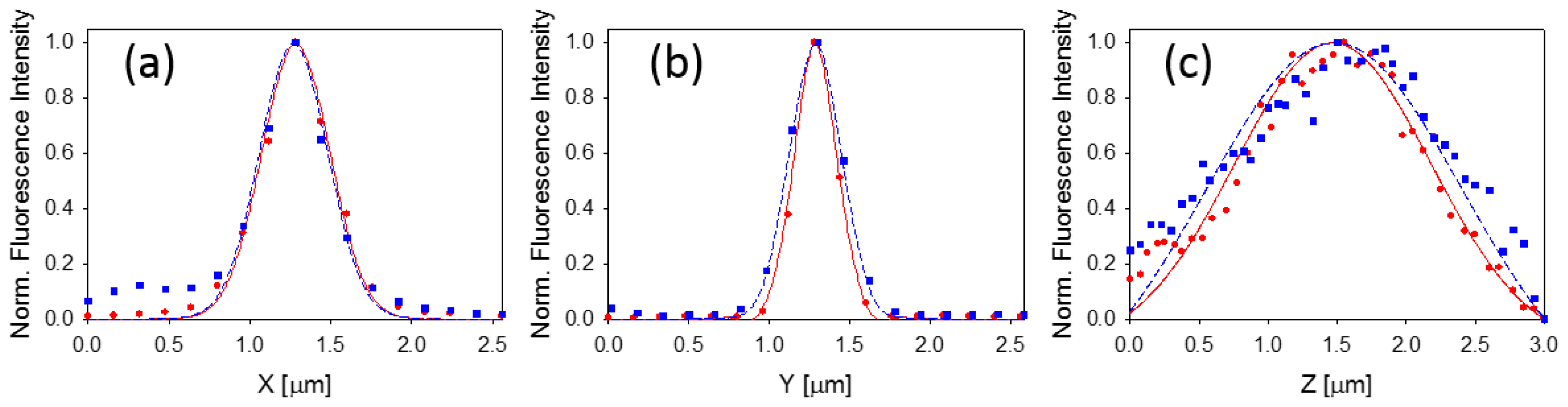
2.3. Characterization of the Point Spread Functions of the Line-Scan and Point-Scan Setups
2.4. Comparison of Sensitivity and Acquisition Speed between the Two Scanning Modes
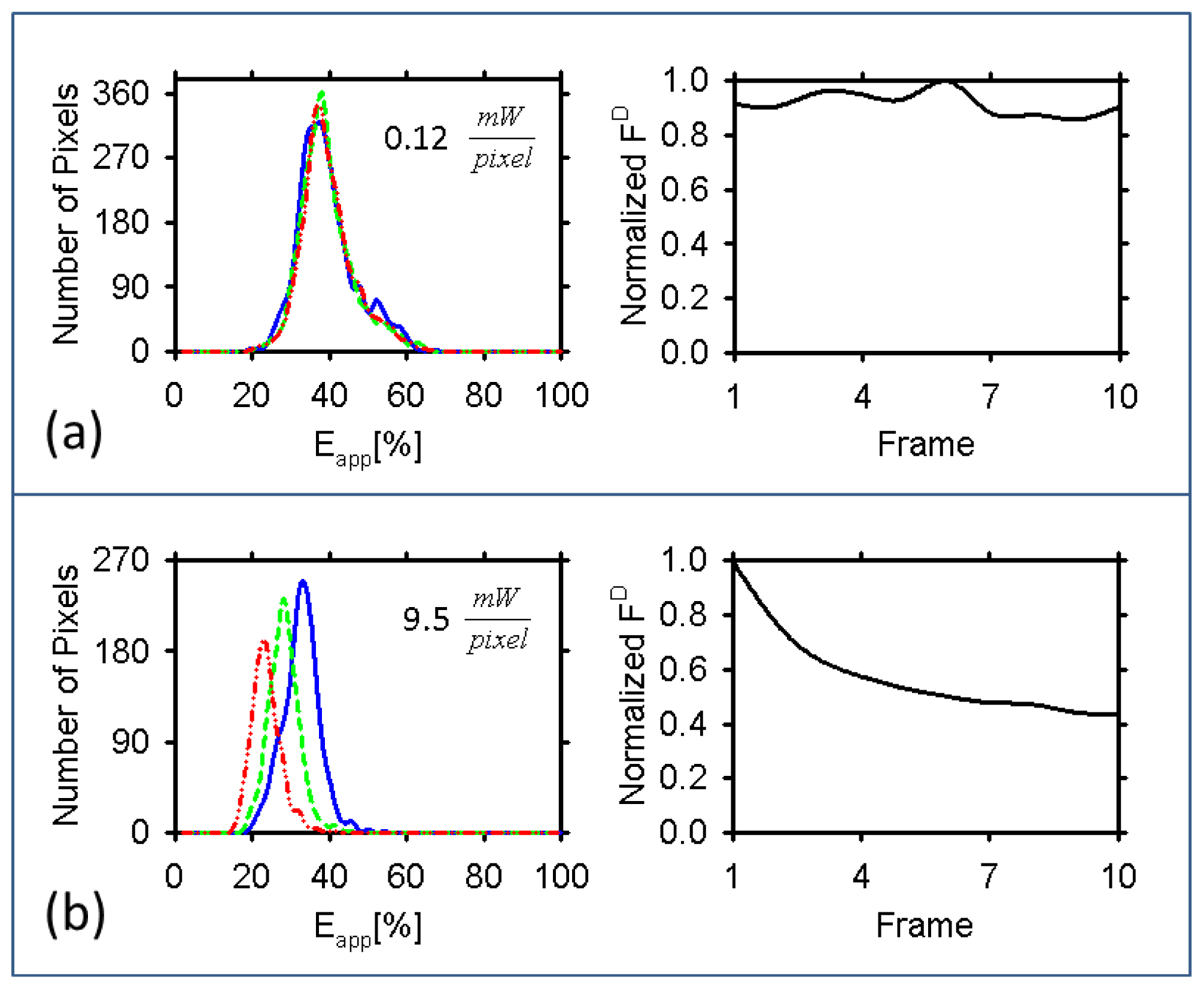
2.5. Quantifying FRET in Biological Cells Using the Two Scanning Methods
3. Methods
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dickinson, M.E.; Bearman, G.; Tille, S.; Lansford, R.; Fraser, S.E. Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques 2001, 31. [Google Scholar]
- Haraguchi, T.; Shimi, T.; Koujin, T.; Hashiguchi, N.; Hiraoka, Y. Spectral imaging fluorescence microscopy. Genes Cells 2002, 7, 881–887. [Google Scholar]
- Neher, R.A.; Neher, E. Applying spectral fingerprinting to the analysis of FRET images. Microsc. Res. Tech 2004, 64, 185–195. [Google Scholar]
- Palero, J. Nonlinear spectral imaging microscopy. Imaging Microscopy 2009, 11, 22–24. [Google Scholar]
- Raicu, V.; Stoneman, M.R.; Fung, R.; Melnichuk, M.; Jansma, D.B.; Pisterzi, L.F.; Rath, S.; Fox, M.; Wells, J.W.; Saldin, D.K. Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nat. Photonics 2009, 3, 107–113. [Google Scholar]
- Mahou, P.; Zimmerley, M.; Loulier, K.; Matho, K.S.; Labroille, G.; Morin, X.; Supatto, W.; Livet, J.; Debarre, D.; Beaurepaire, E. Multicolor two-photon tissue imaging by wavelength mixing. Nat. Methods 2012, 9, 815–818. [Google Scholar]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol 2003, 21, 1369–1377. [Google Scholar]
- Masters, B.R.; So, P.T.C. Antecedents of two-photon excitation laser scanning microscopy. Microsc. Res. Tech 2004, 63, 3–11. [Google Scholar]
- Stoneman, M.; Fox, M.; Zeng, C.; Raicu, V. Real-time monitoring of two-photon photopolymerization for use in fabrication of microfluidic devices. Lab Chip 2009, 9, 819–827. [Google Scholar]
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Mol. Biol 2000, 7, 730–734. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed; Springer: New York, NY, USA, 2006. [Google Scholar]
- Raicu, V.; Popescu, A. Integrated Molecular and Cellular Biophysics; Springer: London, UK, 2008. [Google Scholar]
- Pearce, L.L.; Gandley, R.E.; Han, W.; Wasserloos, K.; Stitt, M.; Kanai, A.J.; McLaughlin, M.K.; Pitt, B.R.; Levitan, E.S. Role of metallothionein in nitric oxide signaling as revealed by a green fluorescent fusion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 477–482. [Google Scholar]
- George, S.R.; O’Dowd, B.F.; Lee, S.P. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov 2002, 1, 808–820. [Google Scholar]
- Heyduk, T. Measuring protein conformational changes by FRET/LRET. Curr. Opin. Biotechnol 2002, 13, 292–296. [Google Scholar]
- Parsons, M.; Vojnovic, B.; Ameer-Beg, S. Imaging protein-protein interactions in cell motility using fluorescence resonance energy transfer (FRET). Biochem. Soc. Trans 2004, 32, 431–433. [Google Scholar]
- Caorsi, V.; Ronzitti, E.; Vicidomini, G.; Krol, S.; McConnell, G.; Diaspro, A. FRET measurements on fuzzy fluorescent nanostructures. Microsc. Res. Tech 2007, 70, 452–458. [Google Scholar]
- An, S.J.; Almers, W. Tracking SNARE complex formation in live endocrine cells. Science 2004, 306, 1042–1046. [Google Scholar]
- Carriba, P.; Navarro, G.; Ciruela, F.; Ferre, S.; Casado, V.; Agnati, L.; Cortes, A.; Mallol, J.; Fuxe, K.; Canela, E.I.; et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 2008, 5, 727–733. [Google Scholar]
- Llères, D.; James, J.; Swift, S.; Norman, D.G.; Lamond, A.I. Quantitative analysis of chromatin compaction in living cells using FLIM–FRET. J. Cell Biol 2009, 187, 481–496. [Google Scholar] [Green Version]
- Demarco, I.A.; Periasamy, A.; Booker, C.F.; Day, R.N. Monitoring dynamic protein interactions with photoquenching FRET. Nat. Methods 2006, 3, 519–524. [Google Scholar]
- Raicu, V.; Jansma, D.B.; Miller, R.J.; Friesen, J.D. Protein interaction quantified in vivo by spectrally resolved fluorescence resonance energy transfer. Biochem. J 2005, 385, 265–277. [Google Scholar]
- Maurel, D.; Comps-Agrar, L.; Brock, C.; Rives, M.L.; Bourrier, E.; Ayoub, M.A.; Bazin, H.; Tinel, N.; Durroux, T.; Prezeau, L.; et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: Application to GPCR oligomerization. Nat. Methods 2008, 5, 561–567. [Google Scholar]
- Pisterzi, L.F.; Jansma, D.B.; Georgiou, J.; Woodside, M.J.; Chou, J.T.; Angers, S.; Raicu, V.; Wells, J.W. Oligomeric size of the M2 muscarinic receptor in live cells as determined by quantitative fluorescence resonance energy transfer (FRET). J. Biol. Chem 2010, 285, 16723–16738. [Google Scholar]
- Chen, L.; Novicky, L.; Merzlyakov, M.; Hristov, T.; Hristova, K. Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc 2010, 132, 3628–3635. [Google Scholar]
- Raicu, V. Efficiency of resonance energy transfer in homo-oligomeric complexes of proteins. J. Biol. Phys 2007, 33, 109–127. [Google Scholar]
- Singh, D.R.; Raicu, V. Comparison between whole distribution- and average-based approaches to the determination of fluorescence resonance energy transfer efficiency in ensembles of proteins in living cells. Biophys. J 2010, 98, 2127–2135. [Google Scholar]
- Singh, D.R.; Mohammad, M.M.; Patowary, S.; Stoneman, M.R.; Oliver, J.A.; Movileanu, L.; Raicu, V. Determination of the quaternary structure of a bacterial ATP-binding cassette (ABC) transporter in living cells. Integr. Biol 2013, 5, 312–323. [Google Scholar]
- Patowary, S.; Alvarez-Curto, E.; Xu, T.R.; Holz, J.D.; Oliver, J.A.; Milligan, G.; Raicu, V. The muscarinic M3 acetylcholine receptor exists as two differently sized complexes at the plasma membrane. Biochem. J 2013, 452, 303–312. [Google Scholar]
- Raicu, V.; Singh, D.R. FRET spectrometry: A new tool for the determination of protein quaternary structure in living cells. Biophys. J 2013, 105, 1937–1945. [Google Scholar]
- Sinclair, M.B.; Timlin, J.A.; Haaland, D.M.; Werner-Washburne, M. Design, construction, characterization, and application of a hyperspectral microarray scanner. Appl. Opt 2004, 43, 2079–2088. [Google Scholar]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett 2002, 531, 245–249. [Google Scholar]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem 1998, 67, 509–544. [Google Scholar]
- Mannan, M.A.; Shadrick, W.R.; Biener, G.; Shin, B.S.; Anshu, A.; Raicu, V.; Frick, D.N.; Dey, M. An Ire1-Phk1 chimera reveals a dispensable role of autokinase activity in endoplasmic reticulum stress response. J. Mol. Biol 2013, 425, 2083–2099. [Google Scholar]
- Bewersdorf, J.; Pick, R.; Hell, S.W. Multifocal multiphoton microscopy. Opt. Lett 1998, 23, 655–657. [Google Scholar]
- Kurtz, R.; Fricke, M.; Kalb, J.; Tinnefeld, P.; Sauer, M. Application of multiline two-photon microscopy to functional in vivo imaging. J. Neurosci. Methods 2006, 151, 276–286. [Google Scholar]
- Kim, K.H.; Buehler, C.; So, P.T.C. High-speed, two-photon scanning microscope. Appl. Opt 1999, 38, 6004–6009. [Google Scholar]
- Kumazaki, S.; Hasegawa, M.; Ghoneim, M.; Shimizu, Y.; Okamoto, K.; Nishiyama, M.; Oh-Oka, H.; Terazima, M. A line-scanning semi-confocal multi-photon fluorescence microscope with a simultaneous broadband spectral acquisition and its application to the study of the thylakoid membrane of a cyanobacterium Anabaena PCC7120. J. Microsc 2007, 228, 240–254. [Google Scholar]
- Brakenhoff, G.J.; Squier, J.; Norris, T.; Bliton, A.C.; Wade, M.H.; Athey, B. Real-time two-photon confocal microscopy using a femtosecond, amplified Ti:sapphire system. J. Microsc 1996, 181, 253–259. [Google Scholar]
- Oron, D.; Tal, E.; Silberberg, Y. Scanningless depth-resolved microscopy. Opt. Express 2005, 13, 1468–1476. [Google Scholar]
- Tal, E.; Oron, D.; Silberberg, Y. Improved depth resolution in video-rate line-scanning multiphoton microscopy using temporal focusing. Opt. Lett 2005, 30, 1686–1688. [Google Scholar]
- Papagiakoumou, E.; Anselmi, F.; Begue, A.; de Sars, V.; Gluckstad, J.; Isacoff, E.Y.; Emiliani, V. Scanless two-photon excitation of channelrhodopsin-2. Nat. Methods 2010, 7, 848–854. [Google Scholar]
- Wolleschensky, R.; Feurer, T.; Sauerbrey, R.; Simon, U. Characterization and optimization of a laser-scanning microscope in the femtosecond regime. Appl. Phys. B 1998, 67, 87–94. [Google Scholar]
- Planchon, T.A.; Gao, L.; Milkie, D.E.; Davidson, M.W.; Galbraith, J.A.; Galbraith, C.G.; Betzig, E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 2011, 8, 417–423. [Google Scholar]
- Cole, R.W.; Jinadasa, T.; Brown, C.M. Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nat. Protocols 2011, 6, 1929–1941. [Google Scholar]
- Shaevitz, J.W.; Fletcher, D.A. Enhanced three-dimensional deconvolution microscopy using a measured depth-varying point-spread function. J. Opt. Soc. Am. A 2007, 24, 2622–2627. [Google Scholar]
- Sheppard, C.J.R. Image formation in three-photon fluorescence microscopy. Bioimaging 1996, 4, 124–128. [Google Scholar]
- Wachsmann-Hogiu, S.; Hwang, J.Y.; Lindsley, E.; Farkas, D.L. Wide-field two-photon microscopy: Features and advantages for biomedical applications. Proc. SPIE 2007, 6441, 1–8. [Google Scholar]
- Raicu, V. FRET-based Determination of Protein Complex Structure at Nanometer Length Scale in Living Cells. In Nanoscopy and Multidimensional Optical Fluorescence Microscopy; Diaspro, A., Ed.; Taylor and Francis: Boca Raton, FL, USA, 2010. [Google Scholar]
- Barzda, V.; Gulbinas, V.; Kananavicius, R.; Cervinskas, V.; van Amerongen, H.; van Grondelle, R.; Valkunas, L. Singlet singlet annihilation kinetics in aggregates and trimers of LHCII. Biophys. J 2001, 80, 2409–2421. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Δx(pixels) | ||
|---|---|---|
| 150 | 1.0 | 96.0 |
| 150 | 103.0 | 1.0 |
| 300 | 1.0 | 186 |
| 300 | 192.0 | 1.0 |
| 512 | 1.0 | 280 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Biener, G.; Stoneman, M.R.; Acbas, G.; Holz, J.D.; Orlova, M.; Komarova, L.; Kuchin, S.; Raicu, V. Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation. Int. J. Mol. Sci. 2014, 15, 261-276. https://doi.org/10.3390/ijms15010261
Biener G, Stoneman MR, Acbas G, Holz JD, Orlova M, Komarova L, Kuchin S, Raicu V. Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation. International Journal of Molecular Sciences. 2014; 15(1):261-276. https://doi.org/10.3390/ijms15010261
Chicago/Turabian StyleBiener, Gabriel, Michael R. Stoneman, Gheorghe Acbas, Jessica D. Holz, Marianna Orlova, Liudmila Komarova, Sergei Kuchin, and Valerică Raicu. 2014. "Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation" International Journal of Molecular Sciences 15, no. 1: 261-276. https://doi.org/10.3390/ijms15010261