EAAC1 Gene Deletion Increases Neuronal Death and Blood Brain Barrier Disruption after Transient Cerebral Ischemia in Female Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
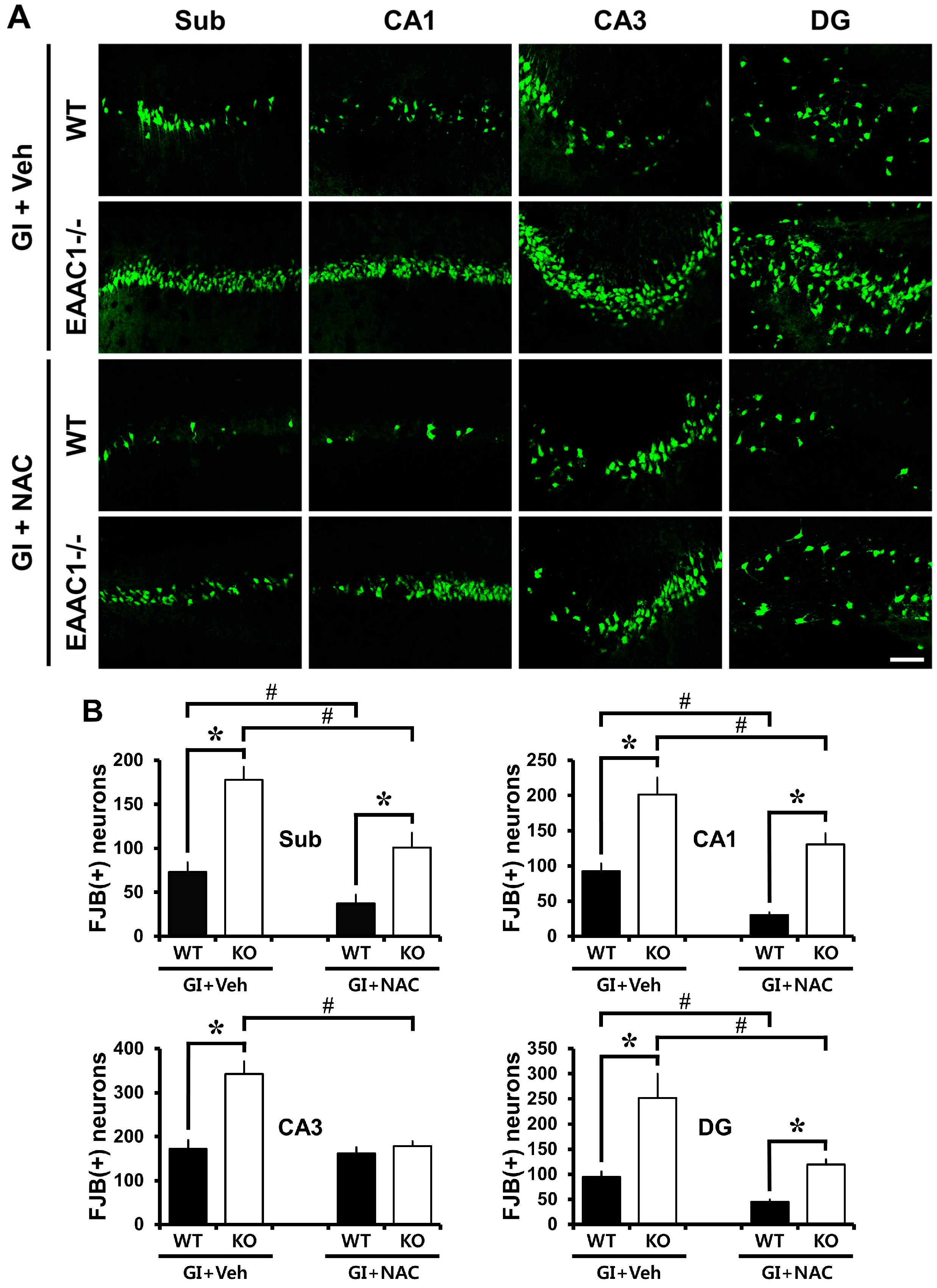
2.1. Ischemia-Induced Neuronal Death Is Increased in EAAC1−/− Female Mice

2.2. NAC Prevents Ischemia-Induced Neuronal Death in EAAC1−/− Female Mice
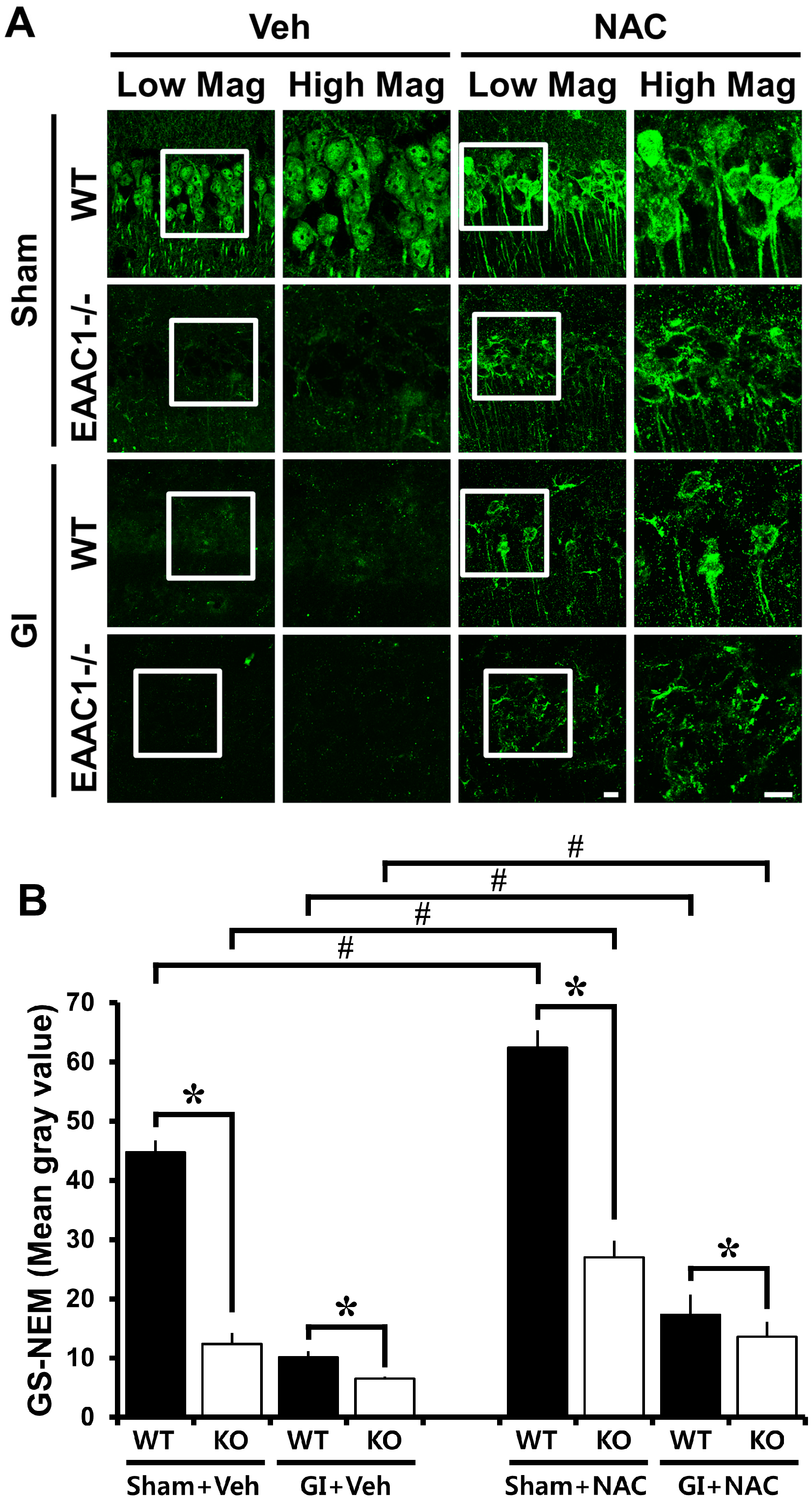
2.3. Ischemia-Induced Neuronal Glutathione (GSH) Loss Is Increased in EAAC1−/− Female Mice
2.4. NAC Up-Regulates GSH Content in EAAC1−/− Neurons

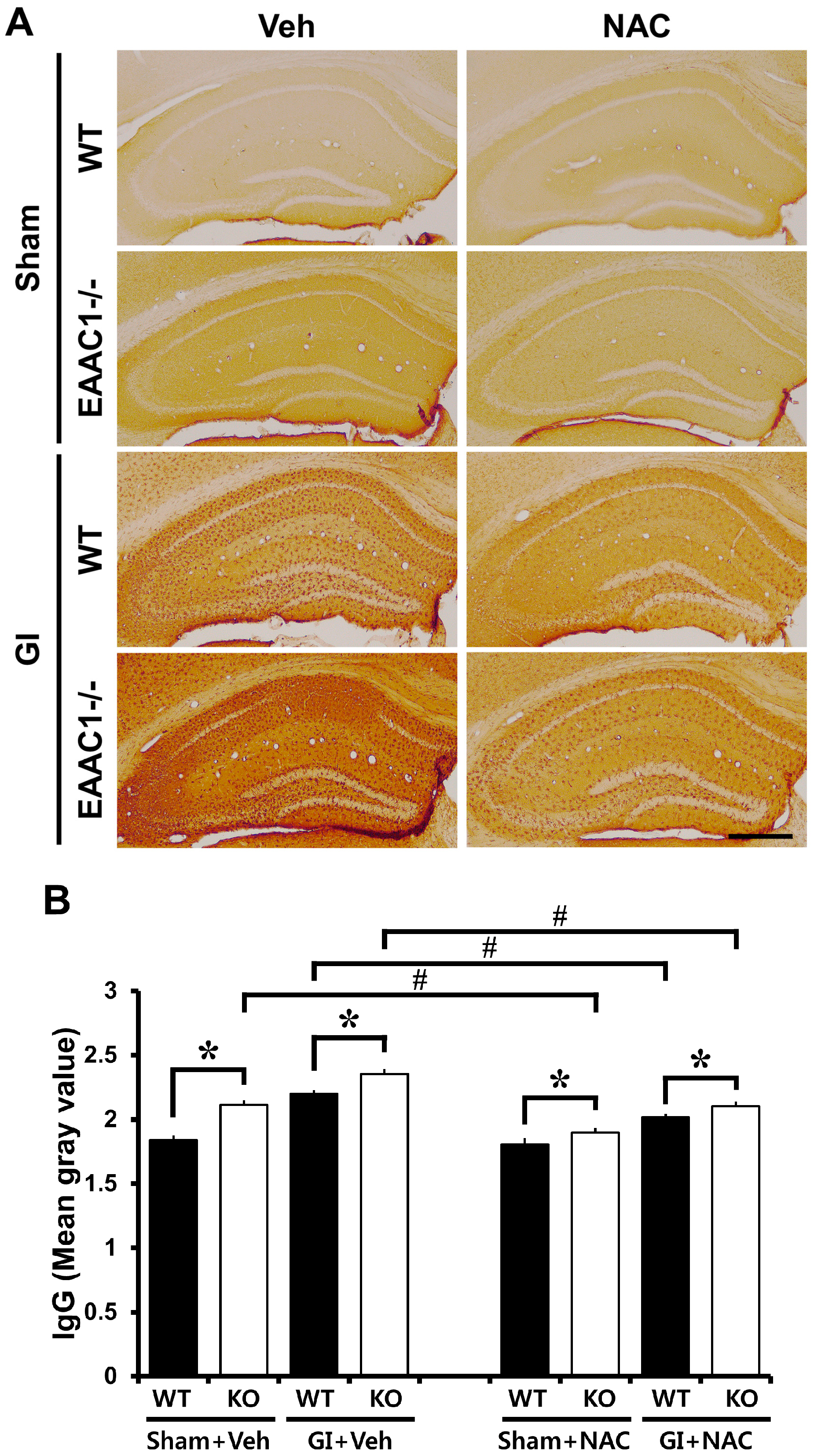
2.5. Ischemia-Induced Blood–Brain Barrier (BBB) Disruption Is Increased in EAAC1−/− Female Mice

2.6. NAC Prevents Ischemia-Induced Blood–Brain Barrier (BBB) Damage
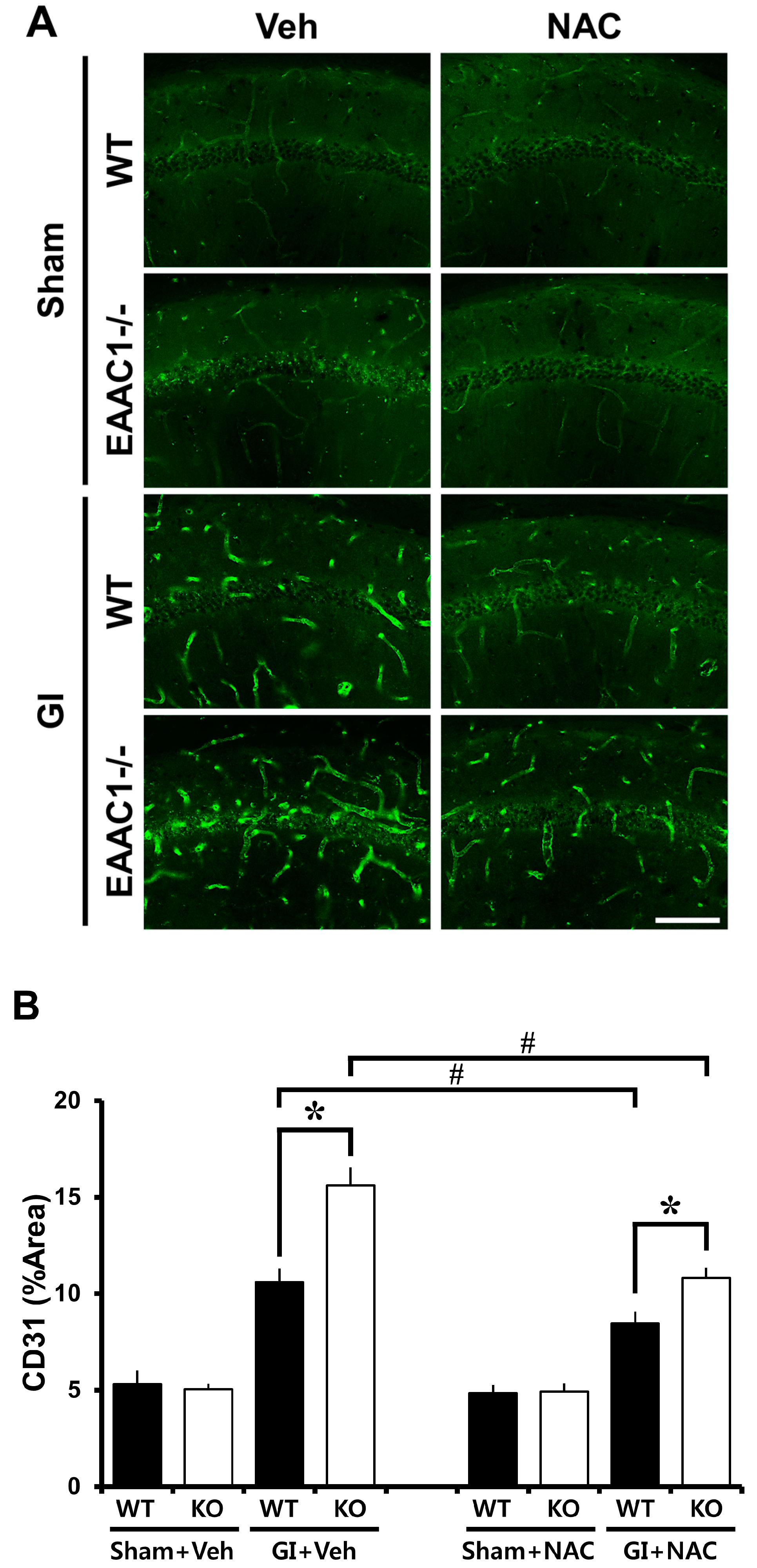
2.7. Ischemia-Induced Vessel Disorganization Is Increased in EAAC1−/− Female Mice

2.8. NAC Prevents Ischemia-Induced Vessel Disorganization in EAAC1−/− Female Mice
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Mouse Colony
4.3. Transient Ischemia
4.4. Neuron Death
4.5. Detection of Reduced GSH
4.6. Detection of Blood–Brain Barrier Disruption by IgG Immunostaining
4.7. Detection of Vessel Disorganization
4.8. N-Acetylcysteine (NAC) Administration
4.9. Confocal Microscopy
4.10. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tuttolomondo, A.; di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Inflammation in ischemic stroke subtypes. Curr. Pharm. Des. 2012, 18, 4289–4310. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm. Des. 2012, 18, 4266–4288. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; di Raimondo, D.; Forte, G.I.; Casuccio, A.; Vaccarino, L.; Scola, L.; Pecoraro, R.; Serio, A.; Clemente, G.; Arnao, V.; et al. Single nucleotide polymorphisms (SNPs) of pro-inflammatory/anti-inflammatory and thrombotic/fibrinolytic genes in patients with acute ischemic stroke in relation to TOAST subtype. Cytokine 2012, 58, 398–405. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, I.G.; Kristal, B.S. The powerhouse takes control of the cell: Is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic. Biol. Med. 2005, 38, 687–697. [Google Scholar]
- Nakanishi, N.; Tu, S.; Shin, Y.; Cui, J.; Kurokawa, T.; Zhang, D.; Chen, H.S.; Tong, G.; Lipton, S.A. Neuroprotection by the NR3A subunit of the NMDA receptor. J. Neurosci. 2009, 29, 5260–5265. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of l-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Watabe, M.; Aoyama, K.; Nakaki, T. A dominant role of GTRAP3–18 in neuronal glutathione synthesis. J. Neurosci. 2008, 28, 9404–9413. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [PubMed]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef]
- Jang, B.G.; Won, S.J.; Kim, J.H.; Choi, B.Y.; Lee, M.W.; Sohn, M.; Song, H.K.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and enhances cortical neuronal injury after transient cerebral ischemia in mice. J. Trace Elem. Med. Biol. 2012, 26, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zuo, Z. Glutamate transporter type 3 knockout reduces brain tolerance to focal brain ischemia in mice. J. Cereb. Blood Flow Metab. 2011, 31, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.; de Flora, S. N-Acetyl-l-cysteine. J. Cell. Biochem. Suppl. 1993, 17F, 270–277. [Google Scholar]
- Mazor, D.; Golan, E.; Philip, V.; Katz, M.; Jafe, A.; Ben-Zvi, Z.; Meyerstein, N. Red blood cell permeability to thiol compounds following oxidative stress. Eur. J. Haematol. 1996, 57, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef]
- Liblau, R.S.; Tisch, R.; Shokat, K.; Yang, X.; Dumont, N.; Goodnow, C.C.; McDevitt, H.O. Intravenous injection of soluble antigen induces thymic and peripheral T-cells apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 3031–3036. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E.; Feinerman, G.S. Foreign and endogenous serum protein extravasation during harmaline tremors or kainic acid seizures in the rat: A comparison. Acta Neuropathol. 1988, 76, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Choi, J.G.; Zuo, Z. Volatile anesthetics attenuate oxidative stress-reduced activity of glutamate transporter type 3. Anesth. Analg. 2009, 109, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Peghini, P.; Janzen, J.; Stoffel, W. Glutamate transporter EAAC1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997, 16, 3822–3832. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Simpson, E.M.; Takahashi, J.S.; Lipp, H.P.; Nakanishi, S.; Wehner, J.M.; Wolfer, D.P. Mutant mice and neuroscience: Recommendations concerning genetic background. Banbury Conference on genetic background in mice. Neuron 1997, 19, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Aoyama, K.; Chen, Y.; Garnier, P.; Matsumori, Y.; Gum, E.; Liu, J.; Swanson, R.A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003, 23, 10681–10690. [Google Scholar] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Yoo, J.H.; Song, H.K.; Sohn, M.; Won, S.J.; Suh, S.W. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS One 2013, 8, e81523. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.N.; Berman, A.E.; Swanson, R.A.; Yenari, M.A. Digitally quantifying cerebral hemorrhage using Photoshop and Image J. J. Neurosci. Methods 2010, 190, 240–243. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. EAAC1 Gene Deletion Increases Neuronal Death and Blood Brain Barrier Disruption after Transient Cerebral Ischemia in Female Mice. Int. J. Mol. Sci. 2014, 15, 19444-19457. https://doi.org/10.3390/ijms151119444
Choi BY, Kim JH, Kim HJ, Lee BE, Kim IY, Sohn M, Suh SW. EAAC1 Gene Deletion Increases Neuronal Death and Blood Brain Barrier Disruption after Transient Cerebral Ischemia in Female Mice. International Journal of Molecular Sciences. 2014; 15(11):19444-19457. https://doi.org/10.3390/ijms151119444
Chicago/Turabian StyleChoi, Bo Young, Jin Hee Kim, Hyun Jung Kim, Bo Eun Lee, In Yeol Kim, Min Sohn, and Sang Won Suh. 2014. "EAAC1 Gene Deletion Increases Neuronal Death and Blood Brain Barrier Disruption after Transient Cerebral Ischemia in Female Mice" International Journal of Molecular Sciences 15, no. 11: 19444-19457. https://doi.org/10.3390/ijms151119444