Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
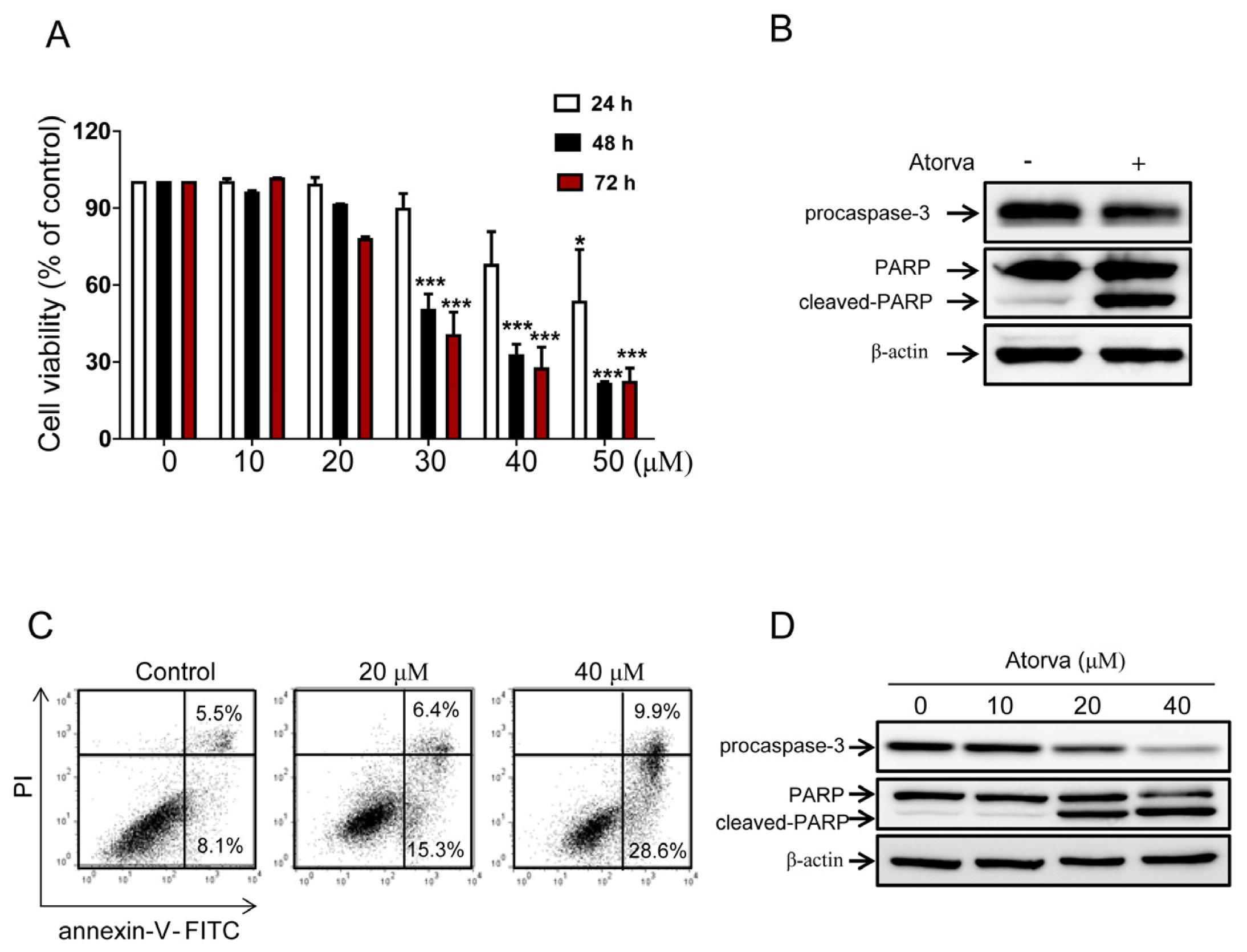
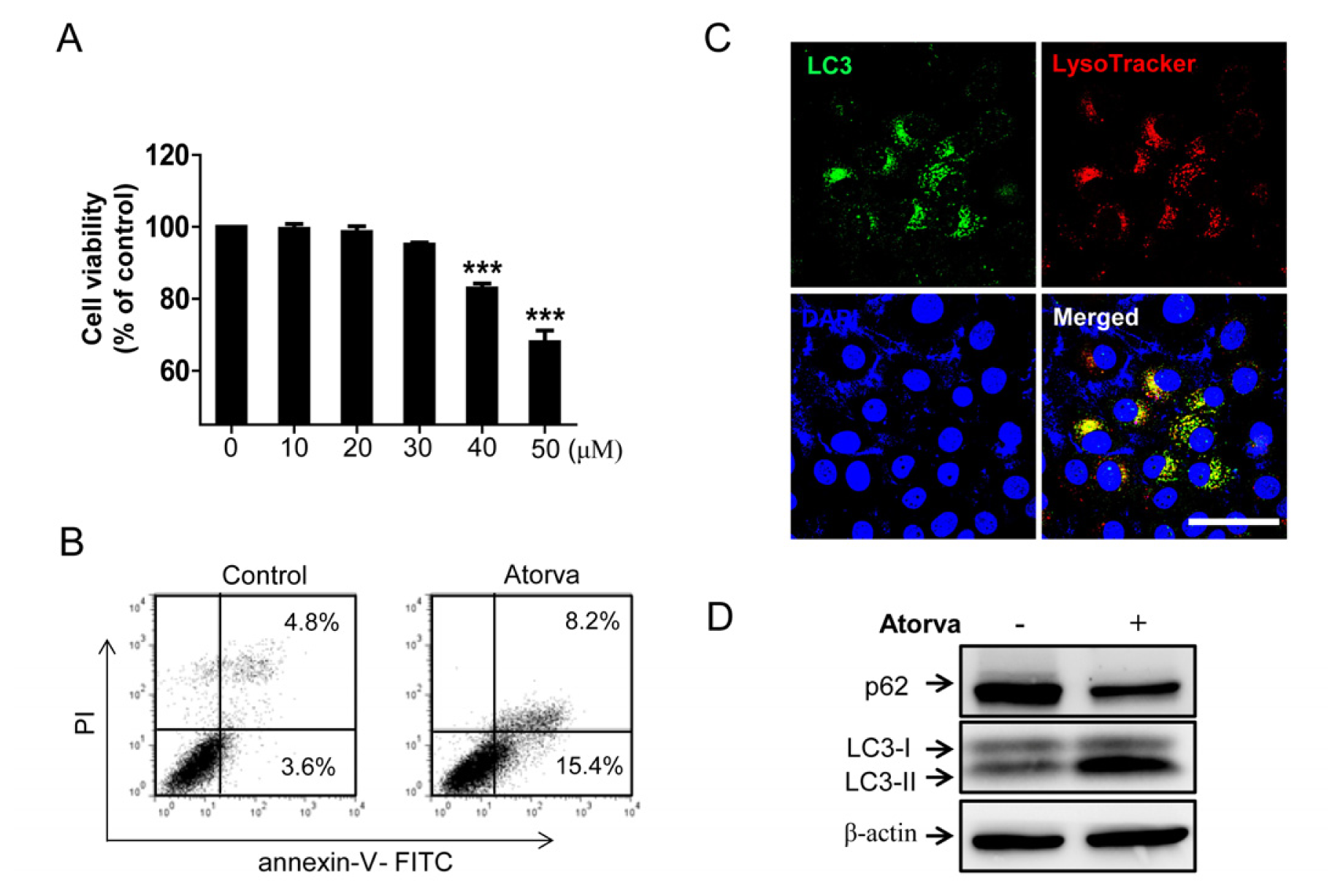
2.1.1. Cytotoxic Effects of Atorvastatin against T24 Human Bladder Cancer Cells
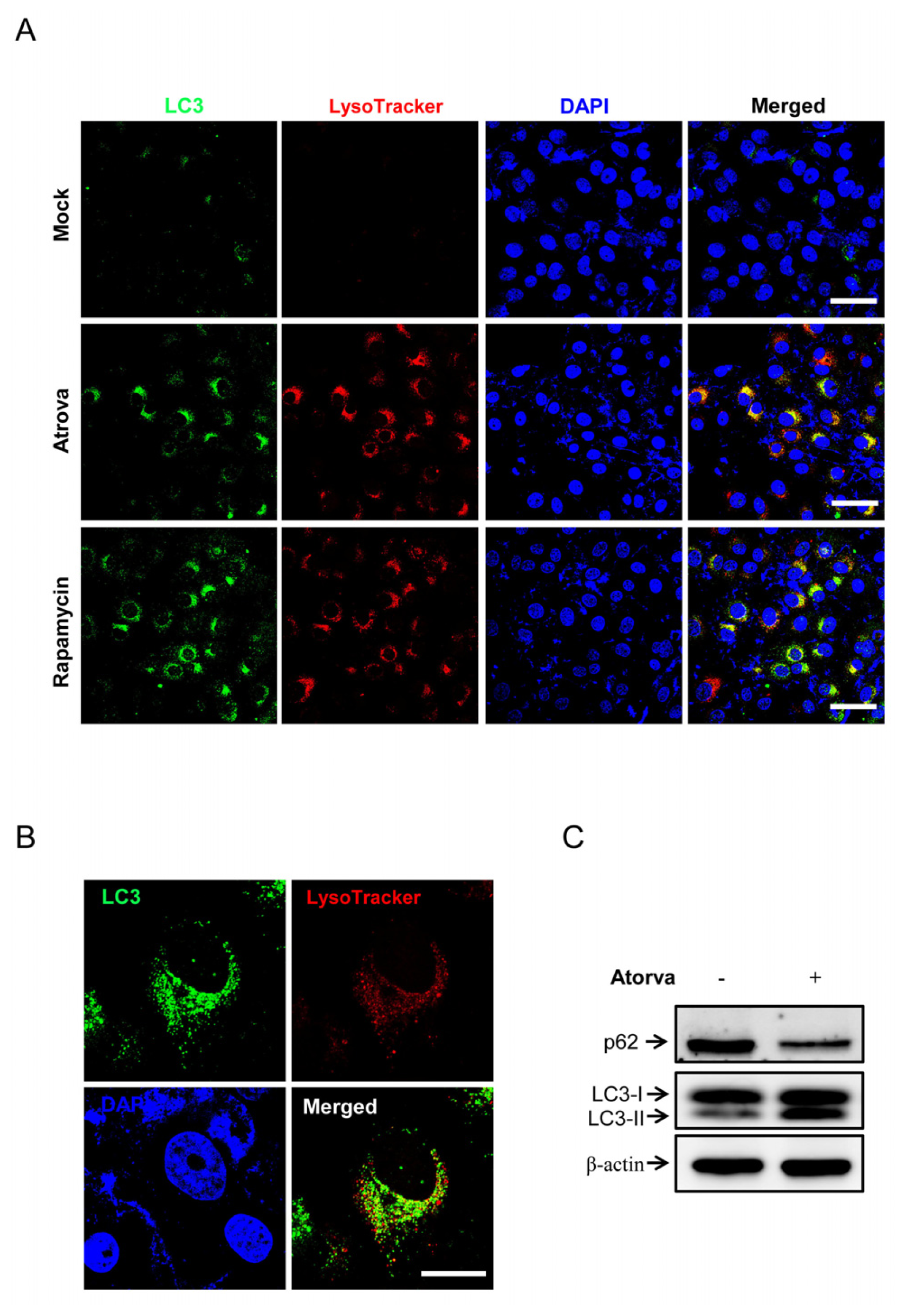
2.1.2. Autophagy Induction by Atorvastatin Treatment in T24 Human Bladder Cancer Cells
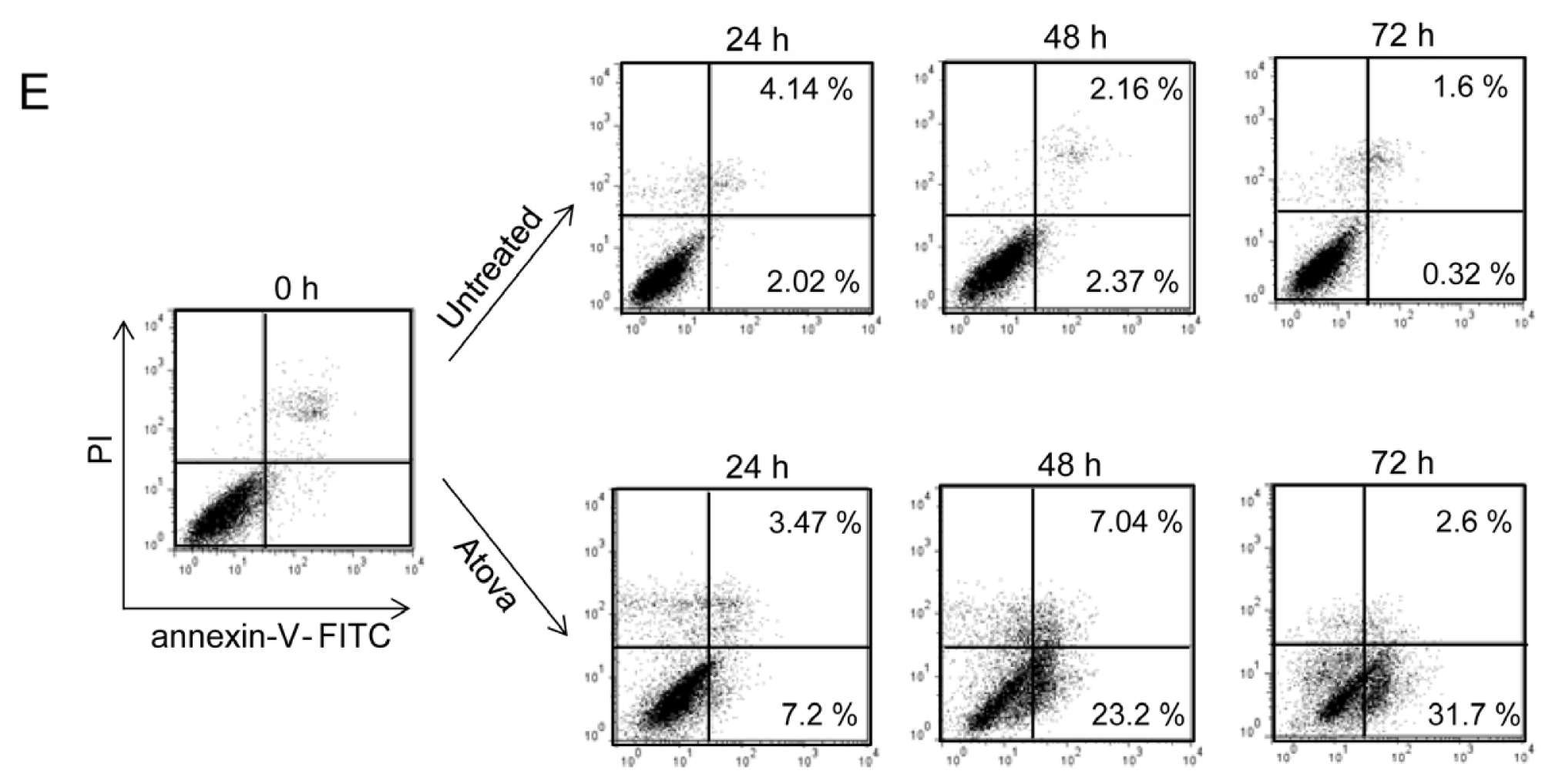
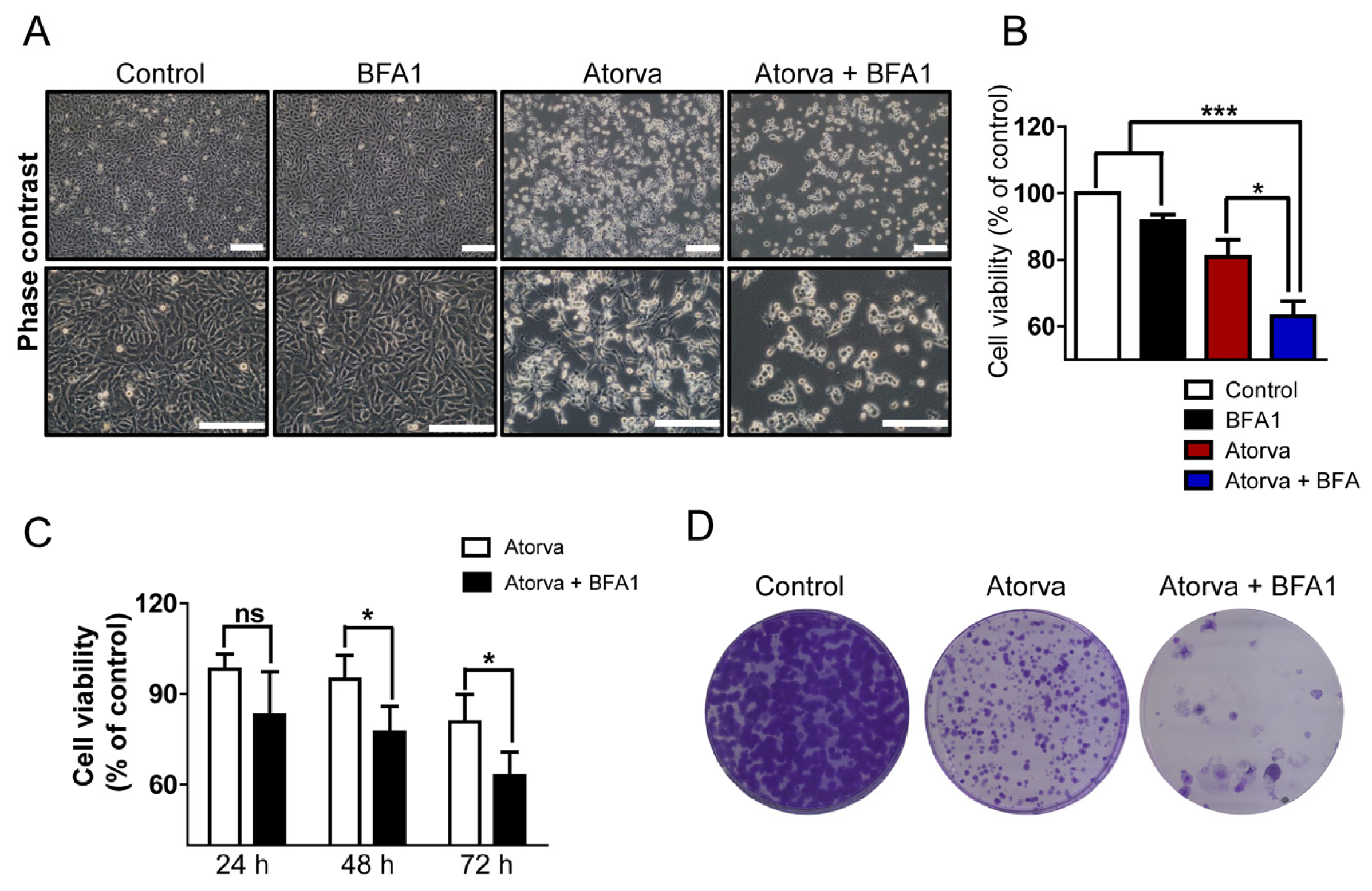
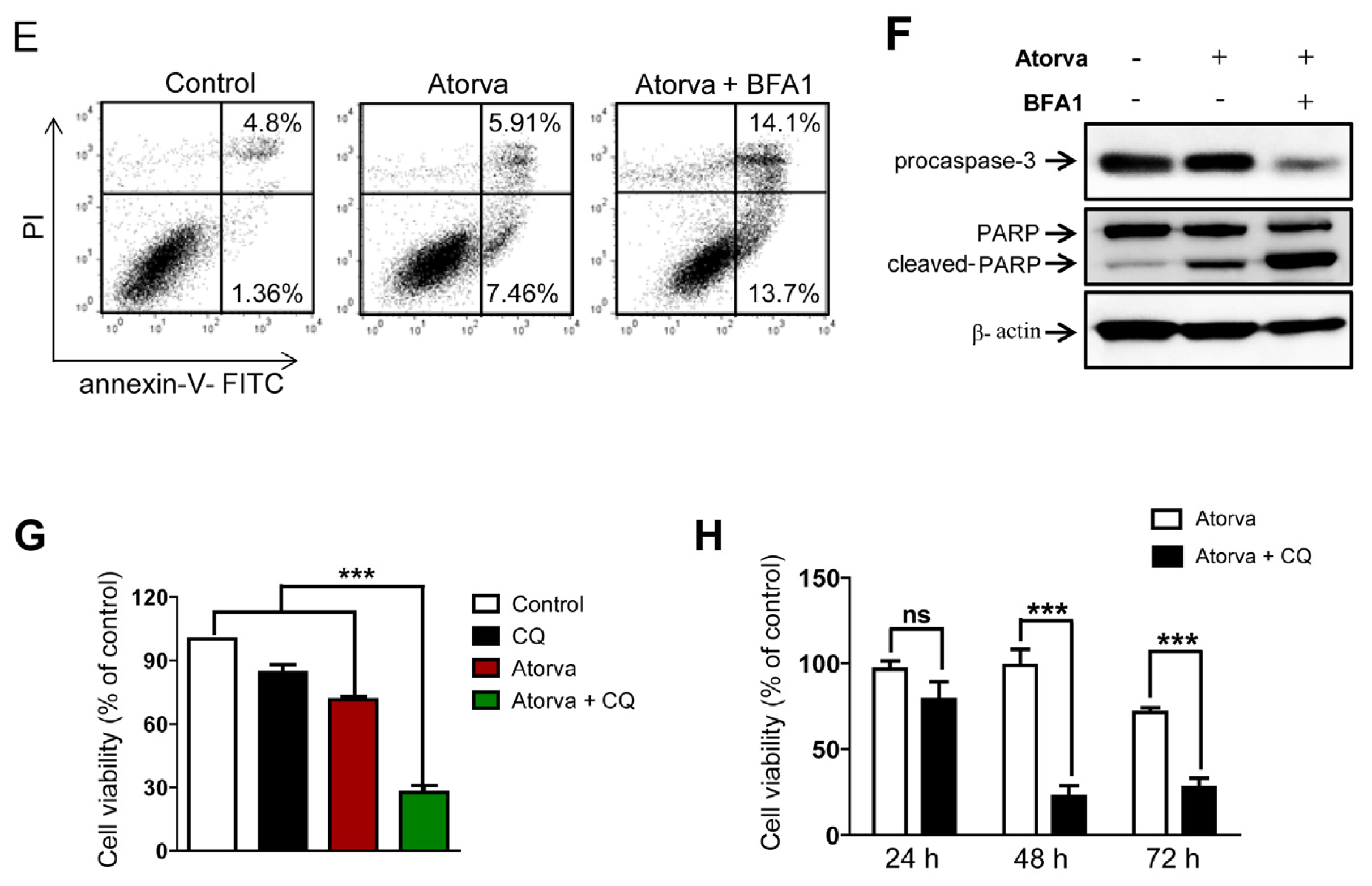
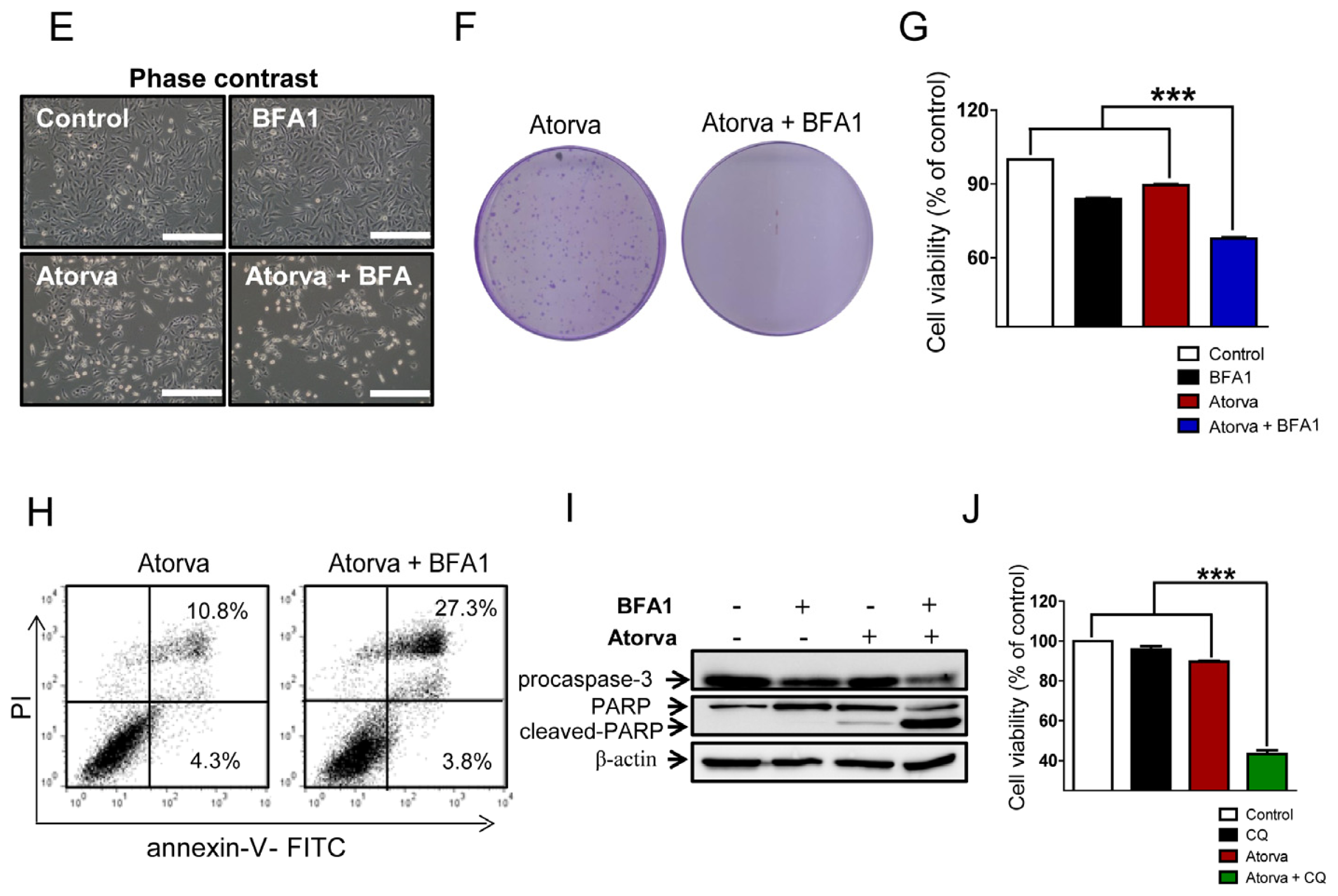
2.1.3. Autophagy Inhibition Enhances Atorvastatin-Induced Apoptotic Cell Death in T24 Bladder Cancer Cells
2.1.4. Inhibition of Autophagy Induced by Atorvastatin Improves Atorvastatin-Induced Apoptosis in J82 Bladder Cancer Cells
2.2. Discussion
3. Experimental Section
3.1. Cell Lines and Culture
3.2. Reagents
3.3. Cell Viability Assay
3.4. Immunocytochemistry
3.5. Western Blot Analysis
3.6. Flow Cytometric Analysis
3.7. Clonogenic (Colony Formation) Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-15-08106-s001.pdfConflicts of Interest
- Author ContributionsStudy conception and design: Minyong Kang, Chang Wook Jeong, Ja Hyeon Ku, and Cheol Kwak; Acquisition of data: Minyong Kang. Analysis and interpretation of data: Minyong Kang, Chang Wook Jeong, Ja Hyeon Ku, Cheol Kwak and Hyeon Hoe Kim; Drafting the manuscript: Minyong Kang; Critical revision: Minyong Kang and Hyeon Hoe Kim.
References
- Parkin, D.M. The global burden of urinary bladder cancer. Scandinavian journal of urology and nephrology. Scand. J. Urol. Nephrol 2008, 218, 12–20. [Google Scholar]
- Shariat, S.F.; Karam, J.A.; Lotan, Y.; Karakiewizc, P.I. Critical evaluation of urinary markers for bladder cancer detection and monitoring. Rev. Urol 2008, 10, 120–135. [Google Scholar]
- Van Rhijn, B.W.; Burger, M.; Lotan, Y.; Solsona, E.; Stief, C.G.; Sylvester, R.J.; Witjes, J.A.; Zlotta, A.R. Recurrence and progression of disease in non-muscle-invasive bladder cancer: from epidemiology to treatment strategy. Eur. Urol 2009, 56, 430–442. [Google Scholar]
- Bellmunt, J.; Orsola, A.; Wiegel, T.; Guix, M.; de Santis, M.; Kataja, V. Bladder cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol 2011, 22, 45–49. [Google Scholar]
- Stenzl, A.; Cowan, N.C.; de Santis, M.; Kuczyk, M.A.; Merseburger, A.S.; Ribal, M.J.; Sherif, A.; Witjes, J.A. Treatment of muscle-invasive and metastatic bladder cancer: Update of the EAU guidelines. Eur. Urol 2011, 59, 1009–1018. [Google Scholar]
- Auer, J.; Berent, R.; Weber, T.; Eber, B. Clinical significance of pleiotropic effects of statins: Lipid reduction and beyond. Curr. Med. Chem 2002, 9, 1831–1850. [Google Scholar]
- Osmak, M. Statins and cancer: Current and future prospects. Cancer Lett 2012, 324, 1–12. [Google Scholar]
- Karp, I.; Behlouli, H.; Lelorier, J.; Pilote, L. Statins and cancer risk. Am. J. Med 2008, 121, 302–309. [Google Scholar]
- Vinogradova, Y.; Coupland, C.; Hippisley-Cox, J. Exposure to statins and risk of common cancers: A series of nested case-control studies. BMC Cancer 2011, 11. [Google Scholar] [CrossRef]
- Hoffmann, P.; Roumeguere, T.; Schulman, C.; van Velthoven, R. Use of statins and outcome of BCG treatment for bladder cancer. N. Engl. J. Med 2006, 355, 2705–2707. [Google Scholar]
- Crivelli, J.J.; Xylinas, E.; Kluth, L.A.; da Silva, R.D.; Chrystal, J.; Novara, G.; Karakiewicz, P.I.; David, S.G.; Scherr, D.S.; Lotan, Y.; et al. Effect of statin use on outcomes of non-muscle-invasive bladder cancer. BJU Int 2013, 112, E4–E12. [Google Scholar]
- Da Silva, R.D.; Xylinas, E.; Kluth, L.; Crivelli, J.J.; Chrystal, J.; Chade, D.; Guglielmetti, G.B.; Pycha, A.; Lotan, Y.; Karakiewicz, P.I.; et al. Impact of statin use on oncologic outcomes in patients with urothelial carcinoma of the bladder treated with radical cystectomy. J. Urol 2013, 190, 487–492. [Google Scholar]
- Kamat, A.M.; Nelkin, G.M. Atorvastatin: A potential chemopreventive agent in bladder cancer. Urology 2005, 66, 1209–1212. [Google Scholar]
- Bil, J.; Zapala, L.; Nowis, D.; Jakobisiak, M.; Golab, J. Statins potentiate cytostatic/cytotoxic activity of sorafenib but not sunitinib against tumor cell lines in vitro. Cancer Lett. 2010, 288, 57–67. [Google Scholar]
- Hindler, K.; Cleeland, C.S.; Rivera, E.; Collard, C.D. The role of statins in cancer therapy. Oncologist 2006, 11, 306–315. [Google Scholar]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med 2013, 368, 1845–1846. [Google Scholar]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res 2006, 66, 9349–9351. [Google Scholar]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res 2009, 15, 5308–5316. [Google Scholar]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011, 25, 460–470. [Google Scholar]
- Zhang, J.; Yang, Z.; Xie, L.; Xu, L.; Xu, D.; Liu, X. Statins, autophagy and cancer metastasis. Int. J. Biochem. Cell Biol 2013, 45, 745–752. [Google Scholar]
- Liu, B.; Wen, X.; Cheng, Y. Survival or death: Disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis 2013, 4. [Google Scholar] [CrossRef]
- Yang, Z.N.J.; Chee, C.E.; Huang, S.B.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther 2011, 10, 1533–1541. [Google Scholar]
- Wu, W.K.K.; Coffelt, S.B.; Cho, C.H.; Wang, X.J.; Lee, C.W.; Chan, F.K.L.; Yu, J.; Sung, J.J.Y. The autophagic paradox in cancer therapy. Oncogene 2012, 31, 939–953. [Google Scholar]
- Jones, R.G. The roles, mechanisms, and controversies of autophagy in mammalian biology. F1000 Biol. Rep 2009, 1. [Google Scholar] [CrossRef]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res 2013, 19, 2995–3007. [Google Scholar]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Yang, P.M.; Liu, Y.L.; Lin, Y.C.; Shun, C.T.; Wu, M.S.; Chen, C.C. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res 2010, 70, 7699–7709. [Google Scholar]
- Wu, Z.; Chang, P.C.; Yang, J.C.; Chu, C.Y.; Wang, L.Y.; Chen, N.T.; Ma, A.H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy blockade sensitizes prostate cancer cells towards src family kinase inhibitors. Genes Cancer 2010, 1, 40–49. [Google Scholar]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Wang, X.; Guo, H.J.; Zhang, B.; Zhang, X.B.; Shi, Z.J.; Yu, L. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy 2013, 9, 595–603. [Google Scholar]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem 2011, 286, 7290–7300. [Google Scholar]
- He, C.C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 2009, 43, 67–93. [Google Scholar]
- Lu, Z.; Luo, R.Z.; Lu, Y.L.; Zhang, X.H.; Yu, Q.H.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.H.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig 2008, 118, 3917–3929. [Google Scholar]







© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kang, M.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro. Int. J. Mol. Sci. 2014, 15, 8106-8121. https://doi.org/10.3390/ijms15058106
Kang M, Jeong CW, Ku JH, Kwak C, Kim HH. Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro. International Journal of Molecular Sciences. 2014; 15(5):8106-8121. https://doi.org/10.3390/ijms15058106
Chicago/Turabian StyleKang, Minyong, Chang Wook Jeong, Ja Hyeon Ku, Cheol Kwak, and Hyeon Hoe Kim. 2014. "Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro" International Journal of Molecular Sciences 15, no. 5: 8106-8121. https://doi.org/10.3390/ijms15058106