Eicosapentaenoic Acid Protects against Palmitic Acid-Induced Endothelial Dysfunction via Activation of the AMPK/eNOS Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
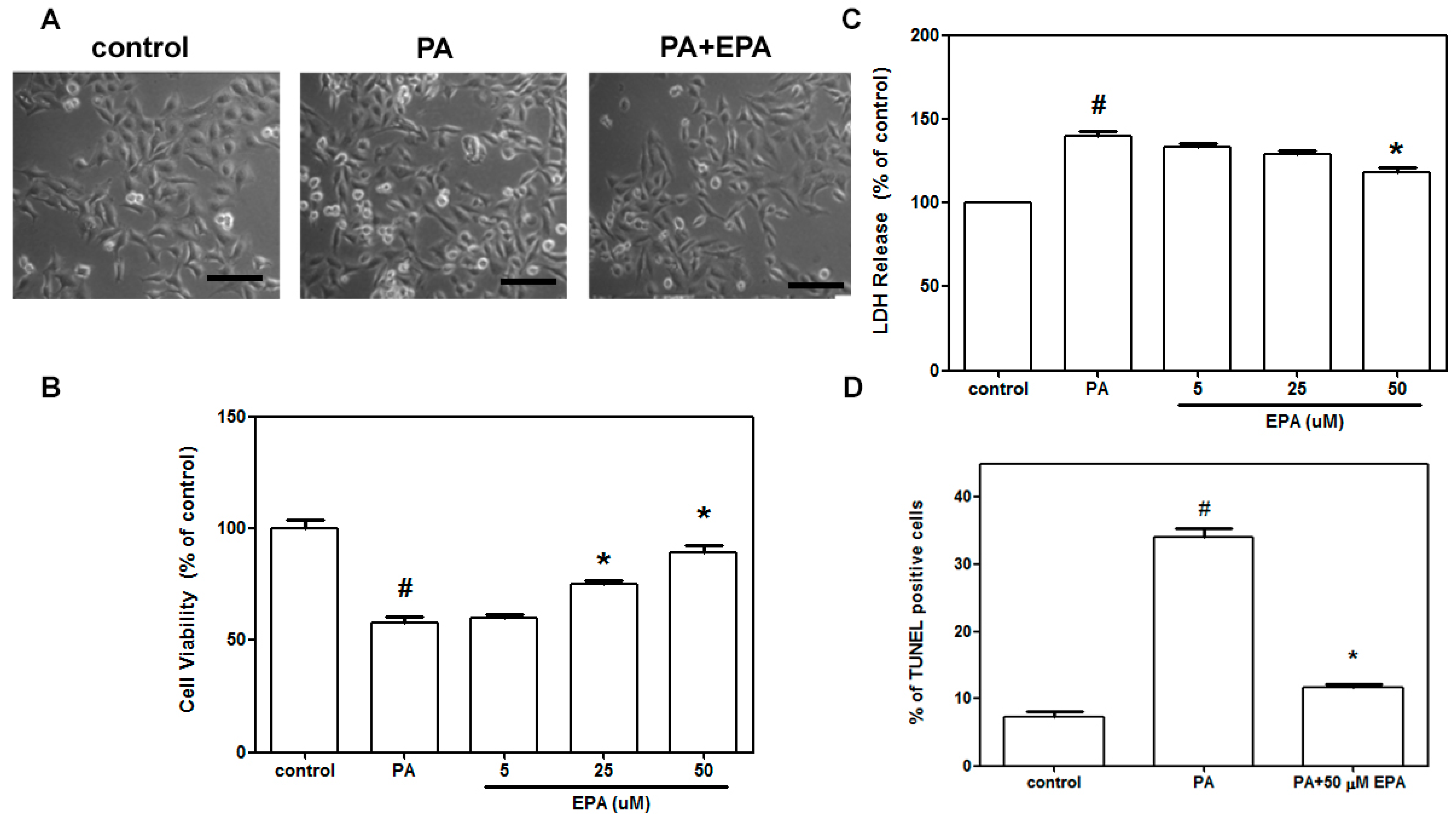
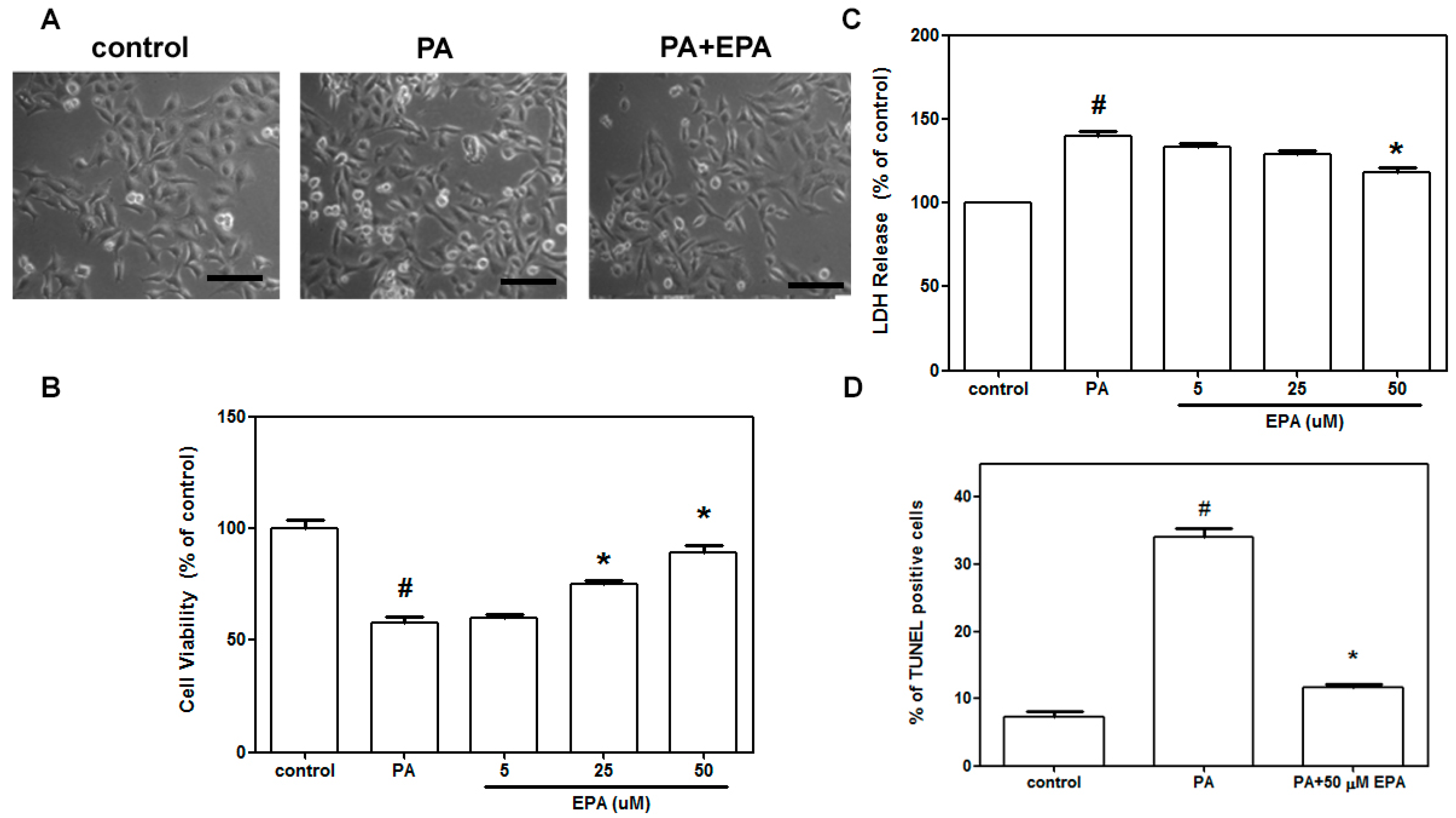
2.1. EPA Blocked Cell Death Induced by PA in HUVECs
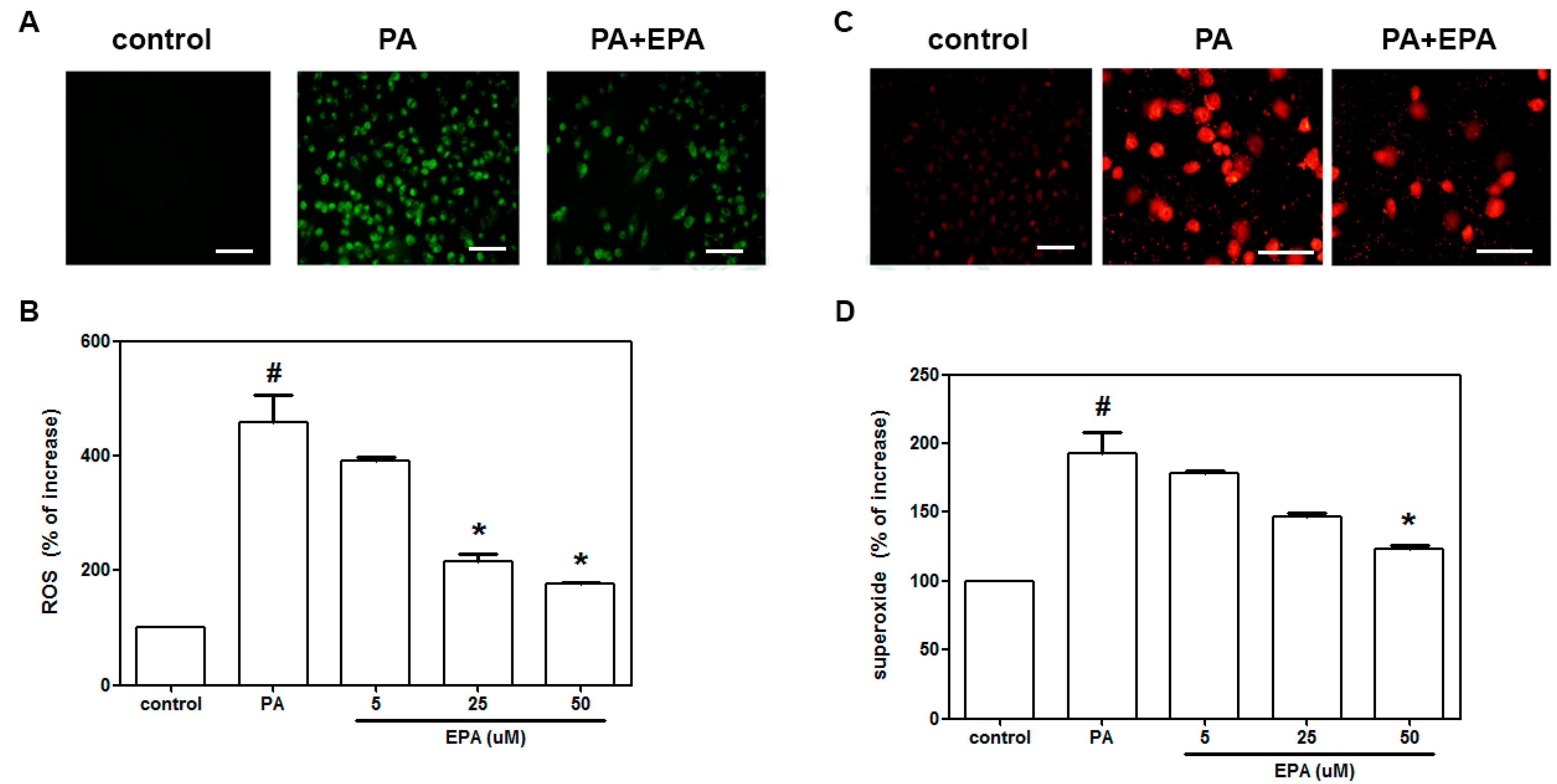
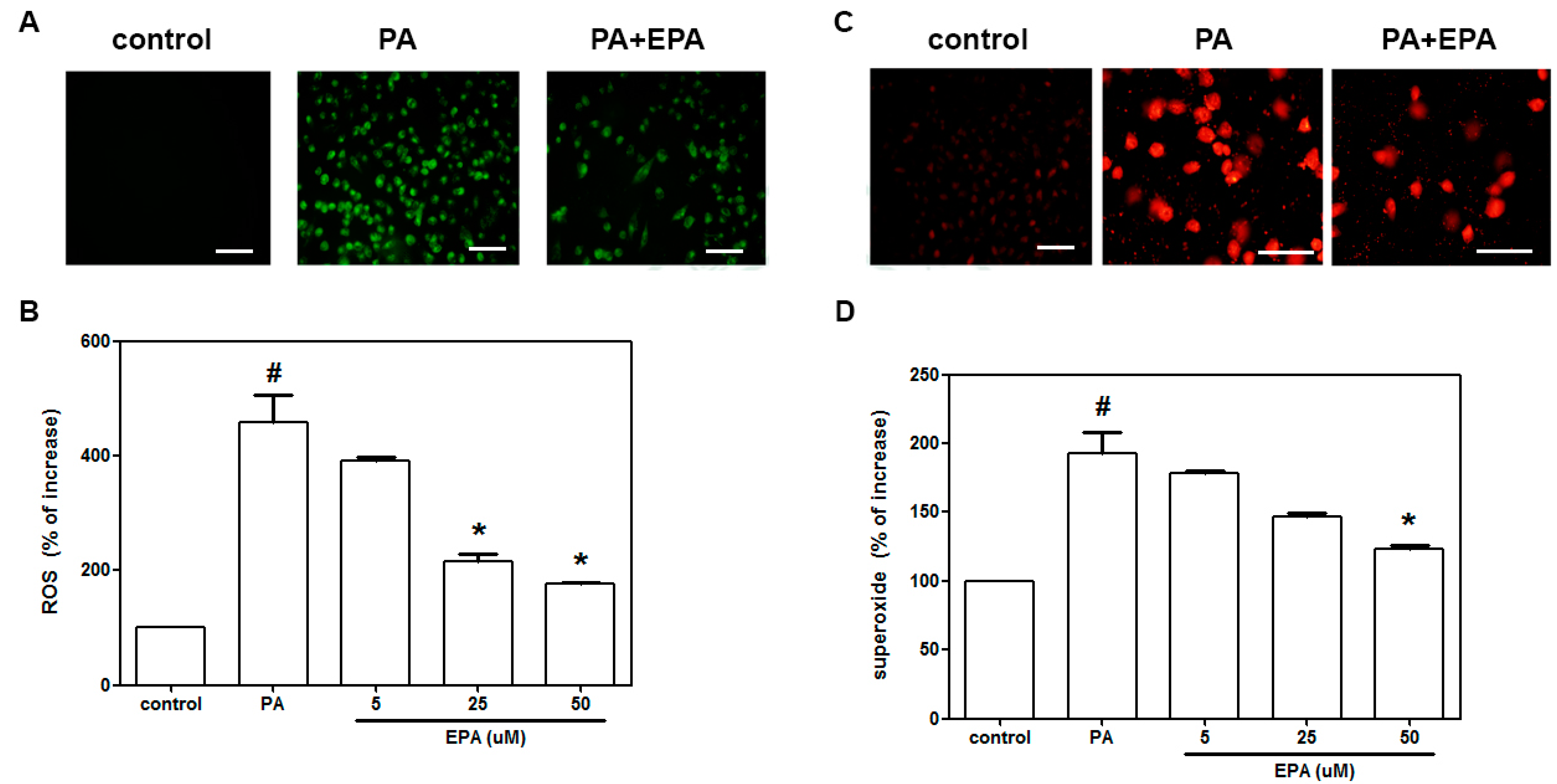
2.2. EPA Inhibited the PA-Induced Intracellular Superoxide Production and ROS Generation
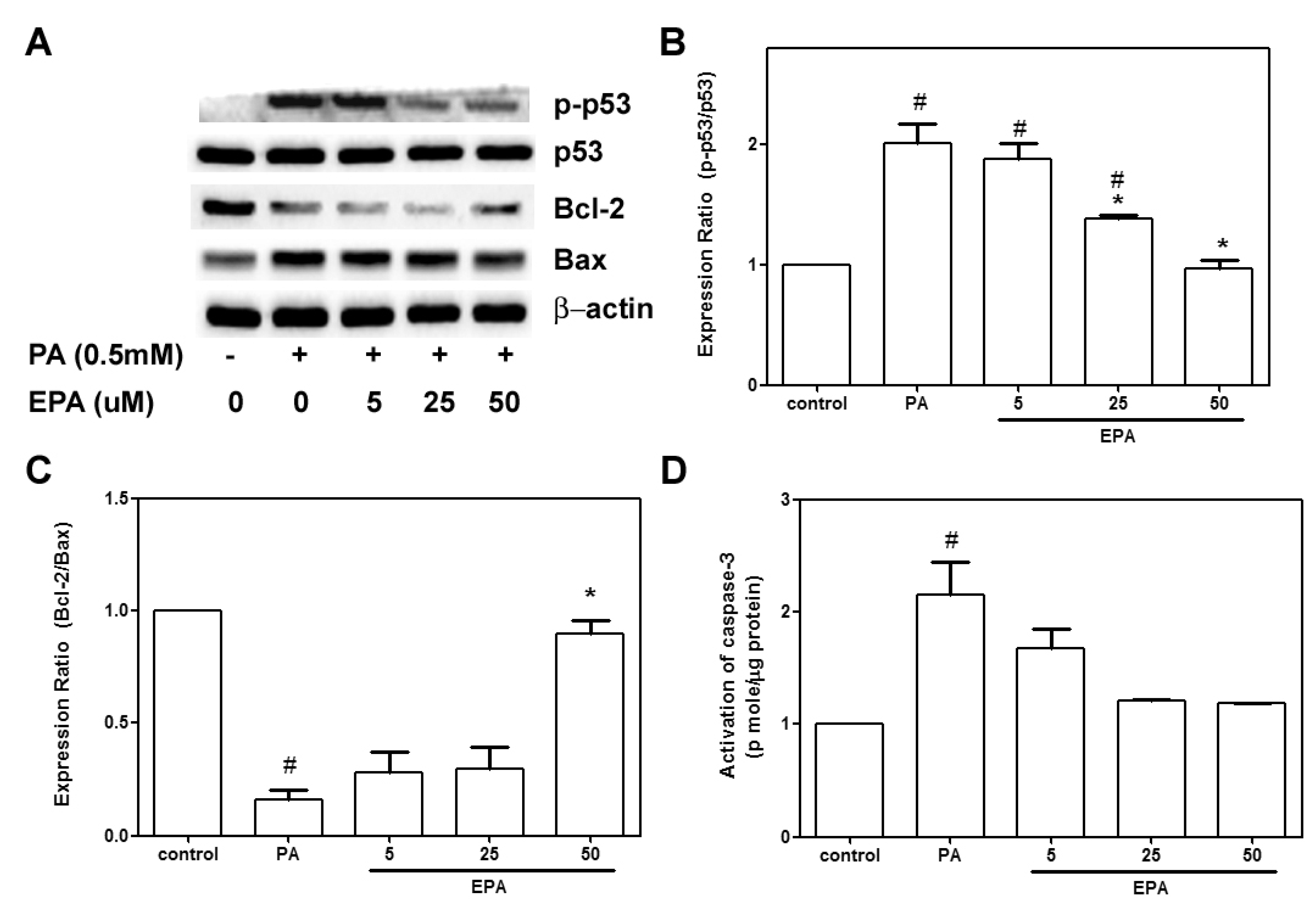
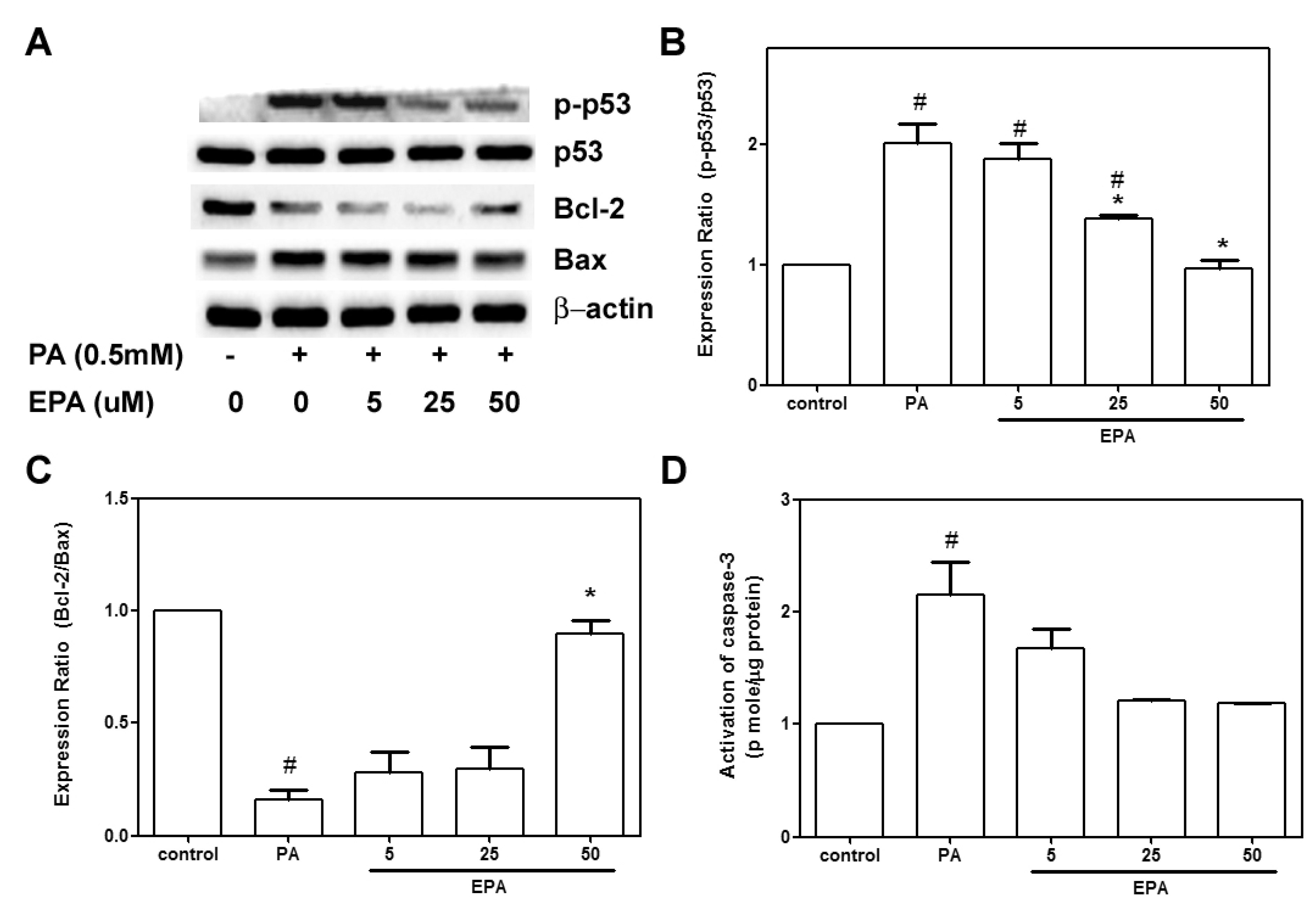
2.3. EPA Protected against the PA-Induced Apoptotic Response
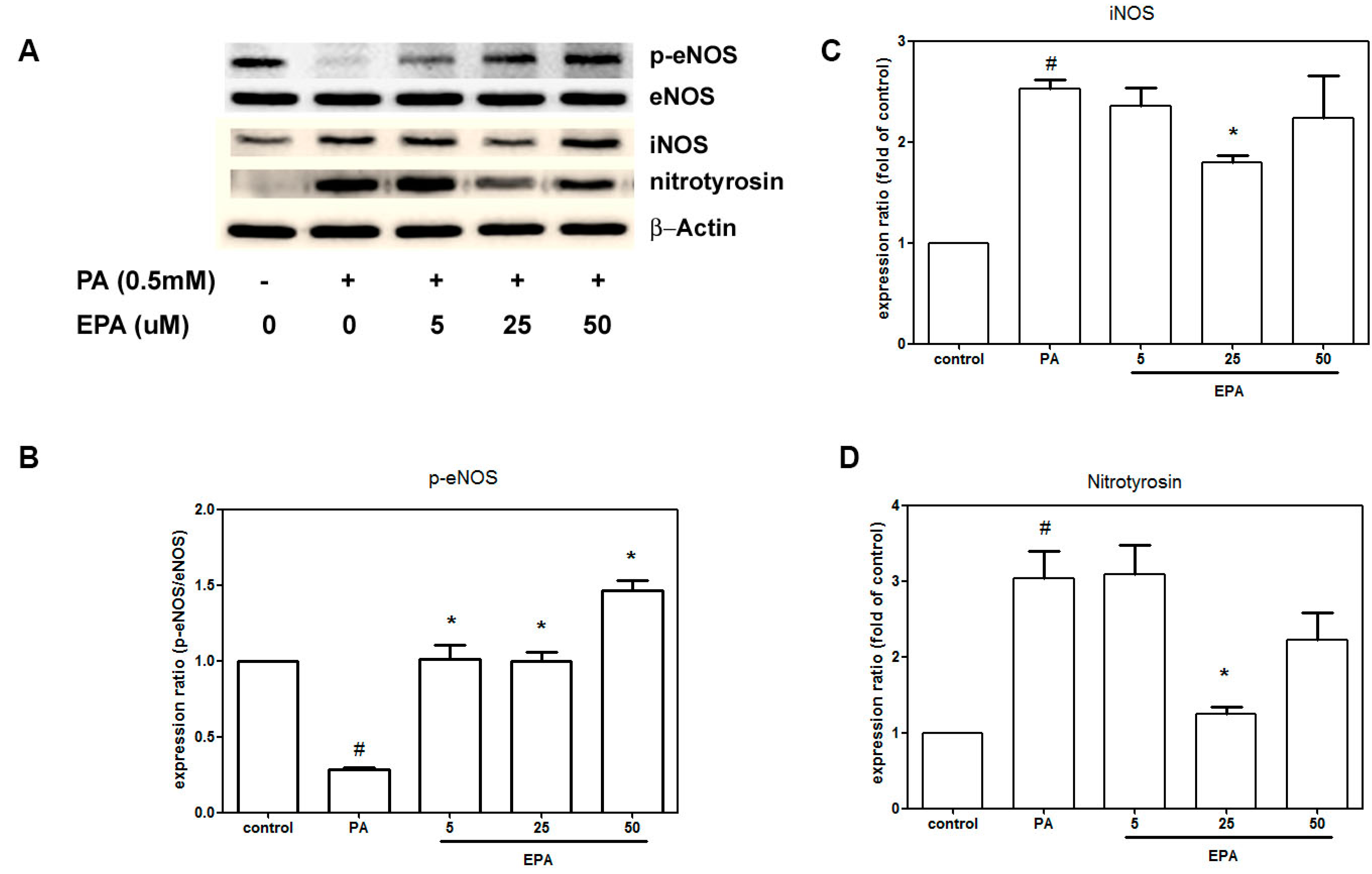
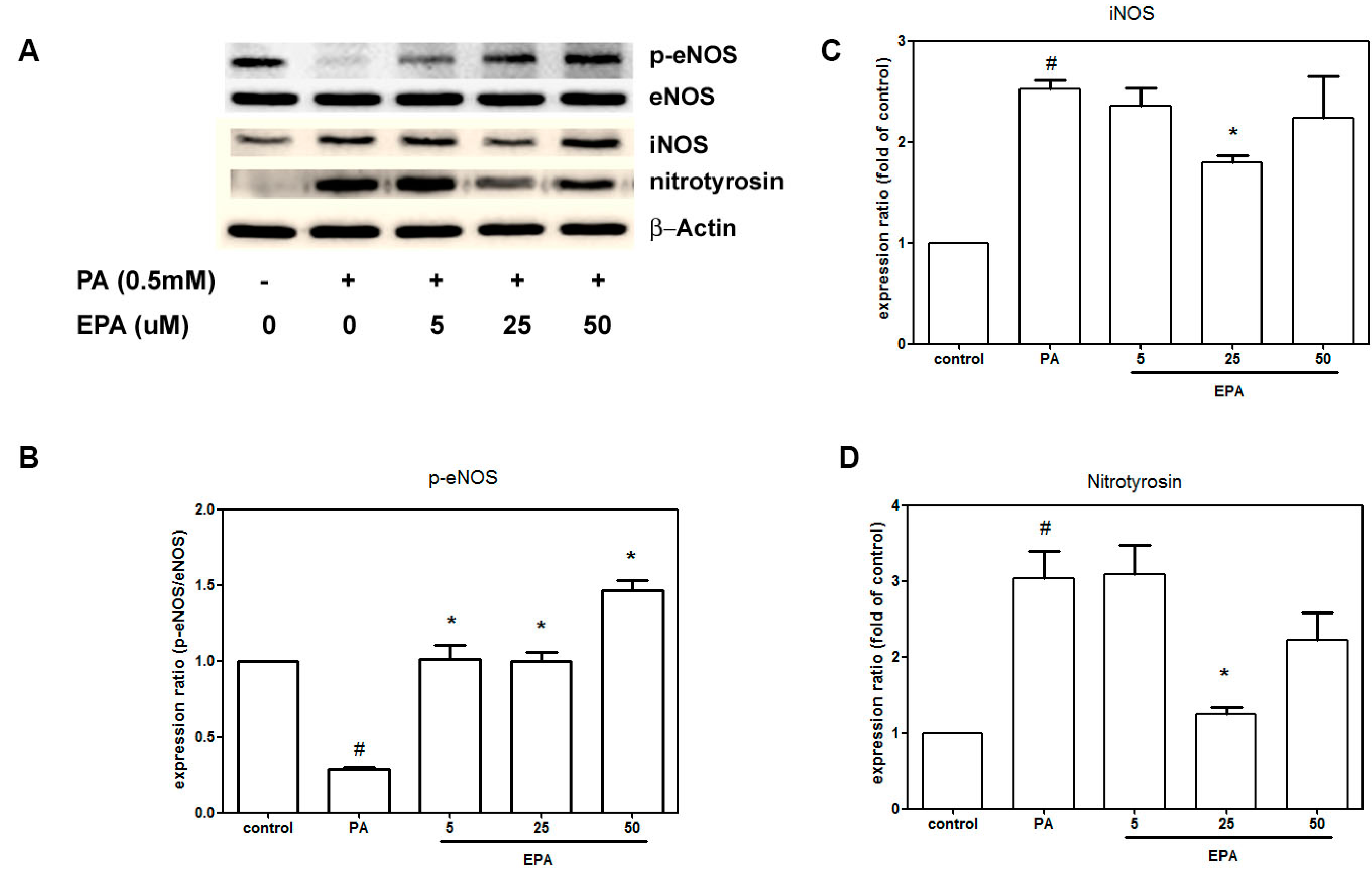
2.4. EPA Suppressed PA-Induced Downregulation of eNOS
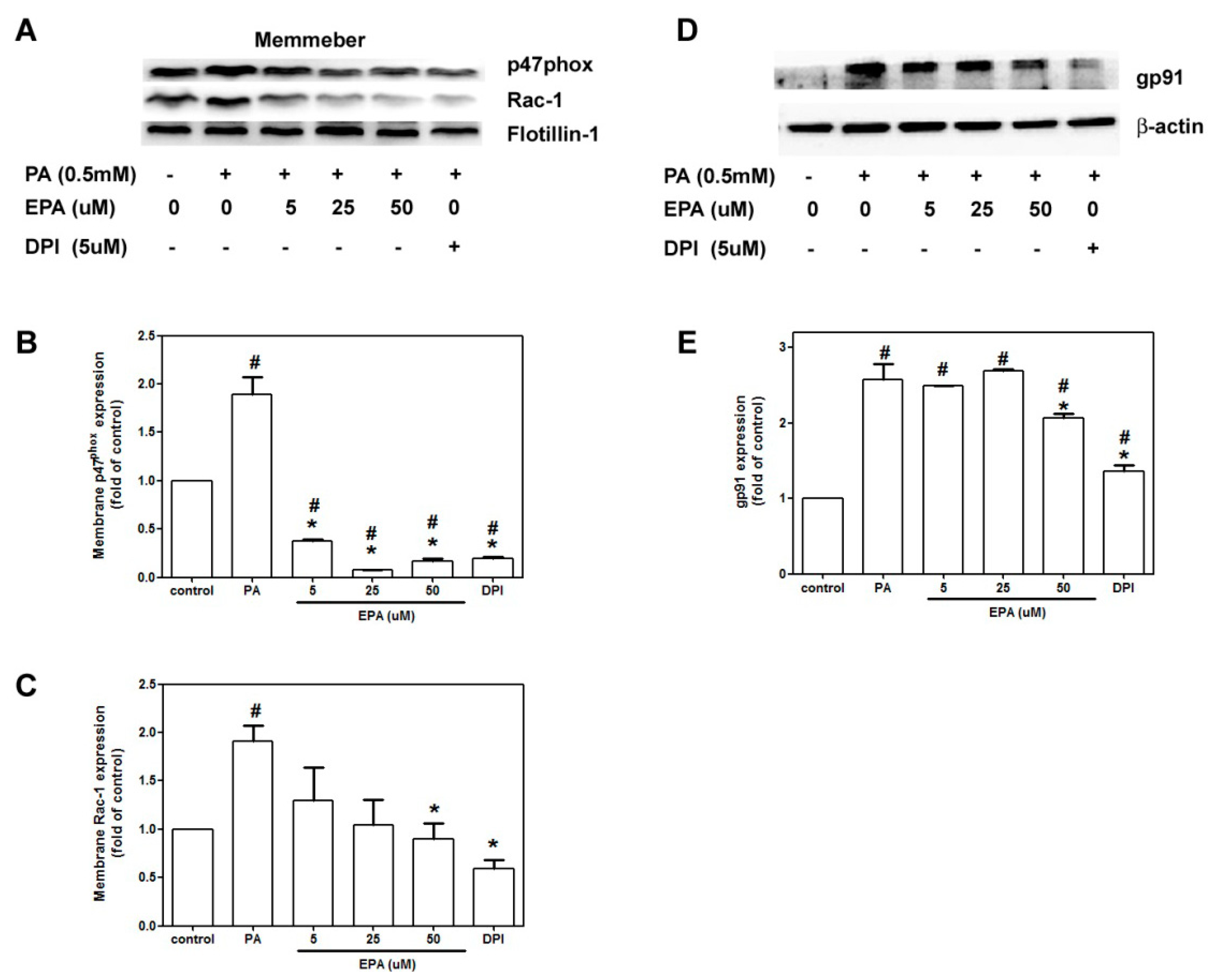
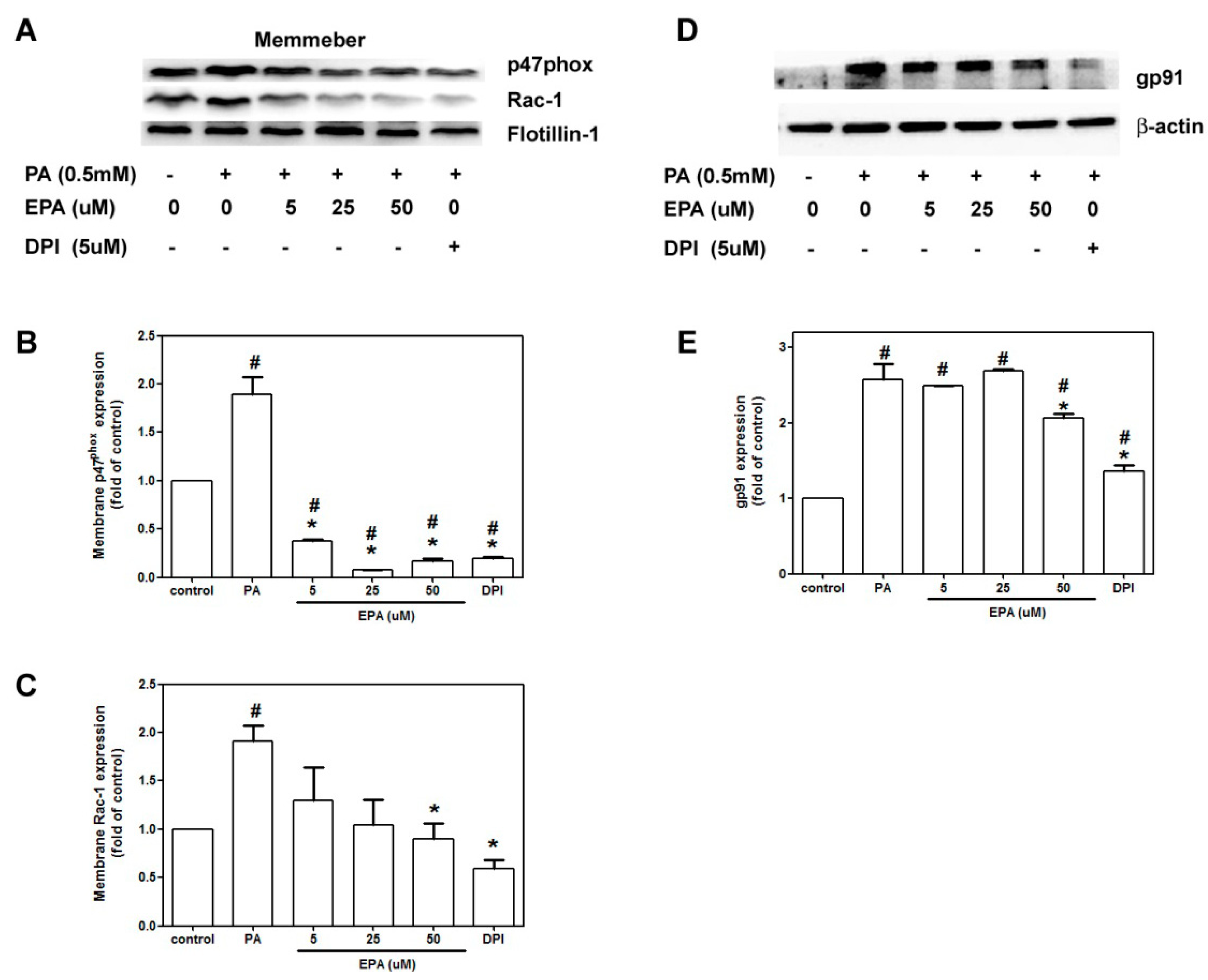
2.5. EPA Attenuated PA-Induced NADPH Oxidase Activation
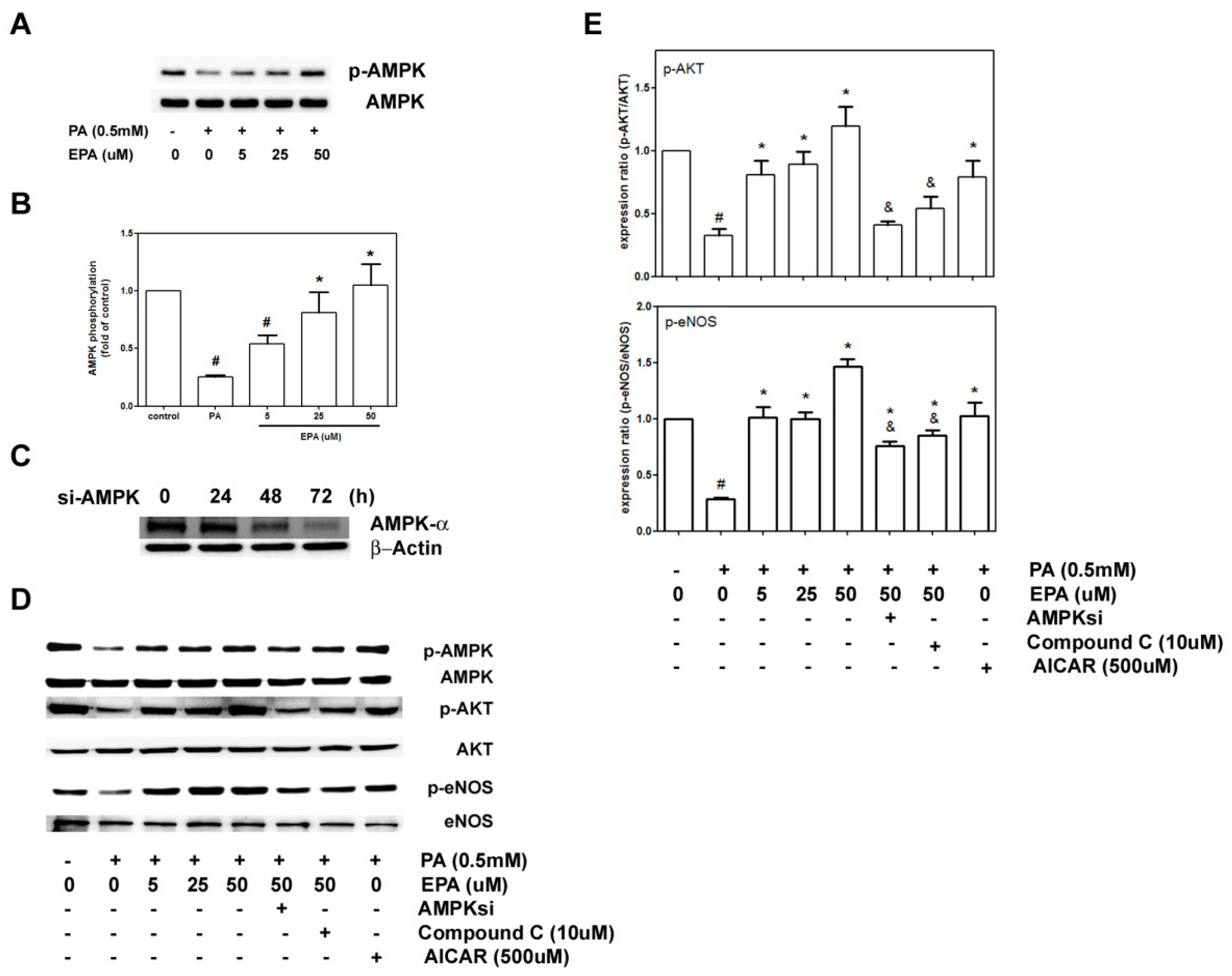
2.6. EPA Protected against PA-Induced Downregulation of eNOS and AKT via AMPK
2.7. EPA Suppressed PA-Induced Activation of p38 MAPK and NF-κB
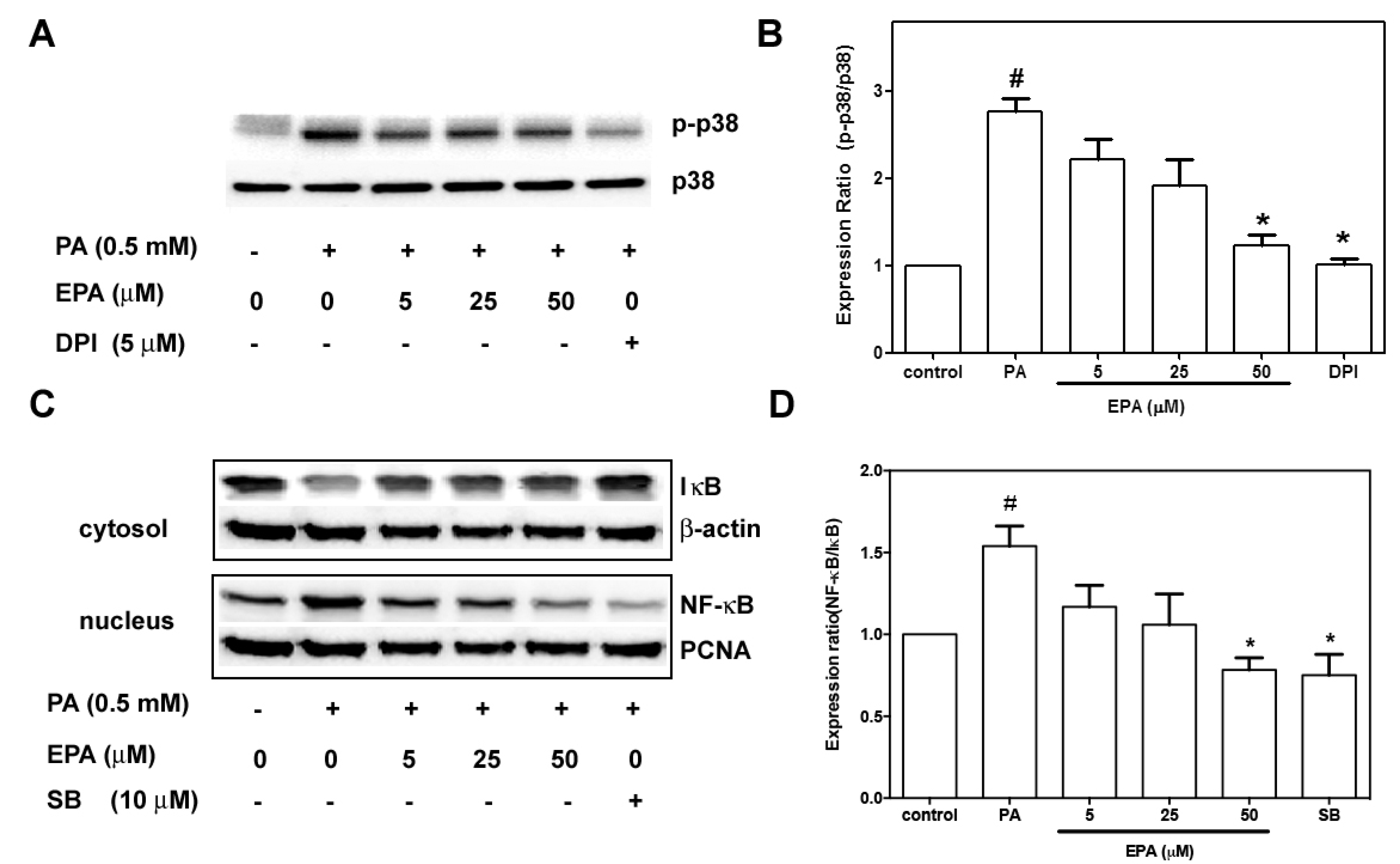
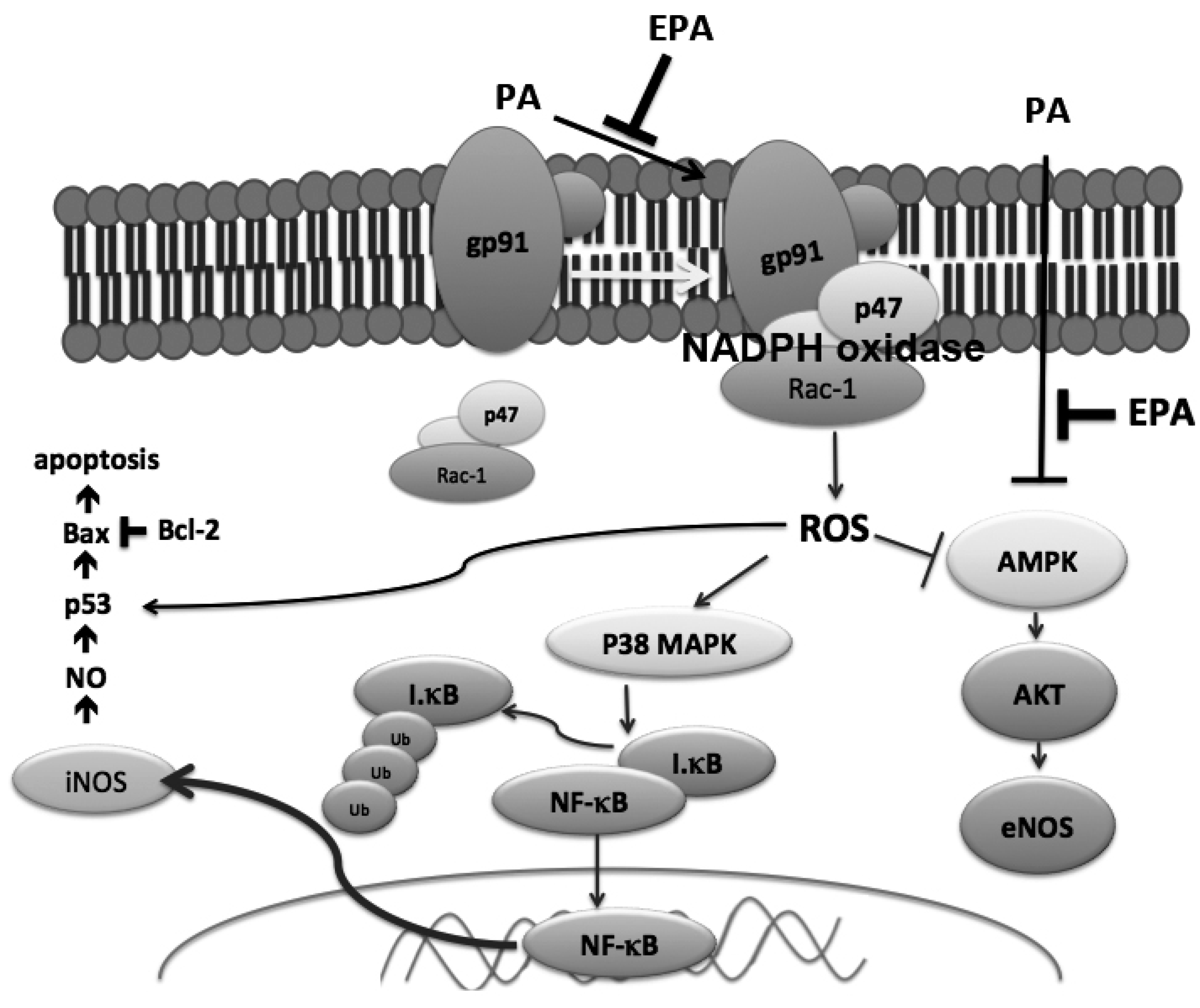
3. Discussion

4. Experimental Section
4.1. Materials
4.2. Cell Cultures
4.3. Determinations of Cell Viability, Cytotoxicity and Apoptosis
4.4. Measurement of Superoxide and ROS Production
4.5. Western Blot Analyses
4.6. Transfection with Small Interfering RNAs (siRNAs)
4.7. Measurement of Active Caspase-3
4.8. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Artwohl, M.; Roden, M.; Waldhausl, W.; Freudenthaler, A.; Baumgartner-Parzer, S.M. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. FASEB J. 2004, 18, 146–148. [Google Scholar]
- Kim, F.; Tysseling, K.A.; Rice, J.; Pham, M.; Haji, L.; Gallis, B.M.; Baas, A.S.; Paramsothy, P.; Giachelli, C.M.; Corson, M.A.; et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef]
- Esenabhalu, V.E.; Schaeffer, G.; Graier, W.F. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid. Redox. Signal. 2003, 5, 147–153. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Khan, M.J.; Rizwan Alam, M.; Waldeck-Weiermair, M.; Karsten, F.; Groschner, L.; Riederer, M.; Hallstrom, S.; Rockenfeller, P.; Konya, V.; Heinemann, A.; et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J. Biol. Chem. 2012, 287, 21110–21120. [Google Scholar] [CrossRef]
- Staiger, H.; Staiger, K.; Stefan, N.; Wahl, H.G.; Machicao, F.; Kellerer, M.; Haring, H.U. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes 2004, 53, 3209–3216. [Google Scholar] [CrossRef]
- Cacicedo, J.M.; Benjachareowong, S.; Chou, E.; Ruderman, N.B.; Ido, Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 2005, 54, 1838–1845. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef]
- Holub, B.J. Dietary fish oils containing eicosapentaenoic acid and the prevention of atherosclerosis and thrombosis. CMAJ 1988, 139, 377–381. [Google Scholar]
- Howe, P.R.; Rogers, P.F.; Lungershausen, Y. Blood pressure reduction by fish oil in adult rats with established hypertension--dependence on sodium intake. Prostaglandins Leukot. Essent. Fatty Acids 1991, 44, 113–117. [Google Scholar] [CrossRef]
- Li, H.; Ruan, X.Z.; Powis, S.H.; Fernando, R.; Mon, W.Y.; Wheeler, D.C.; Moorhead, J.F.; Varghese, Z. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005, 67, 867–874. [Google Scholar] [CrossRef]
- Devaraj, S.; Chien, A.; Rao, B.; Chen, X.; Jialal, I. Modulation of endothelial progenitor cell number and function with n-3 polyunsaturated fatty acids. Atherosclerosis 2013, 228, 94–97. [Google Scholar] [CrossRef]
- Okuda, Y.; Kawashima, K.; Sawada, T.; Tsurumaru, K.; Asano, M.; Suzuki, S.; Soma, M.; Nakajima, T.; Yamashita, K. Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem. Biophys. Res. Commun. 1997, 232, 487–491. [Google Scholar] [CrossRef]
- Balakumar, P.; Taneja, G. Fish oil and vascular endothelial protection: Bench to bedside. Free Radic. Biol. Med. 2012, 53, 271–279. [Google Scholar] [CrossRef]
- Dyerberg, J.; Bang, H.O.; Stoffersen, E.; Moncada, S.; Vane, J.R. Eicosapentaenoic acid and prevention of thrombosis and atherosclerosis? Lancet 1978, 2, 117–119. [Google Scholar]
- Nakajima, K.; Yamashita, T.; Kita, T.; Takeda, M.; Sasaki, N.; Kasahara, K.; Shinohara, M.; Rikitake, Y.; Ishida, T.; Yokoyama, M.; et al. Orally administered eicosapentaenoic acid induces rapid regression of atherosclerosis via modulating the phenotype of dendritic cells in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1963–1972. [Google Scholar] [CrossRef]
- Seifert, R.; Schultz, G. Fatty-acid-induced activation of NADPH oxidase in plasma membranes of human neutrophils depends on neutrophil cytosol and is potentiated by stable guanine nucleotides. Eur. J. Biochem. 1987, 162, 563–569. [Google Scholar]
- Yilmaz, A.; Kliche, S.; Mayr-Beyrle, U.; Fellbrich, G.; Waltenberger, J. p38 MAPK inhibition is critically involved in VEGFR-2-mediated endothelial cell survival. Biochem. Biophys. Res. Commun. 2003, 306, 730–736. [Google Scholar] [CrossRef]
- Xiao, G.F.; Xu, S.H.; Chao, Y.; Xie, L.D.; Xu, C.S.; Wang, H.J. PPARdelta activation inhibits homocysteine-induced p22 expression in EA.hy926 cells through reactive oxygen species/p38MAPK pathway. Eur. J. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Mugabo, Y.; Mukaneza, Y.; Renier, G. Palmitate induces C-reactive protein expression in human aortic endothelial cells. Relevance to fatty acid-induced endothelial dysfunction. Metabolism 2011, 60, 640–648. [Google Scholar] [CrossRef]
- Violi, F.; Basili, S.; Nigro, C.; Pignatelli, P. Role of NADPH oxidase in atherosclerosis. Future Cardiol. 2009, 5, 83–92. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef]
- Ou, H.C.; Lee, W.J.; Wu, C.M.; Chen, J.F.; Sheu, W.H. Aspirin prevents resistin-induced endothelial dysfunction by modulating AMPK, ROS, and Akt/eNOS signaling. J. Vasc. Surg. 2012, 55, 1104–1115. [Google Scholar] [CrossRef]
- Lu, Y.; Qian, L.; Zhang, Q.; Chen, B.; Gui, L.; Huang, D.; Chen, G.; Chen, L. Palmitate induces apoptosis in mouse aortic endothelial cells and endothelial dysfunction in mice fed high-calorie and high-cholesterol diets. Life Sci. 2013, 92, 1165–1173. [Google Scholar] [CrossRef]
- Chai, W.; Liu, Z. p38 mitogen-activated protein kinase mediates palmitate-induced apoptosis but not inhibitor of nuclear factor-kappaB degradation in human coronary artery endothelial cells. Endocrinology 2007, 148, 1622–1628. [Google Scholar] [CrossRef]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am. J. Physiol. Endocrinol. MeTable 2010, 299, E1096–E1105. [Google Scholar] [CrossRef]
- Peng, H.B.; Libby, P.; Liao, J.K. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem. 1995, 270, 14214–14219. [Google Scholar] [CrossRef]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Dong, Y.; Wang, S.; Song, P.; Viollet, B.; Zou, M.H. Activation of the AMP-activated protein kinase by eicosapentaenoic acid (EPA, 20:5 n-3) improves endothelial function in vivo. PLoS One 2012, 7, e35508. [Google Scholar]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Calder, P.C. The role of marine omega-3 (n-3) fatty acids in inflammatory processes, atherosclerosis and plaque stability. Mol. Nutr. Food Res. 2012, 56, 1073–1080. [Google Scholar] [CrossRef]
- Liu, W.; Peng, Y.; Yin, Y.; Zhou, Z.; Zhou, W.; Dai, Y. The Involvement of NADPH Oxidase-Mediated ROS in Cytokine Secretion from Macrophages Induced by Mycobacterium tuberculosis ESAT-6. Inflammation 2014. [Google Scholar] [CrossRef]
- Kuo, W.W.; Huang, C.Y.; Chung, J.G.; Yang, S.F.; Tsai, K.L.; Chiu, T.H.; Lee, S.D.; Ou, H.C. Crude extracts of Solanum lyratum protect endothelial cells against oxidized low-density lipoprotein-induced injury by direct antioxidant action. J. Vasc. Surg. 2009, 50, 849–860. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Huang, H.M.; Ou, H.C.; Hsueh, S.J. Amyloid beta peptide enhanced bradykinin-mediated inositol (1,4,5)trisphosphate formation and cytosolic free calcium. Life Sci. 1998, 63, 195–203. [Google Scholar] [CrossRef]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992, 119, 493–501. [Google Scholar] [CrossRef]
- Wang, L.; Gill, R.; Pedersen, T.L.; Higgins, L.J.; Newman, J.W.; Rutledge, J.C. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J. Lipid Res. 2009, 50, 204–213. [Google Scholar]
- Chang, W.W.; Liu, J.J.; Liu, C.F.; Liu, W.S.; Lim, Y.P.; Cheng, Y.J.; Lee, C.H. An Extract of Rhodobacter sphaeroides Reduces Cisplatin-Induced Nephrotoxicity in Mice. Toxins 2013, 5, 2353–2365. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, C.-H.; Lee, S.-D.; Ou, H.-C.; Lai, S.-C.; Cheng, Y.-J. Eicosapentaenoic Acid Protects against Palmitic Acid-Induced Endothelial Dysfunction via Activation of the AMPK/eNOS Pathway. Int. J. Mol. Sci. 2014, 15, 10334-10349. https://doi.org/10.3390/ijms150610334
Lee C-H, Lee S-D, Ou H-C, Lai S-C, Cheng Y-J. Eicosapentaenoic Acid Protects against Palmitic Acid-Induced Endothelial Dysfunction via Activation of the AMPK/eNOS Pathway. International Journal of Molecular Sciences. 2014; 15(6):10334-10349. https://doi.org/10.3390/ijms150610334
Chicago/Turabian StyleLee, Che-Hsin, Shin-Da Lee, Hsiu-Chung Ou, Su-Chuan Lai, and Yu-Jung Cheng. 2014. "Eicosapentaenoic Acid Protects against Palmitic Acid-Induced Endothelial Dysfunction via Activation of the AMPK/eNOS Pathway" International Journal of Molecular Sciences 15, no. 6: 10334-10349. https://doi.org/10.3390/ijms150610334