Proteomic Analysis of Embryogenesis and the Acquisition of Seed Dormancy in Norway Maple (Acer platanoides L.)



Abstract
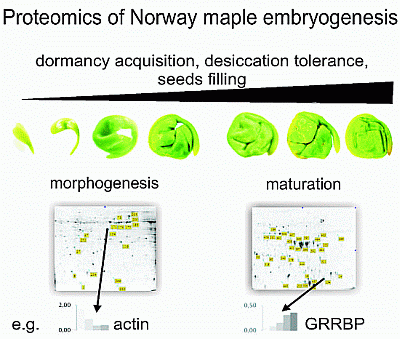
:
1. Introduction
2. Results and Discussion
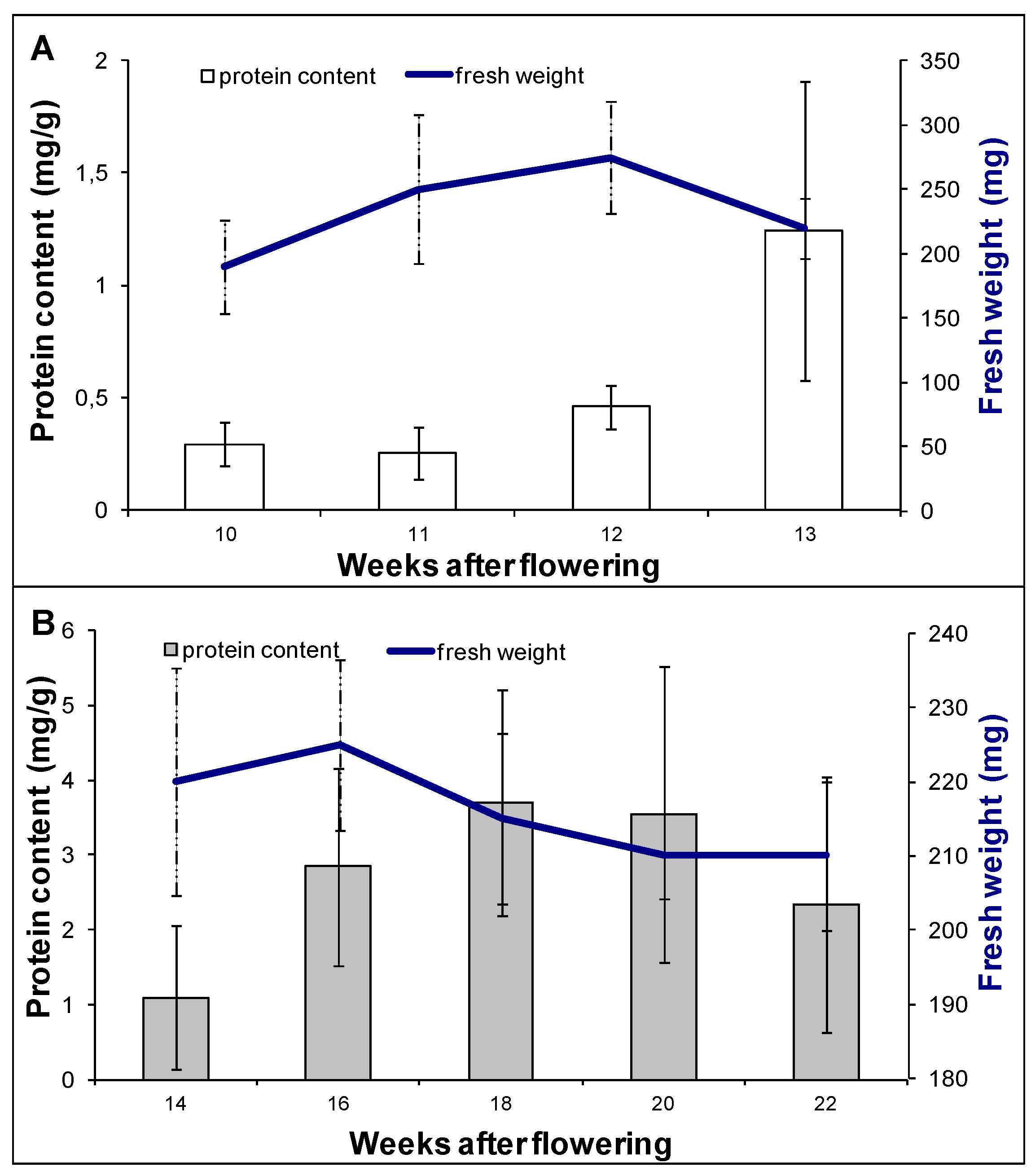
2.1. Proteomic Analysis of Embryo Morphogenesis

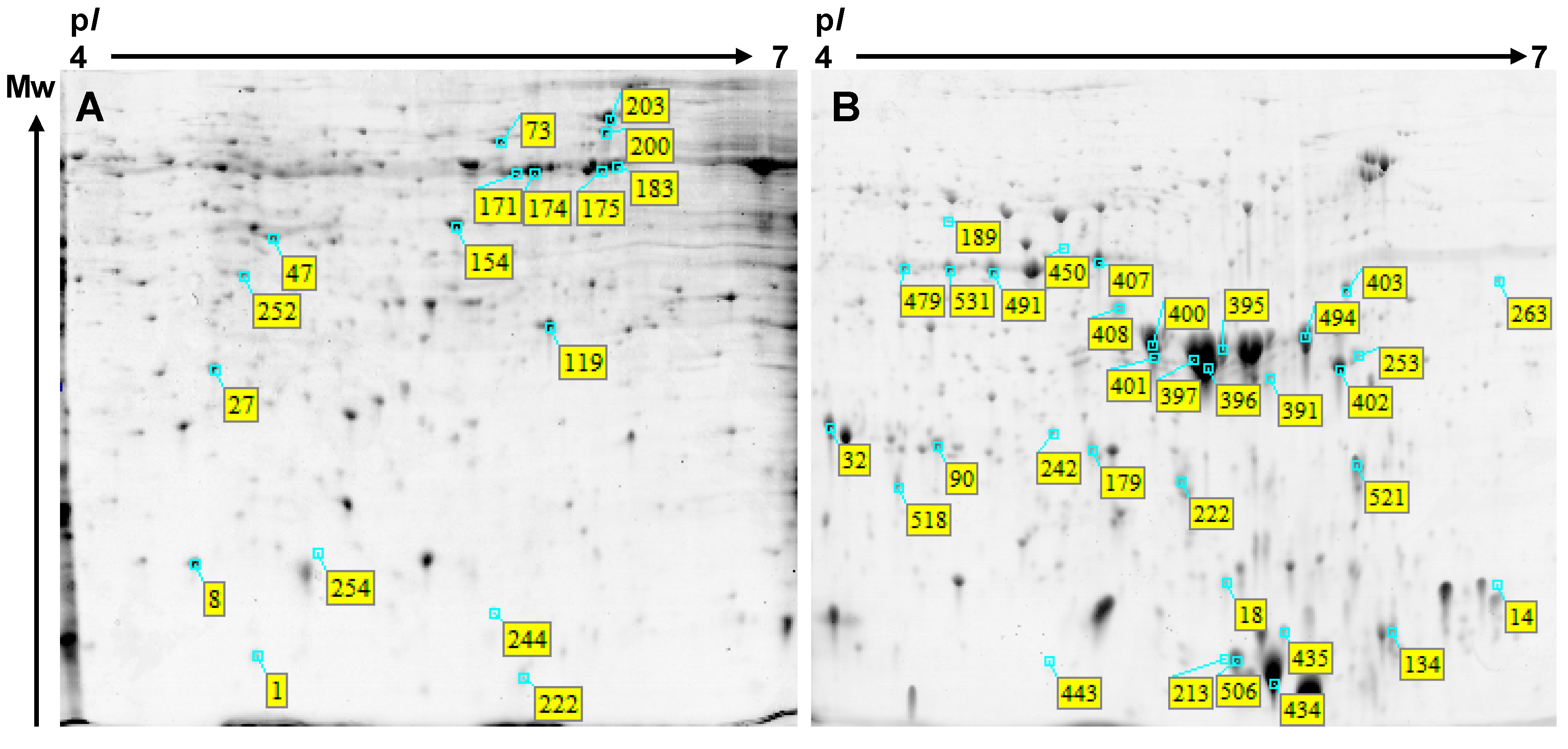

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. a | Assignment | Species | gi No. | Mean % Volume b Weeks After Flowering (WAF) | Observed Mr/pI | Nominal Mr/pI | Score | Cov. (%) c | Pept d | Mean % Volume Graph | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 11 | 12 | 13 | ||||||||||
| Cellular processes | |||||||||||||
| 8 | copper/zinc-superoxide dismutase | Litchi chinensis | gi|164654158 | 1.08 ± 0.09 b | 0.76 ± 0.17 b | 2.19 ± 0.34 a | 1.06 ± 0.31 b | 18/6.33 | 15/5.47 | 483 | 21 | 15 | 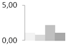 |
| 154 | actin | Boehmeria nivea | gi|255115691 | 1.69 ± 0.23 a | 0.86 ± 0.45 a,b | 0.37 ± 0.45 b | 0.42 ± 0.19 b | 53/5.4 | 42/5.31 | 3161 | 69 | 83 | 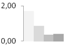 |
| 171 | tubulin α chain | Ricinus communis | gi|255552898 | 1.40 ± 0.11 a | 0.71 ± 0.17 b | 0 c | 0.56 ± 0.21 b | 63/5.19 | 50/4.96 | 3172 | 47 | 65 |  |
| 174 | α tubulin | Betula pendula | gi|214003725 | 2.27 ± 0.04 a | 1.78 ± 0.11 b | 0 d | 0.58 ± 0.20 c | 63/5.13 | 50/4.93 | 7151 | 59 | 175 | 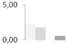 |
| 175 | tubulin β-7 chain | Zea mays | gi|162460038 | 1.06 ± 0.02 a | 0.81 ± 0.18 a | 0 b | 0.12 ± 0.21 b | 64/4.88 | 50/4.72 | 1625 | 43 | 53 | 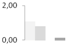 |
| 183 | tubulin β chain | Ricinus communis | gi|255558856 | 1.43 ± 0.30 a | 1.01 ± 0.27 a | 0 b | 0 b | 65/4.82 | 62/4.77 | 3946 | 46 | 132 |  |
| Genetic information processing | |||||||||||||
| 1 | nonspecific lipid transfer protein | Vitis vinifera | gi|359486799 | 0.96 ± 0.45 b | 0.50 ± 0.22 b | 2.03 ± 0.21 a | 0.87 ± 0.33 b | 14/6.11 | 13/9.20 | 225/84 | 9 | 1 | 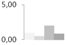 |
| 73 | RuBisCO large subunit-binding protein subunit β, chloroplastic | Pisum sativum | gi|2506277 | 1.08 ± 0.06 a | 0.55 ± 0.21 a,b | 0.26 ± 0.45 b | 0.36 ± 0.16 b | 71/5.24 | 63/6.05 | 6509 | 47 | 161 | 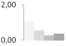 |
| 200 | RuBisCO large subunit-binding protein subunit α, chloroplastic | Brassica napus | gi|1351030 | 0.98 ± 0.24 a,b | 0.61 ± 0.03 a,b | 0 b | 3.03 ± 1.88 a | 74/4.87 | 57/4.84 | 6947 | 22 | 167 | 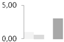 |
| Metabolism | |||||||||||||
| 27 | triosephosphate isomerase, cytosolic | Coptis japonica | gi|136057 | 0.37 ± 0.21 b | 0.82 ± 0.26 a,b | 0.96 ± 0.18 b | 0.48 ± 0.12 a,b | 33/6.26 | 27/5.54 | 1986 | 16 | 48 | 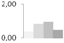 |
| 47 | α-1,4-glucan-protein synthase (UDP-forming) | Ricinus communis | gi|255547137 | 0.70 ± 0.14 a,b | 1.19 ± 0.13 a | 0.80 ± 0.33 a,b | 0.26 ± 0.23 b | 50/6.05 | 41/5.82 | 1259 | 29 | 39 |  |
| 119 | oxygen evolving enhancer protein 1 | Litchi chinensis | gi|326467059 | 0.77 ± 0.04 a,b | 1.52 ± 0.55 a | 0.33 ± 0.57 b | 0.97 ± 0.36 a,b | 38/5.7 | 35/5.86 | 1660 | 30 | 52 |  |
| 222 | cytochrome b6-f complex iron-sulfur subunit, chloroplastic | Pisum sativum | gi|136707 | 0.79 ± 0.30 b | 1.33 ± 0.81 b | 5.51 ± 4.85 a | 6.98 ± 4.13 a | 13/5.16 | 24/8.63 | 108 | 5 | 2 |  |
| 244 | triosephosphate isomerase, cytosolic | Zea mays | gi|226495391 | 0 b | 0.13 ± 0.01 b | 4.41 ± 3.83 a,b | 9.27 ± 4.76 a | 16/5.26 | 27/5.52 | 128 | 10 | 2 |  |
| 252 | fructose-bisphosphate aldolase, putative | Ricinus communis | gi|255581400 | 0.48 ± 0.14 a | 0.31 ± 0.09 a,b | 0.06 ± 0.11 b | 0.21 ± 0.21 a,b | 44/6.16 | 42/6.78 | 1362 | 24 | 42 | 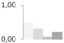 |
| Unclassified | |||||||||||||
| 203 | protein disulfide-isomerase | Vitis vinifera | gi|225459587 | 1.76 ± 0.08 a | 2.61 ± 0.86 a | 0 b | 0 b | 78/4.85 | 56/4.93 | 238 | 11 | 8 | 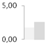 |
| 254 | unknown | Populus/italic> trichocarpa | gi|118484746 | 0 b | 0 b | 0 b | 2.27 ± 0.93 a | 18/5.90 | 41/6.5 | 93 | 4 | 1 | 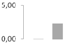 |
| Spot No. a | Assignment | Species | gi No. | Mean % Volume b WAF | Nominal Mr/pI | Observed Mr/pI | Score | Cov. (%) c | Pept d | Mean % Volume Graph | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 16 | 18 | 20 | 22 | ||||||||||
| Metabolism | ||||||||||||||
| 32 | glutathione S-transferase | Catharanthus roseus | gi|67973220 | 0.40 ± 0.15 a,b | 0.15 ± 0.18 b | 0.25 ± 0.12 b | 0.31 ± 0.28 a,b | 0.78 ± 0.11 a | 24/5.6 | 32/6.83 | 499 | 3 | 22 | 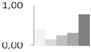 |
| 189 | monodehydroascorbate reductase, chloroplastic | Vitis vinifera | gi|359474156 | 0.00 ± 0.00 b | 0.06 ± 0.01 a | 0.07 ± 0.01 a | 0.07 ± 0.02 a | 0.08 ± 0.02 a | 54/8.34 | 61/6.46 | 1099 | 14 | 27 | 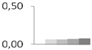 |
| 222 | triosephosphate isomerase | Gossypium hirsutum | gi|295687231 | 0.01 ± 0.02 b | 0.01 ± 0.02 b | 0.06 ± 0.07 b | 0.17 ± 0.03 a,b | 0.36 ± 0.15 a | 33/6.66 | 26/5.58 | 253 | 16 | 5 |  |
| 242 | peptide methionine sulfoxide reductase-like | Vitis vinifera | gi|225454994 | 0.01 ± 0.01 c | 0.05 ± 0.02 b,c | 0.07 ± 0.01 a,b | 0.10 ± 0.01 a,b | 0.13 ± 0.04 a | 28/8.65 | 31/6.02 | 158 | 17 | 6 | 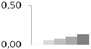 |
| 263 | glyceraldehyde-3-phosphate dehydrogenase, cytosolic | Taxus baccata | gi|3023813 | 0.00 ± 0.01 c | 0.02 ± 0.01 c | 0.03 ± 0.01 b,c | 0.07 ± 0.02 a,b | 0.09 ± 0.02 a | 37/6.41 | 52/4.51 | 120 | 10 | 4 | 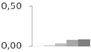 |
| 391 | lactoylglutathione lyase | Vitis vinifera | gi|359483362 | 0.07 ± 0.02 b | 0.22 ± 0.14 a,b | 0.24 ± 0.05 a | 0.25 ± 0.02 a | 0.26 ± 0.01 a | 33/5.5 | 40/5.28 | 288 | 17 | 6 | 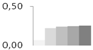 |
| 400 | ascorbate peroxidase | Retama raetam | gi|24496465 | 1.10 ± 0.22 c | 1.78 ± 0.41 b,c | 2.01 ± 0.24 a,b | 2.62 ± 0.34 a | 2.16 ± 0.18 a,b | 39/7.03 | 45/5.68 | 229 | 11 | 4 | 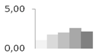 |
| 401 | succinate semialdehyde dehydrogenase, putative | Ricinus communis | gi|255577875 | 0.43 ± 0.03 b | 0.51 ± 0.23 b | 0.45 ± 0.09 b | 0.97 ± 0.07 a | 0.86 ± 0.27 a,b | 66/8.84 | 44/5.67 | 174 | 6 | 3 | 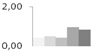 |
| 402 | enolase | Spinacia oleracea | gi|8919731 | 0.36 ± 0.02 c | 0.64 ± 0.10 b | 0.86 ± 0.02 b | 1.22 ± 0.19 a | 1.37 ± 0.08 a | 48/5.49 | 41/5.04 | 381 | 16 | 6 | 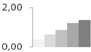 |
| 407 | enolase | Alnus glutinosa | gi|3023685 | 0.28 ± 0.07 b | 0.58 ± 0.24 a,b | 1.04 ± 0.34 a | 0.83 ± 0.25 a,b | 0.77 ± 0.28 a,b | 48/5.41 | 55/5.86 | 392 | 16 | 6 |  |
| 408 | proline iminopeptidase | Vitis vinifera | gi|359475506 | 0.01 ± 0.02 b | 0.28 ± 0.03 a | 0.30 ± 0.12 a | 0.36 ± 0.02 a | 0.38 ± 0.07 a | 44/5.73 | 49/5.79 | 185 | 5 | 4 |  |
| 450 | enolase | Arabidopsis lyrata subsp. lyrata | gi|297820024 | 0.26 ± 0.16 a | 0.09 ± 0.06 a,b | 0.03 ± 0.04 b | 0.03 ± 0.02 b | 0.03 ± 0.03 b | 48/5.57 | 57/5.97 | 323 | 10 | 5 | 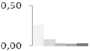 |
| 479 | fructose-bisphosphate aldolase, cytoplasmic | Zea mays | gi|162462282 | 0.01 ± 0.02 b | 0.05 ± 0.05 b | 0.25 ± 0.06 a,b | 0.41 ± 0.08 a | 0.37 ± 0.18 a | 39/7.52 | 54/6.62 | 191 | 10 | 4 |  |
| 491 | isovaleryl-CoA dehydrogenase | β vulgaris | gi|112005099 | 0.16 ± 0.04 b | 0.21 ± 0.03 b | 0.45 ± 0.09 a,b | 0.62 ± 0.20 a | 0.58 ± 0.15 a | 46/6.71 | 54/6.27 | 301 | 13 | 11 |  |
| 531 | cytosolic aldolase | Fragaria x ananassa | gi|10645188 | 0.02 ± 0.03 c | 0.12 ± 0.02 b,c | 0.36 ± 0.17 a,b | 0.48 ± 0.15 a | 0.43 ± 0.13 a,b | 38/6.93 | 54/6.45 | 232 | 16 | 6 |  |
| Genetic information processing | ||||||||||||||
| 14 | 60S ribosomal protein L22, putative | Ricinus communis | gi|255551787 | 0.03 ± 0.05 a | 0.09 ± 0.04 b | 0.30 ± 0.06 c | 0.58 ± 0.16 c,d | 0.85 ± 0.10 d | 14/9.54 | 19/4.52 | 279 | 37 | 7 | 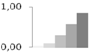 |
| 18 | 40 S Ribosomal protein | Vitis vinifera | gi|225465502 | 0.29 ± 0.05 a | 0.20 ± 0.05 a,b | 0.13 ± 0.12 a,b | 0.06 ± 0.08 b | 0.06 ± 0.08 b | 15/5.84 | 19/5.43 | 510 | 34 | 23 | 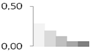 |
| 90 | proteasome subunit α | Ricinus communis | gi|255583952 | 0.01 ± 0.01 c | 0.08 ± 0.07 b,c | 0.12 ± 0.04 b,c | 0.31 ± 0.09 a | 0.24 ± 0.09 a,b | 28/5.84 | 30/6.49 | 179 | 19 | 5 | 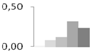 |
| 134 | glycine-rich RNA-binding protein | Prunus avium | gi|34851124 | 0.04 ± 0.04 c | 0.08 ± 0.07 c | 0.15 ± 0.06 b,c | 0.31 ± 0.02 a,b | 0.36 ± 0.11 a | 17/7.82 | 17/4.87 | 127 | 13 | 3 | 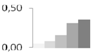 |
| 395 | P0 ribosomal protein-like | Solanum tuberosum | gi|78191424 | 0.33 ± 0.10 c | 0.92 ± 0.31 b,c | 1.11 ± 0.16 a,b | 1.71 ± 0.37 a | 1.36 ± 0.34 a,b | 33/5.12 | 43/5.44 | 200 | 16 | 5 |  |
| 397 | P0 ribosomal protein-like | Solanum tuberosum | gi|78191424 | 0.43 ± 0.12 b | 1.07 ± 0.78 a,b | 2.38 ± 1.05 a,b | 1.90 ± 0.93 a,b | 2.57 ± 0.39 a | 33/5.12 | 43/5.54 | 313 | 21 | 8 | 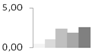 |
| 396 | 60S acidic ribosomal protein P0 | Lupinus luteus | gi|1710585 | 4.35 ± 0.91 b | 6.29 ± 0.81 a,b | 7.01 ± 1.55 a,b | 8.62 ± 1.80 a | 8.26 ± 1.00 a | 34/5.07 | 42/5.49 | 91 | 10 | 3 | 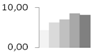 |
| 403 | 26S protease regulatory subunit 6A homolog | Brassica rapa | gi|3024434 | 0.10 ± 0.03 c | 0.15 ± 0.02 c | 0.25 ± 0.02 b | 0.45 ± 0.02 a | 0.45 ± 0.05 a | 48/4.92 | 51/5.02 | 139 | 10 | 3 | 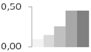 |
| 494 | putative 60S acidic ribosomal protein P0 | Trifolium pratense | gi|84468360 | 0.45 ± 0.18 c | 0.92 ± 0.23 b,c | 1.57 ± 0.36 a,b | 2.22 ± 0.55 a | 1.71 ± 0.51 a,b | 34/5.27 | 44/5.16 | 111 | 12 | 4 |  |
| 518 | proteasome subunit β type-2-A-like | Vitis vinifera | gi|359479647 | 0.03 ± 0.03 c | 0.13 ± 0.03 b | 0.14 ± 0.03 a,b | 0.19 ± 0.05 a,b | 0.23 ± 0.04 a | 22/5.85 | 26/6.64 | 110 | 22 | 5 | 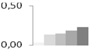 |
| 521 | 60S ribosomal protein L23a | Nicotiana tabacum | gi|585876 | 0.31 ± 0.01 c | 0.28 ± 0.25 c | 0.48 ± 0.07 b,c | 0.68 ± 0.06 a,b | 0.84 ± 0.06 a | 17/10.18 | 28/4.98 | 97 | 14 | 2 | 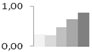 |
| Cellular processes | ||||||||||||||
| 179 | α tubulin | Plantago major | gi|106879605 | 0.02 ± 0.03 c | 0.07 ± 0.01 b,c | 0.11 ± 0.04 b | 0.20 ± 0.03 a | 0.23 ± 0.02 a | 30/4.7 | 29/5.88 | 131 | 6 | 2 | 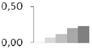 |
| 253 | charged multivesicular body protein 4b, putative | Ricinus communis | gi|255546239 | 0.00 ± 0.01 b | 0.01 ± 0.02 b | 0.04 ± 0.03 b | 0.05 ± 0.01 b | 0.24 ± 0.10 a | 24/4.80 | 44/4.98 | 223 | 27 | 5 | 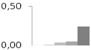 |
| Unclassified | ||||||||||||||
| 213 | type IIIa membrane protein cp-wap13 | Vigna unguiculata | gi|2218152 | 1.84 ± 0.54 a | 2.42 ± 0.42 a | 2.46 ± 0.19 a | 1.81 ± 0.07 a,b | 0.73 ± 0.56 b | 40/6.24 | 15/5.43 | 115 | 7 | 2 | 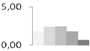 |
| 443 | late embryogenesis abundant protein D-34-like | Brachypodium distachyon | gi|357115298 | 0.12 ± 0.04 b | 0.53 ± 0.77 a,b | 0.72 ± 0.64 a | 0.50 ± 0.84 a,b | 0.03 ± 0.03 b | 6/8.23 | 15/6.03 | 94 | 6 | 1 | 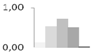 |
| Unknown | ||||||||||||||
| 434 | unknown | 8.31 ± 0.83 a,b | 8.67 ± 0.15 a | 6.93 ± 0.76 b,c | 5.63 ± 0.38 c,d | 4.90 ± 0.29 d | 14/5.27 | 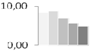 | ||||||
| 435 | unknown | Ricinus communis | gi|255575865 | 1.90 ± 0.16 a | 2.00 ± 0.14 a | 1.23 ± 0.19 b | 0.47 ± 0.11 c | 0.21 ± 0.03 c | 16/10.23 | 17/5.23 | 84 | 19 | 3 |  |
| 506 | unknown | 1.99 ± 0.30 a | 1.47 ± 0.13 a | 0.65 ± 0.04 b | 0.24 ± 0.08 b | 0.43 ± 0.53 b | 15/5.39 | 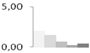 | ||||||
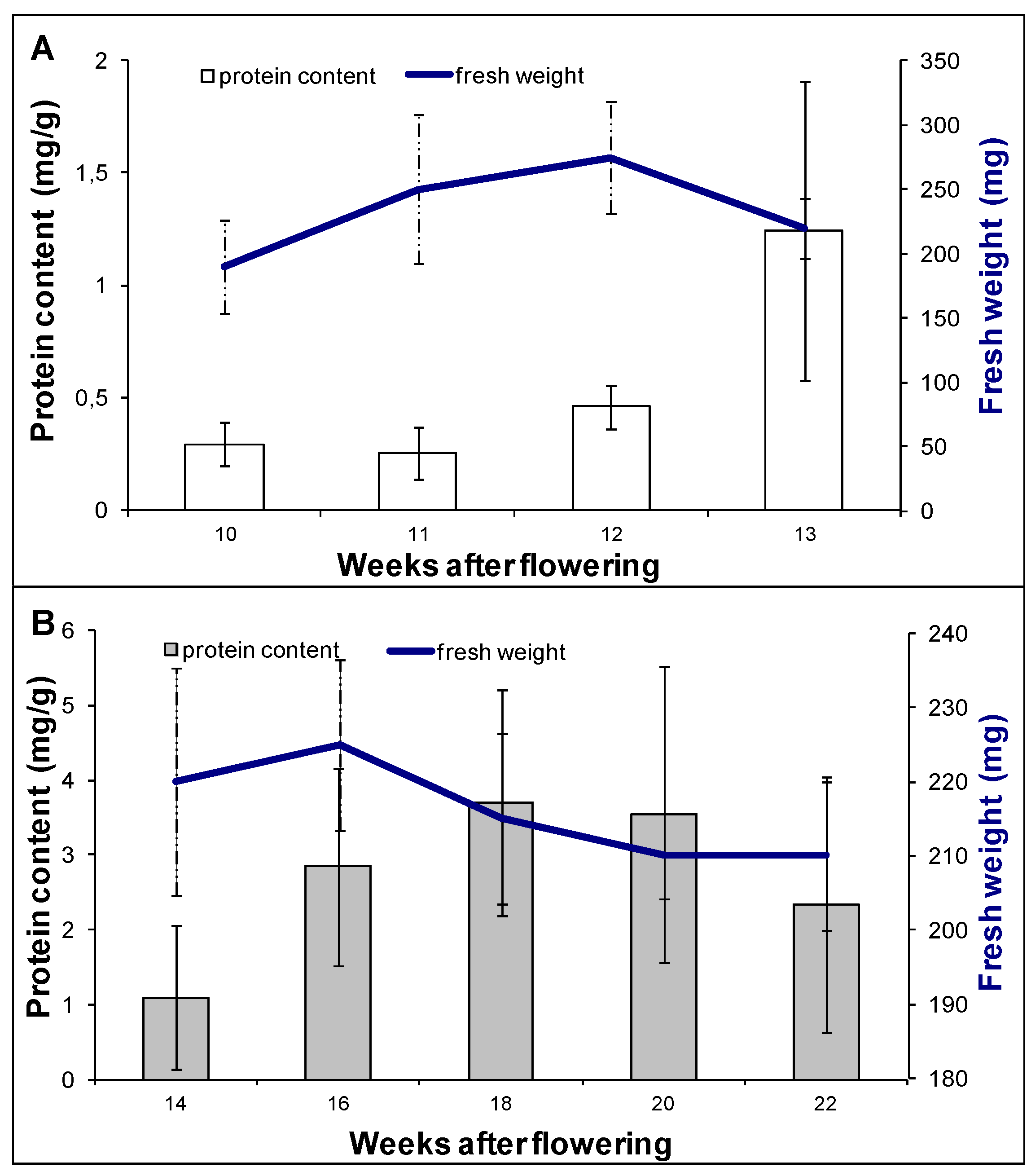
2.2. Proteomic Analysis of Embryo Maturation
2.3. Discussion
2.3.1. Defense Proteins
2.3.2. Metabolic Pathways Activated during Seed Development
2.3.3. Unclassified Proteins
Cell Wall Biosynthesis
Late Embryogenesis Abundant (LEA)
Cellular Processes
Genetic Information Processing
3. Experimental Section
3.1. Plant Material and Experimental Design
3.2. Protein Extraction
3.3. Protein Electrophoresis, 2-DE SDS-PAGE
3.4. Gel Analysis
3.5. Mass Spectrometry (MS)
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bewley, J.D.; Bradford, K.; Hilhorst, H.; Nonogaki, H. Seeds—Physiology of Development, Germination and Dormancy, 3rd ed.; Springer: Berlin, Germany, 2013. [Google Scholar]
- Finch-Savage, W.; Leubner-Metzger, G. Seed dormancy and the control of germination. N. Phytol. 2006, 171, 501–523. [Google Scholar] [CrossRef]
- Baskin, J.; Baskin, C. A classification system for seed dormancy. Seed Sci. Res. 2004, 14, 1–16. [Google Scholar]
- Hilhorst, H.W.M. A critical update on seed dormancy. I. Primary dormancy. Seed Sci. Res. 1995, 5, 61–73. [Google Scholar]
- Black, M.; Bewley, J.D.; Halmer, P. The Encyclopedia of Seeds. Science, Technology and Uses; CABI: Wallingford, UK, 2008. [Google Scholar]
- Tweddle, J.C.; Dickie, J.B.; Baskin, C.C.; Baskin, J.M. Ecological aspects of seed desiccation sensitivity. J. Ecol. 2003, 91, 294–304. [Google Scholar]
- Chibani, K.; Ali-Rachedi, S.; Job, C.; Job, D.; Jullien, M.; Grappin, P. Proteomic analysis of seed dormancy in Arabidopsis. Plant Physiol. 2006, 142, 1493–1510. [Google Scholar] [CrossRef]
- Suszka, B.; Muller, C.; Bonnet-Masimbert, M. Seeds of Forest Broadleaves: From Harvest to Sowing; INRA: Paris, France, 1996. [Google Scholar]
- Pawłowski, T.A. Proteome analysis of Norway maple (Acer platanoides L.) seeds dormancy breaking and germination: Influence of abscisic and gibberellic acids. BMC Plant Biol. 2009, 9. [Google Scholar] [CrossRef]
- Pawłowski, T.A. Proteomics of European beech (Fagus. sylvatica L.) seed dormancy breaking: Influence of abscisic and gibberellic acids. Proteomics 2007, 7, 2246–2257. [Google Scholar] [CrossRef]
- Pawłowski, T.A. Proteomic approach to analyze dormancy breaking of tree seeds. Plant Mol. Biol. 2010, 73, 15–25. [Google Scholar] [CrossRef]
- Chen, S.; Harmon, A.C. Advances in plant proteomics. Proteomics 2006, 6, 5504–5516. [Google Scholar] [CrossRef]
- Gallardo, K.; Firnhaber, C.; Zuber, H.; Hericher, D.; Belghazi, M.; Henry, C.; Kuster, H.; Thompson, R. A combined proteome and transcriptome analysis of developing Medicago. truncatula seeds: Evidence for metabolic specialization of maternal and filial tissues. Mol. Cell. Proteomics 2007, 6, 2165–2179. [Google Scholar] [CrossRef]
- Pawłowski, T.; Kalinowski, A. Qualitative and quantitative changes in proteins in Acer platanoides L. seeds during maturation. Acta Biol. Crac. Ser. Bot. 2003, 45, 139–144. [Google Scholar]
- Dam, S.; Laursen, B.S.; Ørnfelt, J.H.; Jochimsen, B.; Stærfeldt, H.H.; Friis, C.; Nielsen, K.; Goffard, N.; Besenbacher, S.; Krusell, L.; et al. The proteome of seed development in the model legume Lotus japonicus. Plant Physiol. 2009, 149, 1325–1340. [Google Scholar] [CrossRef]
- Nautrup-Pedersen, G.; Dam, S.; Laursen, B.S.; Siegumfeldt, A.L.; Nielsen, K.; Goffard, N.; Stærfeldt, H.H.; Friis, C.; Sato, S.; Tabata, S.; et al. Proteome analysis of pod and seed development in the model legume Lotus japonicus. J. Proteome Res. 2010, 9, 5715–5726. [Google Scholar] [CrossRef]
- Hajduch, M.; Hearne, L.B.; Miernyk, J.A.; Casteel, J.E.; Joshi, T.; Agrawal, G.K.; Song, Z.; Zhou, M.; Xu, D.; Thelen, J.J. Systems analysis of seed filling in Arabidopsis: Using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 2010, 152, 2078–2087. [Google Scholar] [CrossRef]
- Li, W.; Gao, Y.; Xu, H.; Zhang, Y.; Wang, J. A proteomic analysis of seed development in Brassica. campestri L. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Hajduch, M.; Graham, K.; Thelen, J.J. In-depth investigation of the soybean seed-filling proteome and comparison with a parallel study of rapeseed. Plant Physiol. 2008, 148, 504–518. [Google Scholar] [CrossRef]
- Miernyk, J.A.; Hajduch, M. Seed proteomics. J. Proteomics 2011, 74, 389–400. [Google Scholar] [CrossRef]
- Staszak, A.M.; Pawłowski, T. Forest tree research in post genomic era. Introduction to systems biology of broadleaves. Dendrobiology 2012, 68, 113–123. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar]
- Pinfield, N.; Bazaid, S.; Gwarazimba, V. The development of embryo dormancy and testa-imposed dormancy during seed ontogeny in the genus Acer. J. Plant Physiol. 1990, 136, 746–749. [Google Scholar] [CrossRef]
- Pukacka, S. Charakterystyka rozwoju nasion klonu zwyczajnego (Acer platanoides L.) i jaworu (Acer pseudoplatanus L.) (in Polish). Arbor. Kórn. 1998, 43, 97–104. [Google Scholar]
- Bailly, C. Active oxygen species and antioxidants in seed biology. Seed Sci. Res. 2004, 14, 93–107. [Google Scholar] [CrossRef]
- Oracz, K.; Bouteau, H.; Farrant, J.; Cooper, K.; Belghazi, M.; Job, C.; Job, D.; Corbineau, F.; Bailly, C. ROS production and protein oxidation as a novel mechanism for seed dormancy alleviation. Plant J. 2007, 50, 452–465. [Google Scholar] [CrossRef]
- Bailly, C.; El-Maarouf-Bouteau, H.; Corbineau, F. From intracellular signaling networks to cell death: The dual role of reactive oxygen species in seed physiology. C. R. Biol. 2008, 331, 806–814. [Google Scholar] [CrossRef]
- Pukacka, S.; Ratajczak, E. Ascorbate and glutathione metabolism during development and desiccation of orthodox and recalcitrant seeds of the genus Acer. Funct. Plant Biol. 2007, 34, 601–613. [Google Scholar] [CrossRef]
- Lee, Y.P.; Baek, K.-H.; Lee, H.-S.; Kwak, S.-S.; Bang, J.-W.; Kwon, S.-Y. Tobacco seeds simultaneously over-expressing Cu/Zn-superoxide dismutase and ascorbate peroxidase display enhanced seed longevity and germination rates under stress conditions. J. Exp. Bot. 2010, 61, 2499–2506. [Google Scholar] [CrossRef]
- Mohsenzadeh, S.; Esmaeili, M.; Moosavi, F.; Shahrtash, M.; Saffari, B.; Mohabatkar, H. Plant glutathione S-transferase classification, structure and evolution. Afr. J. Biotechnol. 2011, 10, 8160–8165. [Google Scholar]
- Chen, J.-H.; Jiang, H.-W.; Hsieh, E.-J.; Chen, H.-Y.; Chien, C.-T.; Hsieh, H.-L.; Lin, T.-P. Drought and salt stress tolerance of an Arabidopsis glutathione S-transferase U17 knockout mutant are attributed to the combined effect of glutathione and abscisic acid. Plant Physiol. 2012, 158, 340–351. [Google Scholar] [CrossRef]
- Marrs, K.A. The functions and regulation of glutathione S-transferases in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 127–158. [Google Scholar] [CrossRef]
- Rouhier, N.; Kauffmann, B.; Tete-Favier, F.; Palladino, P.; Gans, P.; Branlant, G.; Jacquot, J.-P.; Boschi-Muller, S. Functional and structural aspects of poplar cytosolic and plastidial type A methionine sulfoxide reductases. J. Biol. Chem. 2007, 282, 3367–3378. [Google Scholar] [CrossRef]
- Richard, J. Mechanism for the formation of methylglyoxal from triosephosphates. Biochem. Soc. Trans. 1993, 21, 549–553. [Google Scholar]
- Yadav, S.K.; Singla-Pareek, S.L.; Reddy, M.K.; Sopory, S.K. Transgenic tobacco plants overexpressing glyoxalase enzymes resist an increase in methylglyoxal and maintain higher reduced glutathione levels under salinity stress. FEBS Lett. 2005, 579, 6265–6271. [Google Scholar] [CrossRef]
- Tambasco-Studart, M.; Titiz, O.; Raschle, T.; Forster, G.; Amrhein, N.; Fitzpatrick, T.B. Vitamin B6 biosynthesis in higher plants. Proc. Natl. Acad. Sci. USA 2005, 102, 13687–13692. [Google Scholar]
- Dorion, S.; Clendenning, A.; Jeukens, J.; Salas, J.J.; Parveen, N.; Haner, A.A.; Law, R.D.; Force, E.M.; Rivoal, J. A large decrease of cytosolic triosephosphate isomerase in transgenic potato roots affects the distribution of carbon in primary metabolism. Planta 2012, 236, 1177–1190. [Google Scholar] [CrossRef]
- Konishi, H.; Yamane, H.; Maeshima, M.; Komatsu, S. Characterization of fructose-bisphosphate aldolase regulated by gibberellin in roots of rice seedling. Plant Mol. Biol. 2004, 56, 839–848. [Google Scholar] [CrossRef]
- Kavelaki, K.; Ghanotakis, D. Effect of the manganese complex on the binding of the extrinsic proteins (17, 23 and 33 kDa) of photosystem II. Photosynth. Res. 1991, 29, 149–155. [Google Scholar]
- Basha, S.M.; Mazhar, H.; Vasanthaiah, H.K. N. Proteomics approach to identify unique xylem sap proteins in Pierce’s disease-tolerant Vitis species. Appl. Biochem. Biotechnol. 2010, 160, 932–944. [Google Scholar] [CrossRef]
- Toyokura, K.; Watanabe, K.; Oiwaka, A.; Kusano, M.; Tameshige, T.; Tatematsu, K.; Matsumoto, N.; Tsugeki, R.; Saito, K.; Okada, K. Succinic semialdehyde dehydrogenase is involved in the robust patterning of Arabidopsis leaves along the adaxial-abaxial axis. Plant Cell. Physiol. 2011, 52, 1340–1353. [Google Scholar] [CrossRef]
- Fait, A.; Fromm, H.; Walter, D.; Galili, G.; Fernie, A.R. Highway or byway: The metabolic role of the GABA shunt in plants. Trends Plant Sci. 2008, 13, 14–19. [Google Scholar]
- Popov, V.N.; Eprintsev, A.T.; Fedorin, D.N.; Fomenko, O.Y.; Igamberdiev, A.U. Role of transamination in the mobilization of respiratory substrates in germinating seeds of castor oil plants. Appl. Biochem. Microbiol. 2007, 43, 341–346. [Google Scholar] [CrossRef]
- Engqvist, M.; Drincovich, M.F.; Flügge, U.-I.; Maurino, V.G. Two d-2-hydroxy-acid dehydrogenases in Arabidopsis thaliana with catalytic capacities to participate in the last reactions of the methylglyoxal and β-oxidation pathways. J. Biol. Chem. 2009, 284, 25026–25037. [Google Scholar]
- Araújo, W.L.; Tohge, T.; Ishizaki, K.; Leaver, C.J.; Fernie, A.R. Protein degradation—An alternative respiratory substrate for stressed plants. Trends Plant Sci. 2011, 16, 489–498. [Google Scholar]
- Cunningham, D.F.; O’Connor, B. Proline specific peptidases. Biochim. Biophys. Acta 1997, 1343, 160–186. [Google Scholar] [CrossRef]
- Mattioli, R.; Falasca, G.; Sabatini, S.; Altamura, M.M.; Costantino, P.; Trovato, M. The proline biosynthetic genes P5CS1 and P5CS2 play overlapping roles in Arabidopsis flower transition but not in embryo development. Physiol. Plant 2009, 137, 72–85. [Google Scholar] [CrossRef]
- Shoresh, M.; Harman, G.E. The molecular basis of shoot responses of maize seedlings to Trichoderma. harzianum T22 inoculation of the root: A proteomic approach. Plant Physiol. 2008, 147, 2147–2163. [Google Scholar] [CrossRef]
- Bi, Y.-D.; Wei, Z.-G.; Shen, Z.; Lu, T.-C.; Cheng, Y.-X.; Wang, B.-C.; Yang, C.-P. Comparative temporal analyses of the Pinus. sylvestris L. var. mongolica litv. apical bud proteome from dormancy to growth. Mol. Biol. Rep. 2010, 38, 721–729. [Google Scholar]
- Tunnacliffe, A.; Wise, M.J. The continuing conundrum of the LEA proteins. Naturwissenschaften 2007, 94, 791–812. [Google Scholar] [CrossRef]
- Hundertmark, M.; Hincha, D.K. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics 2008, 9, 10–1186. [Google Scholar]
- Kalemba, E.M.; Pukacka, S. Association of protective proteins with dehydration and desiccation of orthodox and recalcitrant category seeds of three Acer genus species. J. Plant Growth Regul. 2012, 31, 351–362. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, W.; Gao, Y.; Zhao, Y.; Guo, L.; Wang, J. Proteomic analysis of embryo development in rice (Oryza. sativa). Planta 2012, 235, 687–701. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, J.H.; Kim, J.J.; Kim, C.-H.; Jun, S.-S.; Hong, Y.-N. Molecular and functional characterization of CaLEA6, the gene for a hydrophobic LEA protein from Capsicum annuum. Gene 2005, 344, 115–123. [Google Scholar] [CrossRef]
- Battaglia, M.; Olvera-Carrillo, Y.; Garciarrubio, A.; Campos, F.; Covarrubias, A.A. The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008, 148, 6–24. [Google Scholar] [CrossRef]
- Nick, P. The plant cytoskeleton—new jobs for a versatile network. Protoplasma 2007, 230, 125–127. [Google Scholar] [CrossRef]
- Parrotta, L.; Cai, G.; Cresti, M. Changes in the accumulation of α- and β-tubulin during bud development in Vitis. vinifera L. Planta 2010, 231, 277–291. [Google Scholar] [CrossRef]
- Pawłowski, T.A.; Bergervoet, J.H.W.; Bino, R.J.; Groot, S.P.C. Cell cycle activity and β-tubulin accumulation during dormancy breaking of Acer platanoides L. seeds. Biol. Plant 2004, 48, 211–218. [Google Scholar] [CrossRef]
- Elia, N.; Sougrat, R.; Spurlin, T.A.; Hurley, J.H.; Lippincott-Schwartz, J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. USA 2011, 108, 4846–4851. [Google Scholar]
- Nicolás, C.; Rodríguez, D.; Poulsen, F.; Eriksen, E.N.; Nicolás, G. The expression of an abscisic acid-responsive glycine-rich protein coincides with the level of seed dormancy in Fagus. sylvatica. Plant Cell. Physiol. 1997, 38, 1303–1310. [Google Scholar] [CrossRef]
- Mortensen, L.C.; Rodríguez, D.; Nicolás, G.; Eriksen, E.N.; Nicolás, C. Decline in a seed-specific abscisic acid-responsive glycine-rich protein (GRPF1) mRNA may reflect the release of seed dormancy in Fagus. sylvatica during moist prechilling. Seed Sci. Res. 2004, 14, 27–34. [Google Scholar]
- Auld, K.L.; Silver, P.A. Transcriptional regulation by the proteasome as a mechanism for cellular protein homeostasis. Cell Cycle 2006, 5, 1503–1505. [Google Scholar] [CrossRef]
- Smalle, J.; Kurepa, J.; Yang, P.; Emborg, T.J.; Babiychuk, E.; Kushnir, S.; Vierstra, R.D. The pleiotropic role of the 26S proteasome subunit RPN10 in Arabidopsis growth and development supports a substrate-specific function in abscisic acid signaling. Plant Cell 2003, 15, 965–980. [Google Scholar] [CrossRef]
- Liu, F.; Lu, C.-M. An overview of non-specific lipid transfer protein in plant (in Chinese). Yi Chuan/Hered./Zhongguo Yi Chuan Xue Hui Bian Ji 2013, 35, 307–314. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, A.J.; Liu, X.; Zheng, P.; Wilen, R.W.; Nesbitt, N.T.; Gusta, L.V. A lipid transfer protein gene BG-14 is differentially regulated by abiotic stress, ABA, anisomycin, and sphingosine in bromegrass (Bromus inermis). J. Plant Physiol. 2004, 161, 449–458. [Google Scholar] [CrossRef]
- Sterk, P.; Booij, H.; Schellekens, G.A.; van Kammen, A.; de Vries, S.C. Cell-specific expression of the carrot EP2 lipid transfer protein gene. Plant Cell. 1991, 3, 907–921. [Google Scholar] [CrossRef]
- Thoma, S.; Kaneko, Y.; Somerville, C. A non-specific lipid transfer protein from Arabidopsis is a cell wall protein. Plant J. 1993, 3, 427–436. [Google Scholar] [CrossRef]
- Ramagli, L.S.; Rodriguez, L.V. Quantitation of microgram amounts of protein in two-dimensional polyacrylamide gel electrophoresis sample buffer. Electrophoresis 1985, 6, 559–563. [Google Scholar] [CrossRef]
- Neuhoff, V.; Arold, N.; Taube, D.; Ehrhardt, W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 1988, 9, 255–262. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Staszak, A.M.; Pawłowski, T.A. Proteomic Analysis of Embryogenesis and the Acquisition of Seed Dormancy in Norway Maple (Acer platanoides L.). Int. J. Mol. Sci. 2014, 15, 10868-10891. https://doi.org/10.3390/ijms150610868
Staszak AM, Pawłowski TA. Proteomic Analysis of Embryogenesis and the Acquisition of Seed Dormancy in Norway Maple (Acer platanoides L.). International Journal of Molecular Sciences. 2014; 15(6):10868-10891. https://doi.org/10.3390/ijms150610868
Chicago/Turabian StyleStaszak, Aleksandra Maria, and Tomasz Andrzej Pawłowski. 2014. "Proteomic Analysis of Embryogenesis and the Acquisition of Seed Dormancy in Norway Maple (Acer platanoides L.)" International Journal of Molecular Sciences 15, no. 6: 10868-10891. https://doi.org/10.3390/ijms150610868
APA StyleStaszak, A. M., & Pawłowski, T. A. (2014). Proteomic Analysis of Embryogenesis and the Acquisition of Seed Dormancy in Norway Maple (Acer platanoides L.). International Journal of Molecular Sciences, 15(6), 10868-10891. https://doi.org/10.3390/ijms150610868