Association of TNFRSF10D DNA-Methylation with the Survival of Melanoma Patients

Abstract
:1. Introduction
2. Results and Discussion
2.1. Training Set for Selection of Relevant Genes
2.2. Test Set for the Validation of Relevant Genes

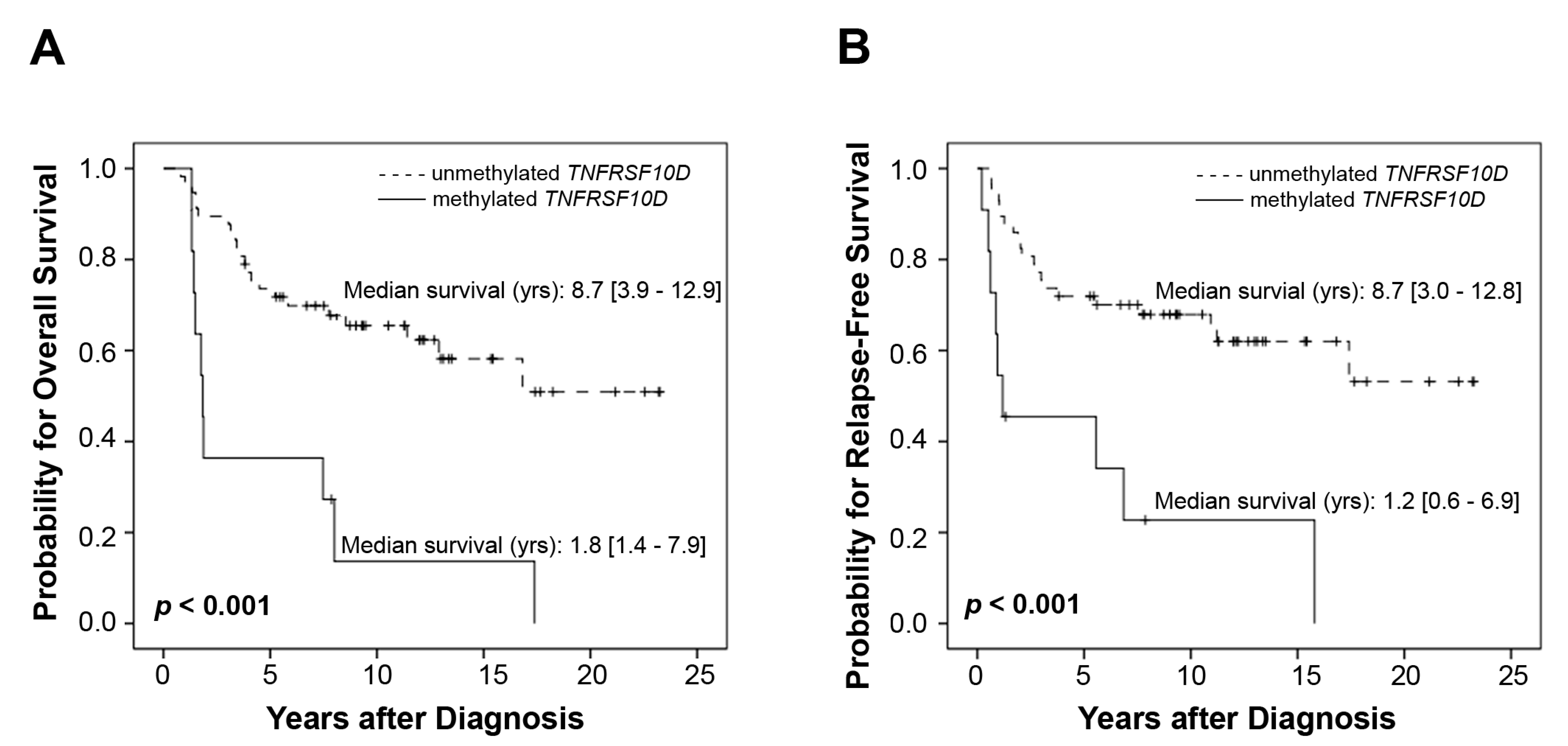{kind=link}
| (A) Training Set | ||||
|---|---|---|---|---|
| Variable | Overall Survival | Relapse-Free Survival | ||
| No. Patients (Died/Total) | p-Value (Log-Rank-Test) | No. Patients (Relapsed/Total) | p-Value (Log-Rank-Test) | |
| Sex | ||||
| Male | 12/24 | 0.712 | 11/24 | 0.717 |
| Female | 5/12 | 5/12 | ||
| Age | ||||
| <60 | 9/24 | 0.049 | 10/24 | 0.239 |
| ≥60 | 8/12 | 6/12 | ||
| Clark level | ||||
| 2/3 | 4/11 | 0.001 | 3/11 | <0.001 |
| 4 | 9/21 | 9/21 | ||
| 5 | 4/4 | 4/4 | ||
| Ulceration | ||||
| No | 7/22 | 0.015 | 7/22 | 0.046 |
| Yes | 10/14 | 9/14 | ||
| Tumor thickness | ||||
| ≤2 mm (T1/T2) | 5/13 | 0.008 | 6/13 | 0.008 |
| 2.01–4 mm (T3) | 2/8 | 1/8 | ||
| ≥4.01 mm (T4) | 10/15 | 9/15 | ||
| Mitotic rate | ||||
| ≤1/mm2 | 3/12 | 0.026 | 3/12 | 0.052 |
| >1/mm2 | 14/24 | 13/24 | ||
| Interferon alpha therapy | ||||
| No | 15/28 | 0.302 | 14/28 | 0.374 |
| Yes | 2/8 | 2/8 | ||
| TNFRSF10D | ||||
| Unmethylated | 11/29 | 0.001 | 11/29 | 0.008 |
| Methylated | 6/7 | 5/7 | ||
| (B) Test Set | ||||
| Sex | ||||
| Male | 5/12 | 0.929 | 5/12 | 0.98 |
| Female | 10/20 | 9/20 | ||
| Age | ||||
| <60 | 7/22 | 0.003 | 7/22 | 0.031 |
| ≥60 | 8/10 | 7/10 | ||
| Clark level | ||||
| 2/3 | 3/7 | <0.001 | 3/7 | <0.001 |
| 4 | 6/18 | 5/18 | ||
| 5 | 6/6 | 6/6 | ||
| Ulceration | ||||
| No | 11/23 | 0.887 | 10/23 | 0.942 |
| Yes | 4/9 | 4/9 | ||
| Tumor thickness | ||||
| ≤2 mm (T1/T2) | 5/12 | 0.919 | 5/12 | 0.992 |
| 2.01–4 mm (T3) | 7/13 | 6/13 | ||
| ≥4.01 mm (T4) | 2/5 | 2/5 | ||
| Mitotic rate | ||||
| ≤1/mm2 | 6/18 | 0.077 | 6/18 | 0.141 |
| >1/mm2 | 9/14 | 8/14 | ||
| Interferon alpha therapy | ||||
| No | 9/24 | 0.003 | 8/24 | 0.002 |
| Yes | 6/8 | 6/8 | ||
| TNFRSF10D | ||||
| Unmethylated | 11/28 | 0.041 | 10/28 | 0.012 |
| Methylated | 4/4 | 4/4 | ||
| Sex | ||||
| Male | 17/36 | 0.683 | 16/36 | 0.734 |
| Female | 15/32 | 14/32 | ||
| Age | ||||
| <60 | 16/46 | <0.001 | 13/22 | 0.013 |
| ≥60 | 16/22 | 23/74 | ||
| Clark level | ||||
| 2/3 | 7/18 | <0.001 | 6/18 | 0.011 |
| 4 | 15/39 | 14/39 | ||
| 5 | 10/10 | 10/10 | ||
| Ulceration | ||||
| No | 18/45 | 0.085 | 17/45 | 0.116 |
| Yes | 14/23 | 13/23 | ||
| Tumor thickness | ||||
| ≤2 mm (T1/T2) | 10/25 | 0.084 | 11/25 | 0.113 |
| 2.01–4 mm (T3) | 9/21 | 7/21 | ||
| ≥4.01 mm (T4) | 12/20 | 11/20 | ||
| Mitotic rate | ||||
| ≤1/mm2 | 9/30 | 0.005 | 9/30 | 0.012 |
| >1/mm2 | 23/38 | 21/38 | ||
| Interferon alpha therapy | ||||
| No | 24/52 | 0.261 | 22/52 | 0.24 |
| Yes | 8/16 | 8/16 | ||
| TNFRSF10D | ||||
| Unmethylated | 22/57 | <0.001 | 21/57 | <0.001 |
| Methylated | 10/11 | 9/11 | ||
2.3. Overall Prognostic Significance Merging the Training and the Test Set

| Variable | Overall Survival | Relapse-Free Survival | ||
|---|---|---|---|---|
| RR of Death (95% CI) | p-Value | RR of Relapse or Progression (95% CI) | p-Value | |
| Sex | ||||
| Male | 0.5 (0.2–1.6) | 0.11 | 0.4 (0.2–0.9) | 0.03 |
| Female | ||||
| Age | ||||
| <60 | 2.1 (0.9–4.6) | 0.07 | 1.6 (0.75–4.1) | 0.20 |
| ≥60 | ||||
| Clark level | ||||
| 2/3 | 3.0 (1.6–5.9) | 0.001 | 4.9 (2.4–10.1) | <0.001 |
| 4 | ||||
| 5 | ||||
| Ulceration | ||||
| No | 1.4 (0.6–3.5) | 0.47 | 1.1 (0.5–2.8) | 0.78 |
| Yes | ||||
| Tumor thickness | ||||
| ≤2 mm (T1/T2) | 0.9 (0.6–1.6) | 0.82 | 0.8 (0.5–1.3) | 0.39 |
| 2.01–4 mm (T3) | ||||
| ≥4.01 mm (T4) | ||||
| Mitotic rate | ||||
| ≤1/mm2 | 1.4 (0.5–3.9) | 0.49 | 1.6 (0.6–4.5) | 0.37 |
| >1/mm2 | ||||
| Interferon alpha therapy | ||||
| No | 2.2 (0.8–5.9) | 0.11 | 2.7 (1.1–7.5) | 0.03 |
| Yes | ||||
| TNFRSF10D | ||||
| Unmethylated | 4.6 (2.0–11.0) | <0.001 | 7.2 (2.8–18.3) | <0.001 |
| Methylated | ||||
2.4. Discussion
3. Experimental Section
3.1. The Patient Study Cohort and Study Design
3.2. DNA Extraction and Bisulfite Conversion from Formalin-Fixed Paraffin-Embedded (FFPE) Tissues
3.3. DNA Methylation Analysis
3.4. Mitotic Rate
3.5. Statistical Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Spatz, A.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2012. Eur. J. Cancer 2012, 48, 2375–2390. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar]
- Weinlich, G.; Eisendle, K.; Hassler, E.; Baltaci, M.; Fritsch, P.O.; Zelger, B. Metallothionein—Overexpression as a highly significant prognostic factor in melanoma: A prospective study on 1270 patients. Br. J. Cancer 2006, 94, 835–841. [Google Scholar]
- Jönsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringnér, M.; Lillehaug, J.R.; Borg, A.; Lønning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Spugnardi, M.; Tommasi, S.; Dammann, R.; Pfeifer, G.P.; Hoon, D.S. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003, 63, 1639–1643. [Google Scholar]
- Hoon, D.S.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004, 23, 4014–4022. [Google Scholar]
- Liu, S.; Ren, S.; Howell, P.; Fodstad, O.; Riker, A.I. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008, 21, 545–558. [Google Scholar] [CrossRef]
- Kuphal, S.; Martyn, A.C.; Pedley, J.; Crowther, L.M.; Bonazzi, V.F.; Parsons, P.G.; Bosserhoff, A.K.; Hayward, N.K.; Boyle, G.M. H-cadherin expression reduces invasion of malignant melanoma. Pigment Cell Melanoma Res. 2009, 22, 296–306. [Google Scholar]
- Muthusamy, V.; Duraisamy, S.; Bradbury, C.M.; Hobbs, C.; Curley, D.P.; Nelson, B.; Bosenberg, M. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res. 2006, 66, 11187–11193. [Google Scholar]
- Bonazzi, V.F.; Irwin, D.; Hayward, N.K. Identification of candidate tumor suppressor genes inactivated by promoter methylation in melanoma. Genes Chromosomes Cancer 2009, 48, 10–21. [Google Scholar] [CrossRef]
- Mori, T.; Martinez, S.R.; O’Day, S.J.; Morton, D.L.; Umetani, N.; Kitago, M.; Tanemura, A.; Nguyen, S.L.; Tran, A.N.; Wang, H.J.; et al. Estrogen receptor-α methylation predicts melanoma progression. Cancer Res. 2006, 66, 6692–6698. [Google Scholar] [CrossRef]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Körner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef]
- Schultz, J.; Ibrahim, S.M.; Vera, J.; Kunz, M. 14-3-3σ gene silencing during melanoma progression and its role in cell cycle control and cellular senescence. Mol. Cancer 2009, 8, 53. [Google Scholar] [CrossRef]
- Bonazzi, V.F.; Nancarrow, D.J.; Stark, M.S.; Moser, R.J.; Boyle, G.M.; Aoude, L.G.; Schmidt, C.; Hayward, N.K. oss-platform array screening identifies COL1A2; THBS1; TNFRSF10D and UCHL1 as genes frequently silenced by methylation in melanoma. PLoS One 2011, 6, e26121. [Google Scholar]
- Shen, L.; Kondo, Y.; Ahmed, S.; Boumber, Y.; Konishi, K.; Guo, Y.; Chen, X.; Vilaythong, J.N.; Issa, J.P. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res. 2007, 67, 11335–11343. [Google Scholar] [CrossRef]
- Wischnewski, F.; Pantel, K.; Schwarzenbach, H. Promoter demethylation and histone acetylation mediate gene expression of MAGE-A1; -A2; -A3; and -A12 in human cancer cells. Mol. Cancer Res. 2006, 4, 339–349. [Google Scholar] [CrossRef]
- Yagyu, S.; Gotoh, T.; Iehara, T.; Miyachi, M.; Katsumi, Y.; Tsubai-Shimizu, S.; Kikuchi, K.; Tamura, S.; Tsuchiya, K.; Imamura, T.; et al. Circulating methylated-DCR2 gene in serum as an indicator of prognosis and therapeutic efficacy in patients with MYCN nonamplified neuroblastoma. Clin. Cancer Res. 2008, 21, 7011–7019. [Google Scholar]
- Shivapurkar, N.; Toyooka, S.; Toyooka, K.O.; Reddy, J.; Miyajima, K.; Suzuki, M.; Shigematsu, H.; Takahashi, T.; Parikh, G.; Pass, H.I.; et al. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int. J. Cancer 2004, 109, 786–792. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Fortunato, C.; Oteri, R.; Teti, D.; Venza, I. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem. Biophys. Res. Commun. 2013, 441, 743–750. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. Statistics subcommittee of the NCI–EORTC working group on cancer diagnostics: Reporting recommendations for tumour marker prognostic studies (REMARK). J. Natl. Cancer Inst. 2005, 97, 1180–1184. [Google Scholar] [CrossRef]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Med. 2012, 10, 51. [Google Scholar] [CrossRef]
- Ehrlich, M.; Jiang, G.; Fiala, E.; Dome, J.S.; Yu, M.C.; Long, T.I.; Youn, B.; Sohn, O.S.; Widschwendter, M.; Tomlinson, G.E.; et al. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene 2002, 21, 6694–6702. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ratzinger, G.; Mitteregger, S.; Wolf, B.; Berger, R.; Zelger, B.; Weinlich, G.; Fritsch, P.; Goebel, G.; Fiegl, H. Association of TNFRSF10D DNA-Methylation with the Survival of Melanoma Patients. Int. J. Mol. Sci. 2014, 15, 11984-11995. https://doi.org/10.3390/ijms150711984
Ratzinger G, Mitteregger S, Wolf B, Berger R, Zelger B, Weinlich G, Fritsch P, Goebel G, Fiegl H. Association of TNFRSF10D DNA-Methylation with the Survival of Melanoma Patients. International Journal of Molecular Sciences. 2014; 15(7):11984-11995. https://doi.org/10.3390/ijms150711984
Chicago/Turabian StyleRatzinger, Gudrun, Simone Mitteregger, Barbara Wolf, Regina Berger, Bernhard Zelger, Georg Weinlich, Peter Fritsch, Georg Goebel, and Heidelinde Fiegl. 2014. "Association of TNFRSF10D DNA-Methylation with the Survival of Melanoma Patients" International Journal of Molecular Sciences 15, no. 7: 11984-11995. https://doi.org/10.3390/ijms150711984