Neighbor Preferences of Amino Acids and Context-Dependent Effects of Amino Acid Substitutions in Human, Mouse, and Dog

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
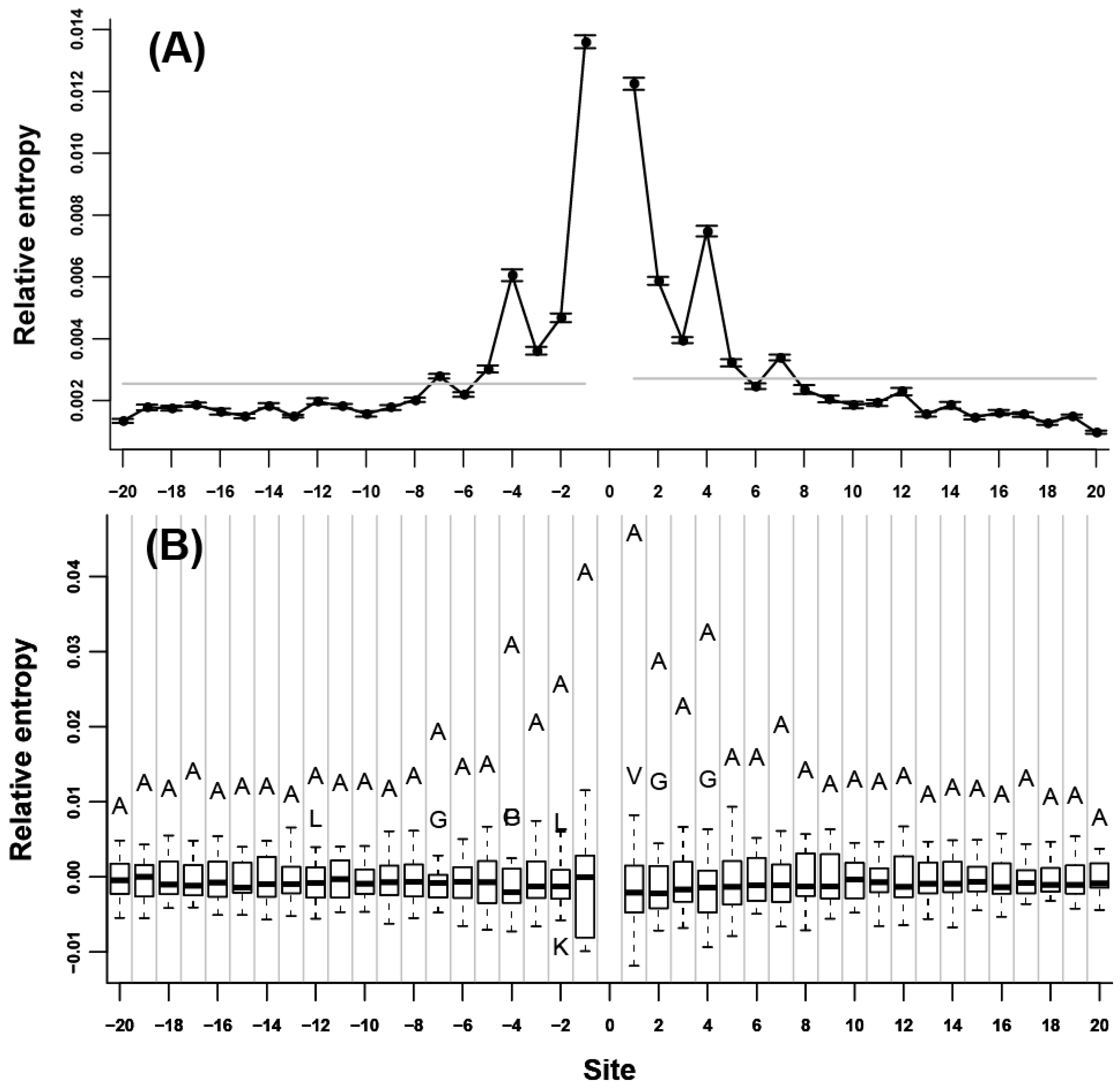
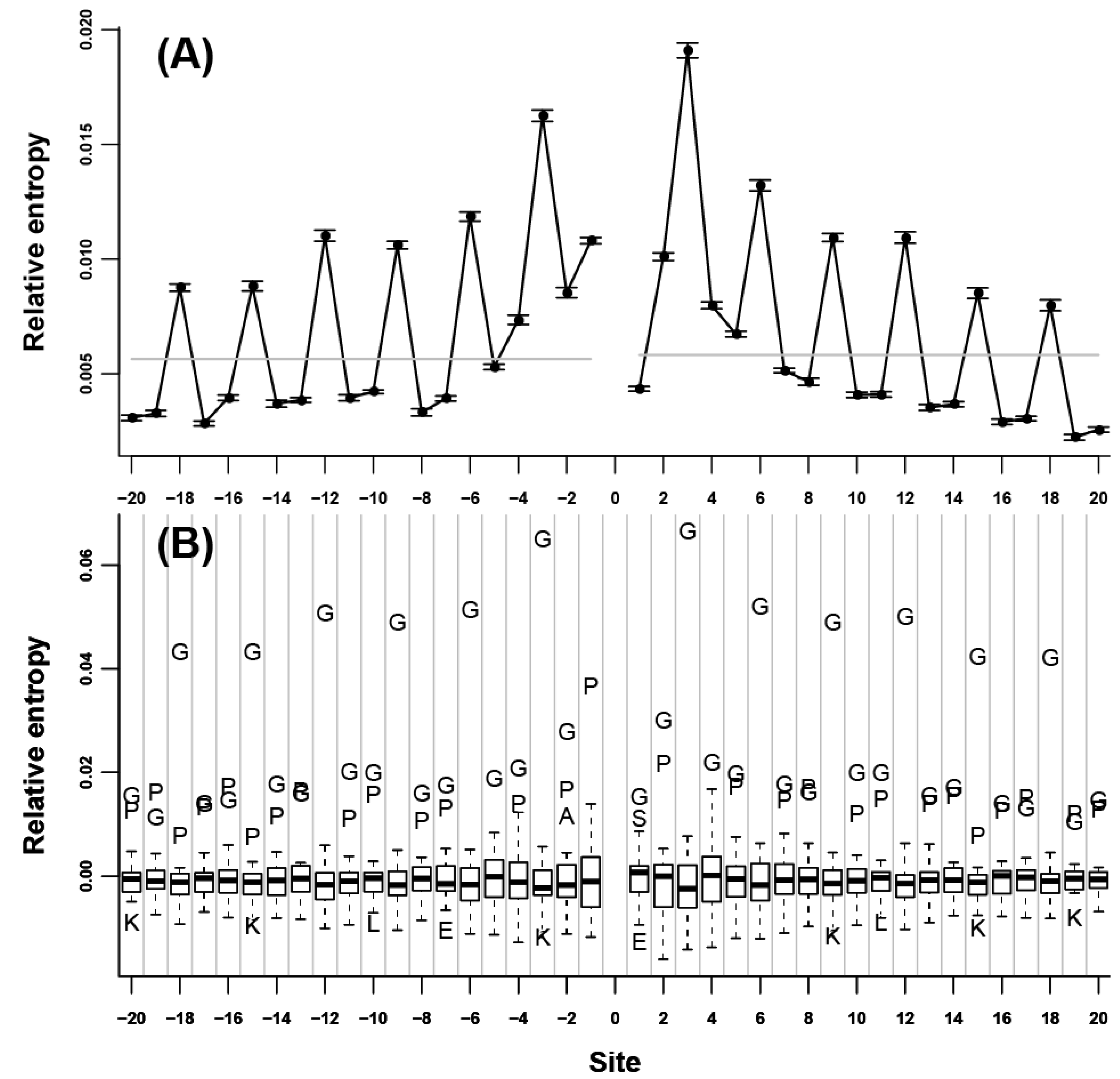
2.1. Neighbor Preferences of Amino Acids
 at each flanking site of the 20 amino acids (Figure 1B, Figure 2B, and Figure 3B). We found that for more than half of the amino acids (A, L, V, P, G, S, T, Q, C, H, K, R, D, and E), the corresponding residues tended to show high propensity at the neighboring sites (e.g., in Figure 1B, amino acid A was a outlier which was above the upper whisker of the boxplot in each flanking site, which demonstrated that amino acid A was a type of preferred amino acid in the neighboring sites of amino acid A). at each flanking site of the amino acid A. The bold line in each box represents the median of the 20 values. The top and bottom lines of each box indicate the upper and lower quartiles, respectively. The upper and lower whiskers represent the largest data point which was less than the sum of the upper quartile plus 1.5 times the interquartile range (IQR), and the lowest data point which was greater than the lower quartile minus 1.5 IQR, respectively. In order to determine which amino acids were apparently preferred (or not preferred) in the neighboring sites, the outliers were represented by the corresponding one letter codes of amino acids.
at each flanking site of the amino acid A. The bold line in each box represents the median of the 20 values. The top and bottom lines of each box indicate the upper and lower quartiles, respectively. The upper and lower whiskers represent the largest data point which was less than the sum of the upper quartile plus 1.5 times the interquartile range (IQR), and the lowest data point which was greater than the lower quartile minus 1.5 IQR, respectively. In order to determine which amino acids were apparently preferred (or not preferred) in the neighboring sites, the outliers were represented by the corresponding one letter codes of amino acids.
at each flanking site of the 20 amino acids (Figure 1B, Figure 2B, and Figure 3B). We found that for more than half of the amino acids (A, L, V, P, G, S, T, Q, C, H, K, R, D, and E), the corresponding residues tended to show high propensity at the neighboring sites (e.g., in Figure 1B, amino acid A was a outlier which was above the upper whisker of the boxplot in each flanking site, which demonstrated that amino acid A was a type of preferred amino acid in the neighboring sites of amino acid A). at each flanking site of the amino acid A. The bold line in each box represents the median of the 20 values. The top and bottom lines of each box indicate the upper and lower quartiles, respectively. The upper and lower whiskers represent the largest data point which was less than the sum of the upper quartile plus 1.5 times the interquartile range (IQR), and the lowest data point which was greater than the lower quartile minus 1.5 IQR, respectively. In order to determine which amino acids were apparently preferred (or not preferred) in the neighboring sites, the outliers were represented by the corresponding one letter codes of amino acids.
at each flanking site of the amino acid A. The bold line in each box represents the median of the 20 values. The top and bottom lines of each box indicate the upper and lower quartiles, respectively. The upper and lower whiskers represent the largest data point which was less than the sum of the upper quartile plus 1.5 times the interquartile range (IQR), and the lowest data point which was greater than the lower quartile minus 1.5 IQR, respectively. In order to determine which amino acids were apparently preferred (or not preferred) in the neighboring sites, the outliers were represented by the corresponding one letter codes of amino acids.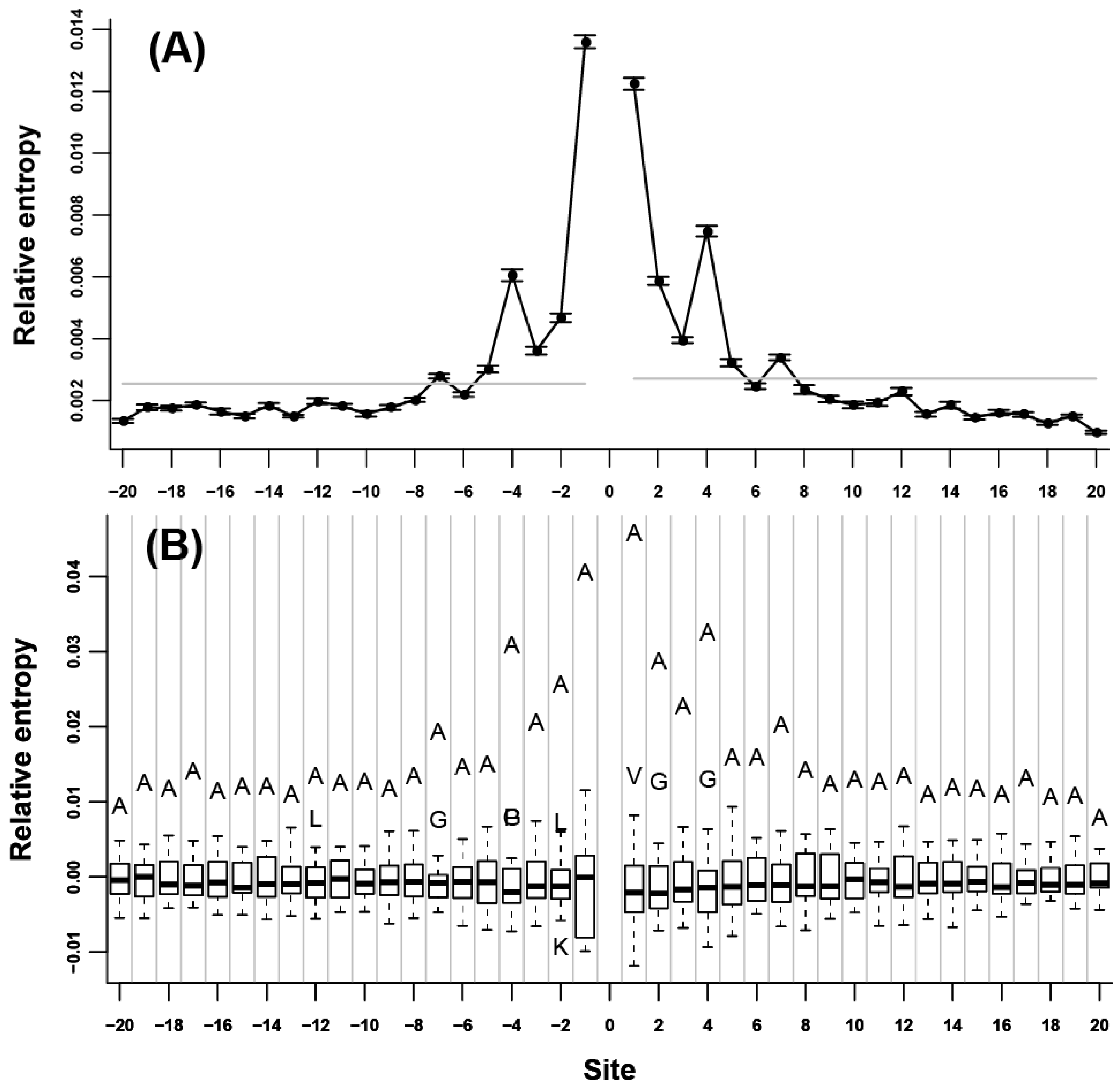

2.2. Neighbor Preferences of Amino Acids in Different Protein Secondary Structures

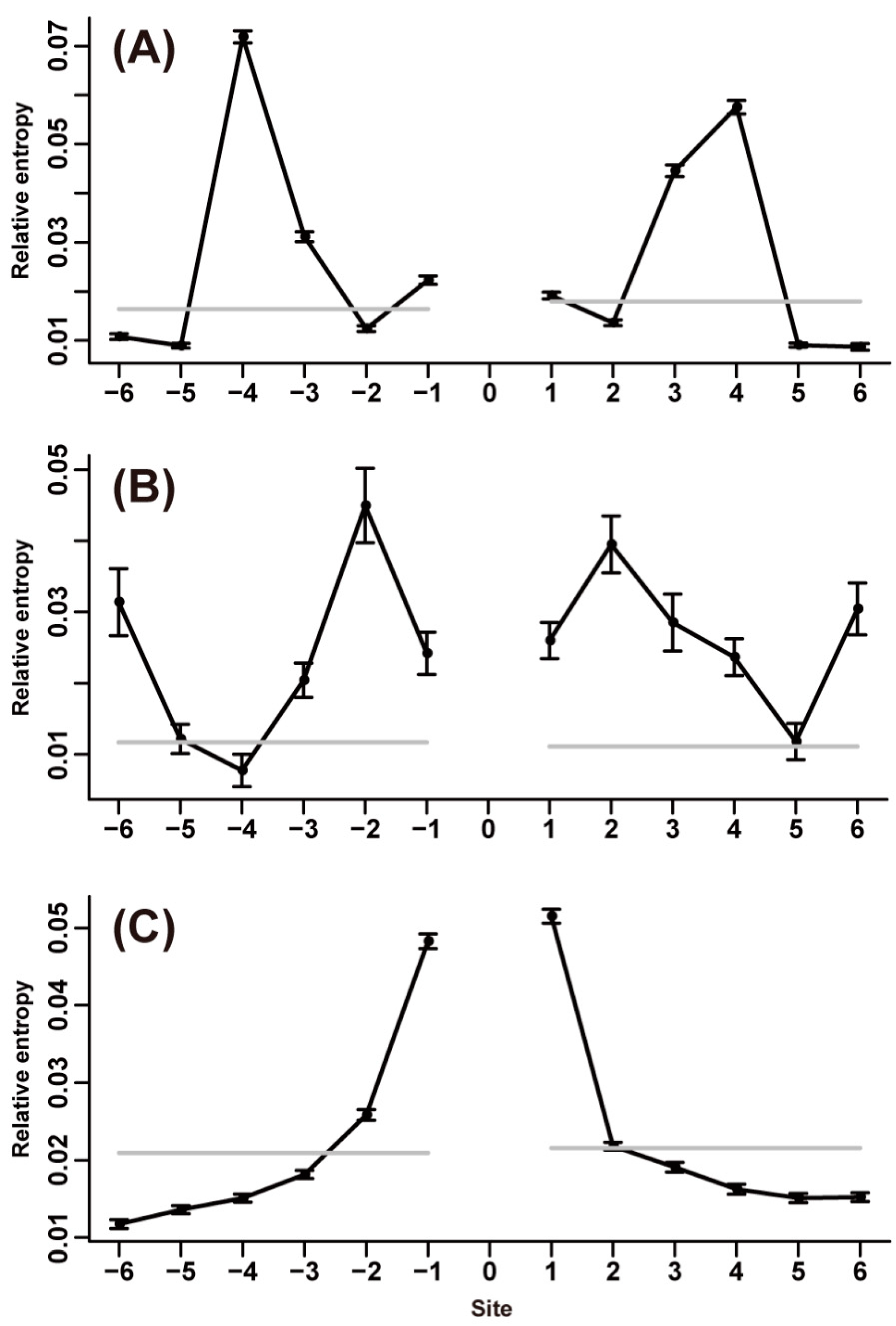
2.3. Context-Dependent Effects of Amino Acid Substitutions

2.4. Context-Dependent Effects of Amino Acid Substitutions in Different Protein Secondary Structures

3. Discussion
4. Experimental Section
4.1. Neighbor Preferences of Amino Acids
4.2. Context-Dependent Effects of Amino Acid Substitutions
4.3. Calculation of Relative Entropy


4.4. Prediction of Protein Secondary Structures
4.5. Prediction of Protein Cellular Locations
5. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dwyer, D.S. Nearest-neighbor effects and structural preferences in dipeptides are a function of the electronic properties of amino acid side-chains. Proteins 2006, 63, 939–948. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar] [CrossRef]
- Chou, P.Y. Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 47, 45–148. [Google Scholar]
- Chou, P.Y.; Fasman, G.D. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978, 47, 251–276. [Google Scholar] [CrossRef]
- Chakrabartty, A.; Kortemme, T.; Baldwin, R.L. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 1994, 3, 843–852. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Baldwin, R.L. Helix-stabilizing interaction between tyrosine and leucine or valine when the spacing is i, i + 4. J. Mol. Biol. 1994, 241, 706–713. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Baldwin, R.L. Tests for helix-stabilizing interactions between various nonpolar side chains in alanine-based peptides. Protein Sci. 1994, 3, 1992–1997. [Google Scholar] [CrossRef]
- Andrew, C.D.; Penel, S.; Jones, G.R.; Doig, A.J. Stabilizing nonpolar/polar side-chain interactions in the α-helix. Proteins Struct. Funct. Bioinform. 2001, 45, 449–455. [Google Scholar] [CrossRef]
- Wang, J.; Feng, J.A. Exploring the sequence patterns in the α-helices of proteins. Protein Eng. 2003, 16, 799–807. [Google Scholar] [CrossRef]
- Fonseca, N.A.; Camacho, R.; Magalhães, A.L. Amino acid pairing at the N- and C-termini of helical segments in proteins. Proteins Struct. Funct. Bioinform. 2008, 70, 188–196. [Google Scholar]
- De Sousa, M.M.; Munteanu, C.R.; Pazos, A.; Fonseca, N.A.; Camacho, R.; Magalhães, A. Amino acid pair-and triplet-wise groupings in the interior of α-helical segments in proteins. J. Theor. Biol. 2011, 271, 136–144. [Google Scholar]
- Wouters, M.A.; Curmi, P.M. An analysis of side chain interactions and pair correlations within antiparallel beta-sheets: The differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins 1995, 22, 119–131. [Google Scholar] [CrossRef]
- Hutchinson, E.G.; Sessions, R.B.; Thornton, J.M.; Woolfson, D.N. Determinants of strand register in antiparallel β-sheets of proteins. Protein Sci. 1998, 7, 2287–2300. [Google Scholar] [CrossRef]
- Fooks, H.; Martin, A.; Woolfson, D.; Sessions, R.; Hutchinson, E. Amino acid pairing preferences in parallel β-sheets in proteins. J. Mol. Biol. 2006, 356, 32–44. [Google Scholar] [CrossRef]
- Crasto, C.J.; Feng, J. Sequence codes for extended conformation: A neighbor-dependent sequence analysis of loops in proteins. Proteins 2001, 42, 399–413. [Google Scholar] [CrossRef]
- George, R.A.; Heringa, J. An analysis of protein domain linkers: Their classification and role in protein folding. Protein Eng. 2002, 15, 871–879. [Google Scholar] [CrossRef]
- Blake, R.D.; Hess, S.T.; Nicholson-Tuell, J. The influence of nearest neighbors on the rate and pattern of spontaneous point mutations. J. Mol. Evol. 1992, 34, 189–200. [Google Scholar] [CrossRef]
- Zhao, Z.; Boerwinkle, E. Neighboring-nucleotide effects on single nucleotide polymorphisms: A study of 2.6 million polymorphisms across the human genome. Genome Res. 2002, 12, 1679–1686. [Google Scholar] [CrossRef]
- Nevarez, P.A.; DeBoever, C.M.; Freeland, B.J.; Quitt, M.A.; Bush, E.C. Context dependent substitution biases vary within the human genome. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef]
- Panchin, A.; Mitrofanov, S.; Alexeevski, A.; Spirin, S.; Panchin, Y. New words in human mutagenesis. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef]
- Baele, G.; van de Peer, Y.; Vansteelandt, S. Modelling the ancestral sequence distribution and model frequencies in context-dependent models for primate non-coding sequences. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Baele, G. Context-dependent evolutionary models for non-coding sequences: An overview of several decades of research and an analysis of laurasiatheria and primate evolution. Evol. Biol. 2012, 39, 61–82. [Google Scholar] [CrossRef]
- Baele, G.; van de Peer, Y.; Vansteelandt, S. Using non-reversible context-dependent evolutionary models to study substitution patterns in primate non-coding sequences. J. Mol. Evol. 2010, 71, 34–50. [Google Scholar] [CrossRef]
- Hwang, D.G.; Green, P. Bayesian markov chain monte carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 13994–14001. [Google Scholar] [CrossRef]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar]
- Miyata, T.; Miyazawa, S.; Yasunaga, T. Two types of amino acid substitutions in protein evolution. J. Mol. Evol. 1979, 12, 219–236. [Google Scholar] [CrossRef]
- Dayhoff, M.O. A model of evolutionary change in proteins. Atlas Protein Seq. Struct. 1978, 5, 345–352. [Google Scholar]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Overington, J.; Johnson, M.S.; Sali, A.; Blundell, T.L. Tertiary structural constraints on protein evolutionary diversity: Templates, key residues and structure prediction. Proc. R. Soc. Lond. B 1990, 241, 132–145. [Google Scholar] [CrossRef]
- Luthy, R.; McLachlan, A.D.; Eisenberg, D. Secondary structure-based profiles: Use of structure-conserving scoring tables in searching protein sequence databases for structural similarities. Proteins 1991, 10, 229–239. [Google Scholar] [CrossRef]
- Overington, J.; Donnelly, D.; Johnson, M.S.; Šali, A.; Blundell, T.L. Environment-specific amino acid substitution tables: Tertiary templates and prediction of protein folds. Protein Sci. 1992, 1, 216–226. [Google Scholar]
- Topham, C.M.; McLeod, A.; Eisenmenger, F.; Overington, J.P.; Johnson, M.S.; Blundell, T.L. Fragment ranking in modelling of protein structure: Conformationally constrained environmental amino acid substitution tables. J. Mol. Biol. 1993, 229, 194–220. [Google Scholar] [CrossRef]
- Thorne, J.L.; Goldman, N.; Jones, D.T. Combining protein evolution and secondary structure. Mol. Biol. Evol. 1996, 13, 666–673. [Google Scholar] [CrossRef]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar]
- Robinson, D.M.; Jones, D.T.; Kishino, H.; Goldman, N.; Thorne, J.L. Protein evolution with dependence among codons due to tertiary structure. Mol. Biol. Evol. 2003, 20, 1692–1704. [Google Scholar] [CrossRef]
- Rodrigue, N.; Lartillot, N.; Bryant, D.; Philippe, H. Site interdependence attributed to tertiary structure in amino acid sequence evolution. Gene 2005, 347, 207–217. [Google Scholar] [CrossRef]
- Kleinman, C.L.; Rodrigue, N.; Lartillot, N.; Philippe, H. Statistical potentials for improved structurally constrained evolutionary models. Mol. Biol. Evol. 2010, 27, 1546–1560. [Google Scholar]
- Rastogi, S.; Reuter, N.; Liberles, D.A. Evaluation of models for the evolution of protein sequences and functions under structural constraint. Biophys. Chem. 2006, 124, 134–144. [Google Scholar] [CrossRef]
- Grahnen, J.; Nandakumar, P.; Kubelka, J.; Liberles, D. Biophysical and structural considerations for protein sequence evolution. BMC Evol. Biol. 2011, 11, 361. [Google Scholar] [CrossRef]
- Rodrigue, N.; Philippe, H.; Lartillot, N. Assessing site-interdependent phylogenetic models of sequence evolution. Mol. Biol. Evol. 2006, 23, 1762–1775. [Google Scholar] [CrossRef]
- Liberles, D.A.; Teichmann, S.A.; Bahar, I.; Bastolla, U.; Bloom, J.; Bornberg-Bauer, E.; Colwell, L.J.; de Koning, A.P.; Dokholyan, N.V.; Echave, J.; et al. The interface of protein structure, protein biophysics, and molecular evolution. Protein Sci. Publ. Protein Soc. 2012, 21, 769–785. [Google Scholar] [CrossRef]
- Wang, G.Z.; Chen, L.L.; Zhang, H.Y. Neighboring-site effects of amino acid mutation. Biochem. Biophys. Res. Commun. 2007, 353, 531–534. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Nishizawa, M.; Nishizawa, K. Local-scale repetitiveness in amino acid use in eukaryote protein sequences: A genomic factor in protein evolution. Proteins Struct. Funct. Bioinform. 1999, 37, 284–292. [Google Scholar] [CrossRef]
- Mularoni, L.; Veitia, R.A.; Alba, M.M. Highly constrained proteins contain an unexpectedly large number of amino acid tandem repeats. Genomics 2007, 89, 316–325. [Google Scholar] [CrossRef]
- Haerty, W.; Golding, G.B. Genome-wide evidence for selection acting on single amino acid repeats. Genome Res. 2010, 20, 755–760. [Google Scholar] [CrossRef]
- Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Evolution and function of cag/polyglutamine repeats in protein–protein interaction networks. Nucleic Acids Res. 2012, 40, 4273–4287. [Google Scholar] [CrossRef]
- Toll-Riera, M.; Radó-Trilla, N.; Martys, F.; Albà, M.M. Role of low-complexity sequences in the formation of novel protein coding sequences. Mol. Biol. Evol. 2012, 29, 883–886. [Google Scholar] [CrossRef]
- Katti, M.V.; Sami-Subbu, R.; Ranjekar, P.K.; Gupta, V.S. Amino acid repeat patterns in protein sequences: Their diversity and structural-functional implications. Protein Sci. 2000, 9, 1203–1209. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef]
- Giddu, S.; Xu, F.; Nanda, V. Sequence recombination improves target specificity in a redesigned collagen peptide abc-type heterotrimer. Proteins Struct. Funct. Bioinform. 2013, 81, 386–393. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. David-ws: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef]
- DAVID. Available online: http://david.abcc.ncifcrf.gov/home.jsp (accessed on 27 August 2014).
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef]
- Mandel-Gutfreund, Y.; Gregoret, L.M. On the significance of alternating patterns of polar and non-polar residues in beta-strands. J. Mol. Biol. 2002, 323, 453–461. [Google Scholar] [CrossRef]
- Misawa, K.; Kikuno, R.F. Evaluation of the effect of cpg hypermutability on human codon substitution. Gene 2009, 431, 18–22. [Google Scholar] [CrossRef]
- Duncan, B.K.; Miller, J.H. Mutagenic deamination of cytosine residues in DNA. Nature 1980, 287, 560–561. [Google Scholar] [CrossRef]
- Hubbard, T.; Barker, D.; Birney, E.; Cameron, G.; Chen, Y.; Clark, L.; Cox, T.; Cuff, J.; Curwen, V.; Down, T.; et al. The ensembl genome database project. Nucleic Acids Res. 2002, 30, 38–41. [Google Scholar]
- Ensembl. Available online: http://www.ensembl.org/index.html (accessed on 27 August 2014).
- Waterhouse, R.M.; Tegenfeldt, F.; Li, J.; Zdobnov, E.M.; Kriventseva, E.V. Orthodb: A hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013, 41, D358–D365. [Google Scholar]
- OrthoDB. Available online: http://orthodb.org/orthodb7 (accessed on 27 August 2014).
- Higgins, D.G.; Sharp, P.M. Clustal: A package for performing multiple sequence alignment on a microcomputer. Gene 1988, 73, 237–244. [Google Scholar] [CrossRef]
- Plotree, D.; Plotgram, D. PHYLIP-phylogeny inference package (version 3.2). Cladistics 1989, 5, 163–166. [Google Scholar] [CrossRef]
- Yang, Z. Paml: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi (accessed on 27 August 2014).
- Yang, Z. Computational molecular evolution; Oxford University Press: New York, NY, USA, 2006; Volume 284. [Google Scholar]
- Ma, L.; Zhang, T.; Huang, Z.; Jiang, X.; Tao, S. Patterns of nucleotides that flank substitutions in human orthologous genes. BMC Genomics 2010, 11, 416. [Google Scholar] [CrossRef]
- Cole, C.; Barber, J.D.; Barton, G.J. The jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008, 36, W197–W201. [Google Scholar] [CrossRef]
- Cuff, J.A.; Barton, G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins Struct. Funct. Bioinform. 2000, 40, 502–511. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. Scop: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar]
- Consortium, U. The universal protein resource (uniprot). Nucleic Acids Res. 2008, 36, D190–D195. [Google Scholar] [CrossRef]
- Advanced Jpred. Available online: http://www.compbio.dundee.ac.uk/www-jpred/advanced.html (accessed on 27 August 2014).
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- WoLF PSORT. Available online: http://www.genscript.com/psort/wolf_psort.html (accessed on 27 August 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fu, M.; Huang, Z.; Mao, Y.; Tao, S. Neighbor Preferences of Amino Acids and Context-Dependent Effects of Amino Acid Substitutions in Human, Mouse, and Dog. Int. J. Mol. Sci. 2014, 15, 15963-15980. https://doi.org/10.3390/ijms150915963
Fu M, Huang Z, Mao Y, Tao S. Neighbor Preferences of Amino Acids and Context-Dependent Effects of Amino Acid Substitutions in Human, Mouse, and Dog. International Journal of Molecular Sciences. 2014; 15(9):15963-15980. https://doi.org/10.3390/ijms150915963
Chicago/Turabian StyleFu, Mingchuan, Zhuoran Huang, Yuanhui Mao, and Shiheng Tao. 2014. "Neighbor Preferences of Amino Acids and Context-Dependent Effects of Amino Acid Substitutions in Human, Mouse, and Dog" International Journal of Molecular Sciences 15, no. 9: 15963-15980. https://doi.org/10.3390/ijms150915963