
Autophagy Induction by Silibinin Positively Contributes to Its Anti-Metastatic Capacity via AMPK/mTOR Pathway in Renal Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
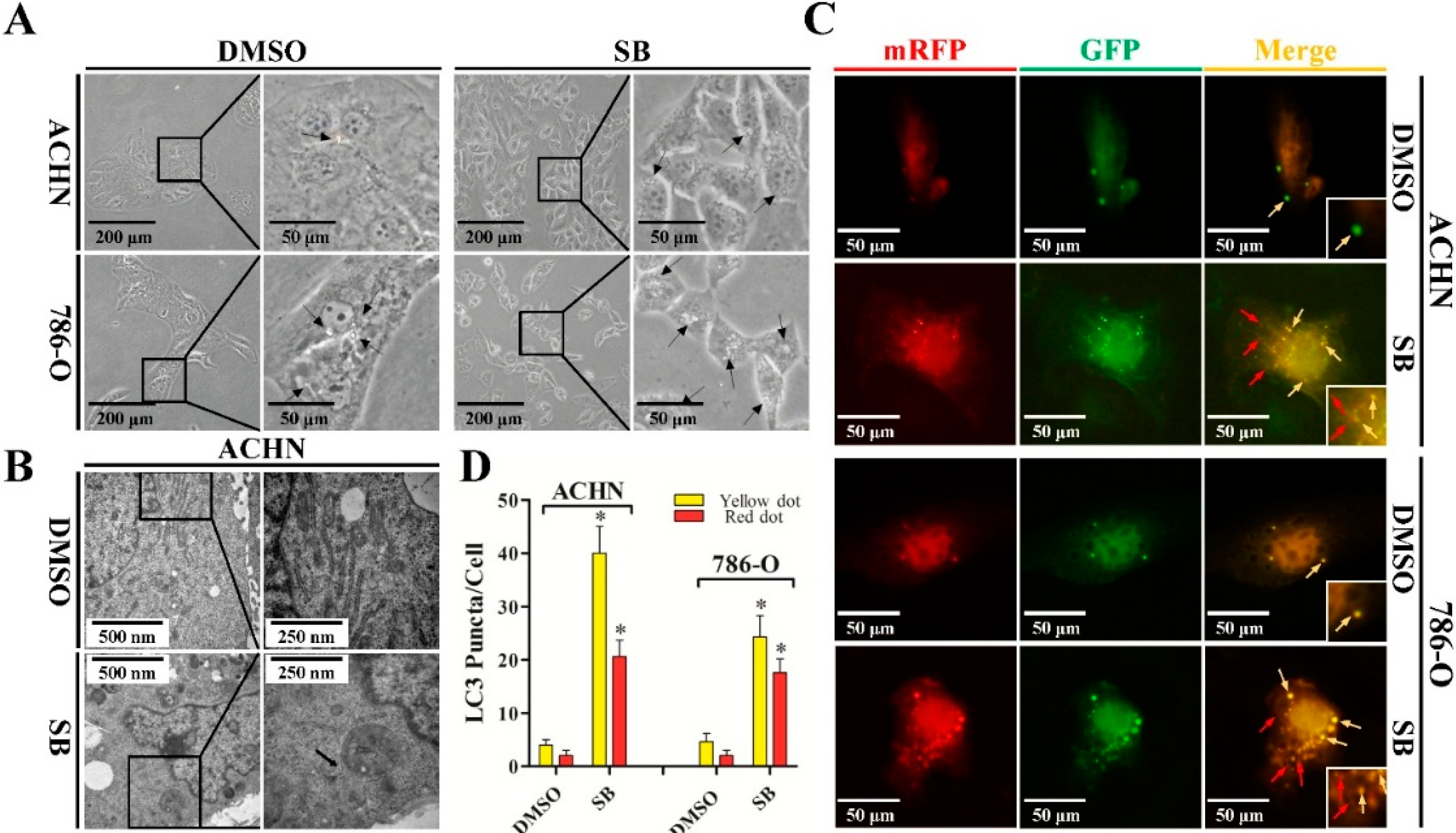
2.1. Silibinin Induces Autophagic Vacuoles in Renal Cell Carcinoma (RCC) ACHN and 786-O Cell Lines

2.2. Silibinin Induces Autophagic Flux in RCC ACHN and 786-O Cells

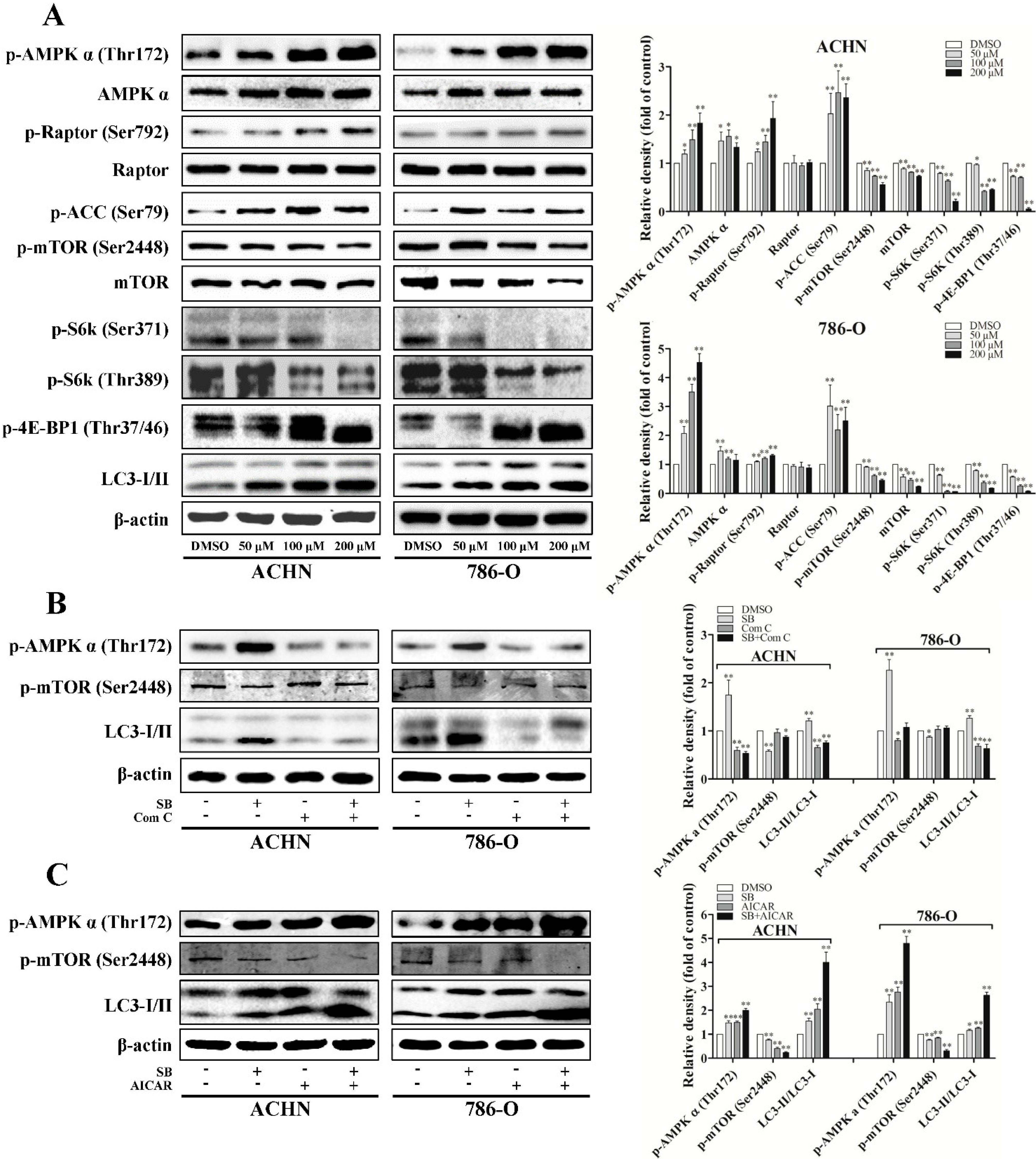
2.3. Silibinin Induces Autophagy through AMPK/Mammalian Target of Rapamycin (mTOR) Pathway

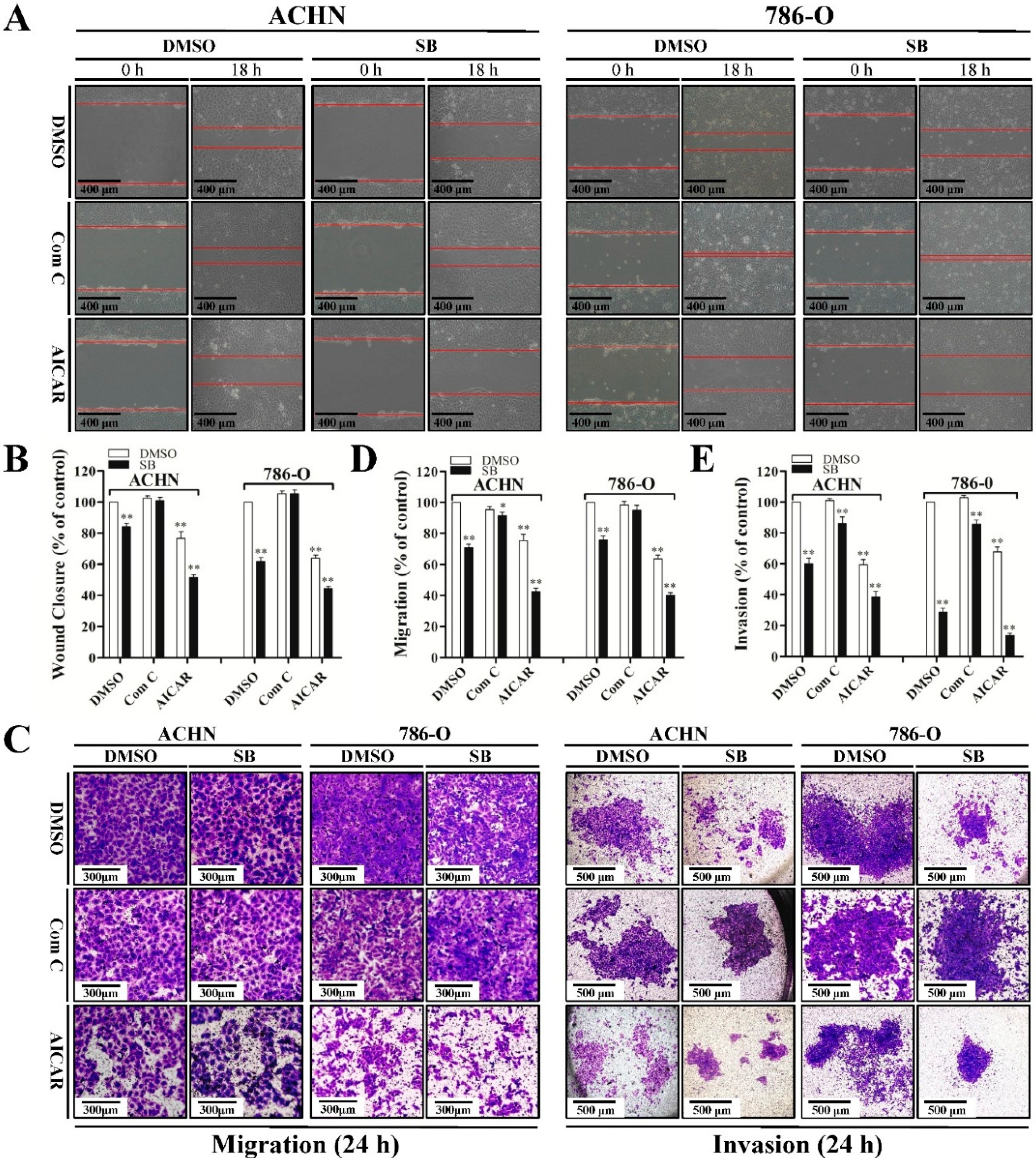
2.4. Autophagy Induced by Silibinin Contributes to Its Anti-Metastatic Effects

3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Regents, Antibodies, and Plasmid
4.3. Tandem Fluorescence Microscopy
4.4. Transmission Electron Microscopy
4.5. Western Blot
4.6. Wound Healing Assay
4.7. Trans-Well Migration and Invasion Assay
4.8. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef]
- Bhatt, J.R.; Finelli, A. Landmarks in the diagnosis and treatment of renal cell carcinoma. Nat. Rev. Urol. 2014, 11, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Cho, Y.M.; Lee, J.L.; Ahn, J.H.; Lee, D.H.; Song, C.; Hong, J.H.; Kim, C.S.; Ahn, H. Prognostic factors of metastatic renal cell carcinoma with extensive sarcomatoid component. J. Cancer Res. Clin. Oncol. 2013, 139, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Gao, J.; Rathmell, W.K. Renal cell carcinoma. BMJ 2014, 349, g4797. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.C.; Hamming, L.; Soetekouw, P.M.; Aarts, M.J.; Veeck, J.; van Engeland, M.; Tjan-Heijnen, V.C. Resistance to sunitinib in renal cell carcinoma: From molecular mechanisms to predictive markers and future perspectives. Biochim. Biophys. Acta 2014, 1855, 1–16. [Google Scholar] [PubMed]
- Rini, B.I. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.K.; Agarwal, C.; Chan, D.C.; Agarwal, R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol. Rep. 2004, 11, 493–499. [Google Scholar] [PubMed]
- Kumar, S.; Raina, K.; Agarwal, C.; Agarwal, R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell-enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget 2014, 5, 4972–4989. [Google Scholar] [PubMed]
- Mateen, S.; Raina, K.; Agarwal, R. Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutr. Cancer 2013, 65, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Sun, Y.; Wu, K.; Li, L.; Zhang, G.; Yang, Z.; Wang, Z.; Zhang, D.; Xue, Y.; Chen, Y.; et al. Chemopreventive and chemotherapeutic effects of intravesical silibinin against bladder cancer by acting on mitochondria. Mol. Cancer Ther. 2011, 10, 104–116. [Google Scholar]
- Li, L.; Gao, Y.; Zhang, L.; Zeng, J.; He, D.; Sun, Y. Silibinin inhibits cell growth and induces apoptosis by caspase activation, down-regulating survivin and blocking EGFR-ERK activation in renal cell carcinoma. Cancer Lett. 2008, 272, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Zeng, J.; Zhu, G.D.; Zhang, L.L.; Zhang, D.; Li, L.; Fan, J.H.; Wang, X.Y.; He, D.L. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol. Sin. 2009, 30, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Chen, P.N.; Yang, S.F.; Sun, Y.S.; Wu, S.W.; Hung, T.W.; Lian, J.D.; Chu, S.C.; Hsieh, Y.S. Silibinin inhibits the invasion and migration of renal carcinoma 786-O cells in vitro, inhibits the growth of xenografts in vivo and enhances chemosensitivity to 5-fluorouracil and paclitaxel. Mol. Carcinog. 2011, 50, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Li, L.; Zeng, J.; Gao, Y.; Chen, Y.L.; Wang, Z.Q.; Wang, X.Y.; Chang, L.S.; He, D. Inhibitory effect of silibinin on EGFR signal-induced renal cell carcinoma progression via suppression of the EGFR/MMP-9 signaling pathway. Oncol. Rep. 2012, 28, 999–1005. [Google Scholar] [PubMed]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, R.L.; Timpson, P.; Thorburn, J.; Anderson, K.I.; Thorburn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2014, 11, 2022–2029. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; Del, R.L.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.J.; Li, Q.S.; Xia, M.Y.; Tashiro, S.; Onodera, S.; Ikejima, T. Silibinin activated p53 and induced autophagic death in human fibrosarcoma HT1080 cells via reactive oxygen species-p38 and c-Jun N-terminal kinase pathways. Biol. Pharm. Bull. 2011, 34, 47–53. [Google Scholar] [CrossRef]
- Duan, W.; Jin, X.; Li, Q.; Tashiro, S.; Onodera, S.; Ikejima, T. Silibinin induced autophagic and apoptotic cell death in HT1080 cells through a reactive oxygen species pathway. J. Pharmacol. Sci. 2010, 113, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Li, L.; Chen, S.; Yu, Y.; Qi, M.; Tashiro, S.; Onodera, S.; Ikejima, T. Silibinin induced-autophagic and apoptotic death is associated with an increase in reactive oxygen and nitrogen species in HeLa cells. Free Radic. Res. 2011, 45, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yepuri, G.; Forbiteh, M.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy 2014, 10, 2223–2238. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Lippens, S.; Vanden, B.T.; Romagnoli, A.; Fimia, G.M.; Piacentini, M.; Vandenabeele, P. Beclin1: A role in membrane dynamics and beyond. Autophagy 2012, 8, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Yin, J.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Huang, S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKα leading to apoptosis of neuronal cells. Lab. Investig. 2010, 90, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Otkur, W.; Li, L.; Wang, Q.; He, H.; Ye, Y.; Zhang, Y.; Hayashi, T.; Tashiro, S.; Onodera, S.; et al. Autophagy induced by silibinin protects human epidermoid carcinoma A431 cells from UVB-induced apoptosis. J. Photochem. Photobiol. B 2013, 123, 23–31. [Google Scholar]
- Liu, W.; Otkur, W.; Zhang, Y.; Li, Q.; Ye, Y.; Zang, L.; He, H.; Hayashi, T.; Tashiro, S.; Onodera, S.; et al. Silibinin protects murine fibroblast L929 cells from UVB-induced apoptosis through the simultaneous inhibition of ATM-p53 pathway and autophagy. FEBS J. 2013, 280, 4572–4584. [Google Scholar]
- Raina, K.; Agarwal, C.; Wadhwa, R.; Serkova, N.J.; Agarwal, R. Energy deprivation by silibinin in colorectal cancer cells: A double-edged sword targeting both apoptotic and autophagic machineries. Autophagy 2013, 9, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Choi, C.H.; Kim, T.H.; Kwon, C.H.; Woo, J.S.; Kim, Y.K. Silibinin inhibits glioma cell proliferation via Ca2+/ROS/MAPK-dependent mechanism in vitro and glioma tumor growth in vivo. Neurochem. Res. 2009, 34, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Hua, F.; Hu, Z.W. DEDD, a novel tumor repressor, reverses epithelial-mesenchymal transition by activating selective autophagy. Autophagy 2012, 8, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhu, H.; Gu, D.; Pan, X.; Qian, L.; Xue, B.; Yang, D.; Zhou, J.; Shan, Y. MiRNA-30a-mediated autophagy inhibition sensitizes renal cell carcinoma cells to sorafenib. Biochem. Biophys. Res. Commun. 2015, 459, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar]
- Santoni, M.; Pantano, F.; Amantini, C.; Nabissi, M.; Conti, A.; Burattini, L.; Zoccoli, A.; Berardi, R.; Santoni, G.; Tonini, G.; et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim. Biophys. Acta 2014, 1845, 221–231. [Google Scholar]
- Santoni, M.; de Giorgi, U.; Iacovelli, R.; Conti, A.; Burattini, L.; Rossi, L.; Luca Burgio, S.; Berardi, R.; Muzzonigro, G.; Cortesi, E.; et al. Pre-treatment neutrophil-to-lymphocyte ratio may be associated with the outcome in patients treated with everolimus for metastatic renal cell carcinoma. Br. J. Cancer 2013, 109, 1755–1759. [Google Scholar]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Ma, Z.; Guan, Z.; Chen, Y.; Wu, K.; Guo, P.; Wang, X.; He, D.; Zeng, J. Autophagy Induction by Silibinin Positively Contributes to Its Anti-Metastatic Capacity via AMPK/mTOR Pathway in Renal Cell Carcinoma. Int. J. Mol. Sci. 2015, 16, 8415-8429. https://doi.org/10.3390/ijms16048415
Li F, Ma Z, Guan Z, Chen Y, Wu K, Guo P, Wang X, He D, Zeng J. Autophagy Induction by Silibinin Positively Contributes to Its Anti-Metastatic Capacity via AMPK/mTOR Pathway in Renal Cell Carcinoma. International Journal of Molecular Sciences. 2015; 16(4):8415-8429. https://doi.org/10.3390/ijms16048415
Chicago/Turabian StyleLi, Feng, Zhenkun Ma, Zhenfeng Guan, Yule Chen, Kaijie Wu, Peng Guo, Xinyang Wang, Dalin He, and Jin Zeng. 2015. "Autophagy Induction by Silibinin Positively Contributes to Its Anti-Metastatic Capacity via AMPK/mTOR Pathway in Renal Cell Carcinoma" International Journal of Molecular Sciences 16, no. 4: 8415-8429. https://doi.org/10.3390/ijms16048415
APA StyleLi, F., Ma, Z., Guan, Z., Chen, Y., Wu, K., Guo, P., Wang, X., He, D., & Zeng, J. (2015). Autophagy Induction by Silibinin Positively Contributes to Its Anti-Metastatic Capacity via AMPK/mTOR Pathway in Renal Cell Carcinoma. International Journal of Molecular Sciences, 16(4), 8415-8429. https://doi.org/10.3390/ijms16048415