
Identification of miRNAs and Their Targets in Cotton Inoculated with Verticillium dahliae by High-Throughput Sequencing and Degradome Analysis

Abstract
:
1. Introduction
2. Results
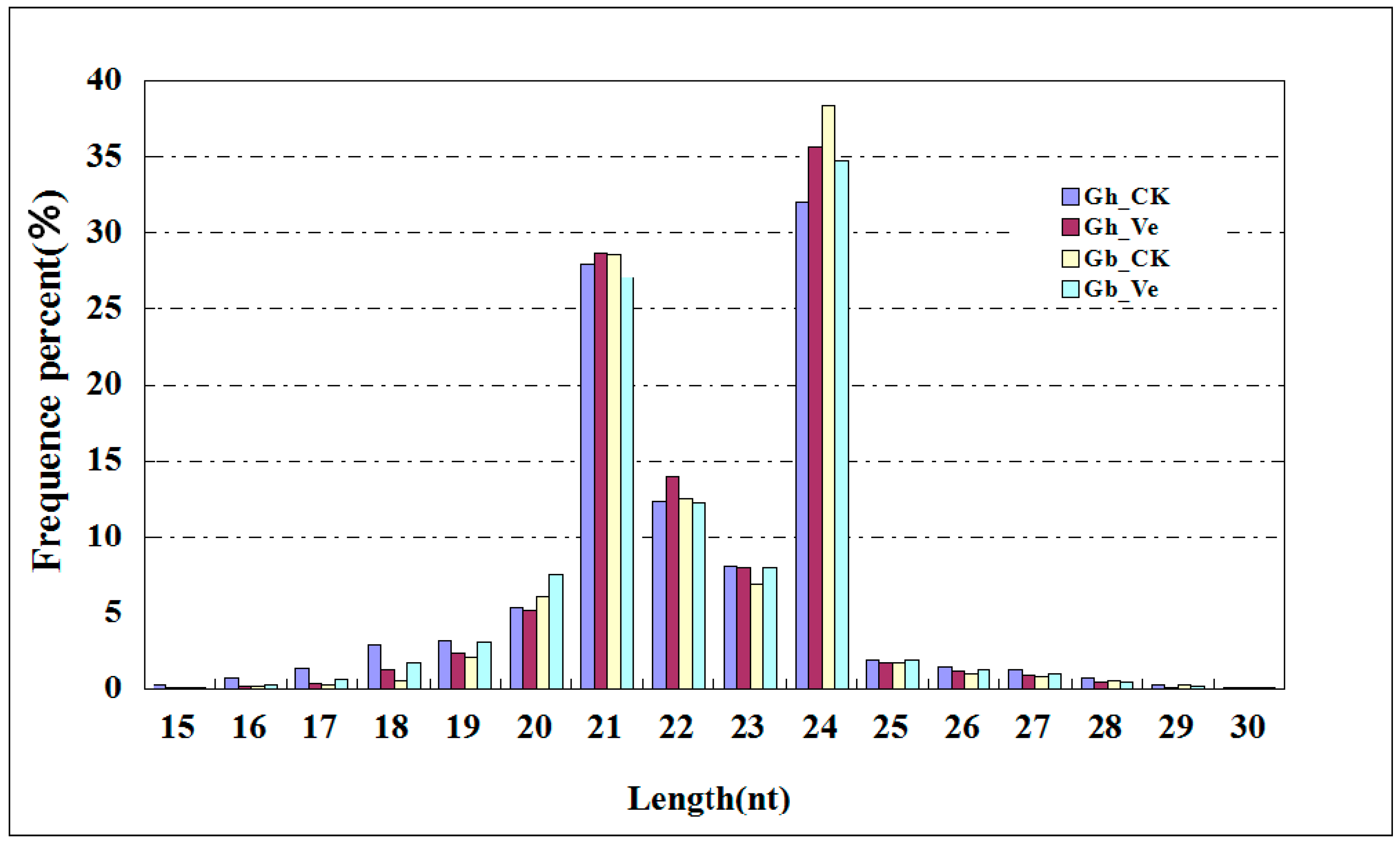
2.1. Overview of Small RNA (sRNA) Library Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Library Name | |||
|---|---|---|---|---|
| Gh_CK | Gh_Ve | Gb_CK | Gb_Ve | |
| Total reads | 26,495,780 | 19,511,637 | 20,609,252 | 19,965,211 |
| High quality | 26,409,519 | 19,450,186 | 20,548,220 | 19,903,524 |
| Clean reads | 25,679,650 | 19,296,249 | 20,389,102 | 19,638,276 |
| Unique sRNAs | 7,181,742 | 6,033,991 | 6,729,005 | 6,415,595 |
| Unique sRNA mapping to genome | 2,235,867 | 1,855,605 | 2,078,150 | 1,988,015 |
| miRNA | 37,936 | 29,080 | 32,658 | 34,630 |
| Unannotated | 6,264,557 | 5,281,380 | 5,889,075 | 5,583,553 |

2.2. Identification of Known MicroRNAs (miRNAs) by sRNA Sequencing
2.3. Identification of Novel miRNAs in G. hirsutum and G. barbadense
| miRNA | Mature Sequence | LM | Arm | LP | G + C (%) | MFE | Reads Per Million | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Gh_CK | Gh_Ve | Gb_CK | Gb_Ve | |||||||
| novel_miR_1 | TTGACAAGTAAAAGAACATA | 20 | 3p | 147 | 29.25 | −23.44 | 0 | 0.98 | 0.74 | 1.07 |
| novel_miR_2 | CGAAGTCTTGGAAGAGAGTAA | 21 | 3p | 91 | 37.36 | −39.7 | 49.65 | 0 | 24.82 | 30.3 |
| novel_miR_3 | TGGTATTGGAGTGAAGGGAGC | 21 | 5p | 70 | 37.14 | −21.1 | 50.9 | 73.64 | 68.57 | 44.5 |
| novel_miR_4 | TGATTGAGCCGTGCCAATATC | 21 | 3p | 101 | 42.57 | −53.9 | 115.81 | 141.22 | 231.64 | 163.25 |
| novel_miR_5 | GTGGGCGTGCCGGAGTGGTTA | 21 | 5p | 74 | 56.76 | −26.8 | 110.52 | 96.7 | 34.04 | 36.76 |
| novel_miR_6 | CGGACTCTCAAACAGTGGAGGTA | 23 | 5p | 256 | 45.31 | −108.08 | 7.63 | 5.44 | 0 | 12.78 |
| novel_miR_7 | CTGGACTGTCAATGGCCGGCAC | 22 | 3p | 95 | 45.26 | −35.4 | 0 | 0 | 2.45 | 1.63 |
| novel_miR_8 | ATTAGATACTATAGCATGAGACA | 23 | 5p | 289 | 24.57 | −46.7 | 3.66 | 0 | 0 | 9.01 |
| novel_miR_9 | CTGTCGCAGGGGAGATGGCTCGT | 23 | 5p | 141 | 58.16 | −66.1 | 9.27 | 8.24 | 0 | 0.51 |
| novel_miR_10 | TCTAATAGAAGAATGACAAATCA | 23 | 5p | 84 | 20.24 | −22 | 0 | 0.93 | 1.52 | 0.81 |
| novel_miR_11 | TGGGAAATGATGACAGCTTA | 20 | 3p | 193 | 29.53 | −44.9 | 4.24 | 0 | 0 | 1.22 |
| novel_miR_12 | GCAGATGATGATAAGAATAGACA | 23 | 5p | 140 | 40.71 | −33.3 | 2.26 | 2.23 | 1.77 | 0.66 |
| novel_miR_13 | GGGCGCCTCTTACTTGGCAGG | 21 | 5p | 132 | 44.7 | −56.5 | 0.9 | 4.3 | 0 | 7.13 |
| novel_miR_14 | ATTAGATACTATAGCGTGAGACA | 23 | 3p | 226 | 37.17 | −61.1 | 0 | 0 | 0 | 7.69 |
| novel_miR_15 | GCTCACTTCTCTTTCTGTCAGTT | 23 | 3p | 107 | 44.86 | −57.7 | 5.33 | 9.74 | 11.87 | 7.84 |
| novel_miR_16 | TGCCTGGCTCCCTGAATGCCA | 21 | 5p | 104 | 50 | −53.3 | 1.75 | 0.73 | 0.49 | 1.32 |
| novel_miR_17 | AGGTGCAGGTGCAGGCGCAGC | 21 | 3p | 146 | 40.41 | −49.62 | 0 | 0 | 7.9 | 1.53 |
| novel_miR_18 | CGGACCCTCTAACAGTGGAGG | 21 | 5p | 214 | 50.47 | −90.3 | 56.93 | 37.47 | 0 | 38.9 |
| novel_miR_19 | TGAGAAAGTGGAGATGGGTGG | 21 | 3p | 128 | 45.31 | −63.9 | 2.18 | 1.71 | 3.87 | 2.6 |
| novel_miR_20 | CTCTGGTTTGACTCATTTGTA | 21 | 5p | 181 | 33.7 | −66.8 | 3.12 | 0 | 0 | 1.63 |
| novel_miR_21 | CAAATGAGTTAGGCGAGAGGT | 21 | 3p | 170 | 33.53 | −63.56 | 0 | 13.42 | 0 | 7.08 |
| novel_miR_22 | TCAAGCAAATCAAGGAAAGGCC | 22 | 3p | 344 | 38.66 | −113.5 | 0.55 | 0.47 | 1.13 | 0.36 |
| novel_miR_23 | TCTGAAAAGCAATAAAGAACACA | 23 | 3p | 94 | 27.66 | −25.7 | 0 | 0 | 2.55 | 1.78 |
| novel_miR_24 | GTGGAAGTTAGGCTGACTTAGGC | 23 | 5p | 79 | 45.57 | −21.7 | 0 | 0 | 0 | 4.12 |
| novel_miR_25 | AGGCAGTCACCTTGGCTAAC | 20 | 5p | 180 | 45.56 | −64.6 | 0 | 4.98 | 0 | 1.68 |
| novel_miR_26 | TTGGATGGGCGGTGTGTTTACTT | 23 | 3p | 246 | 28.46 | −49.4 | 0 | 0 | 0.64 | 1.93 |
| novel_miR_27 | GGCAAGTTGTCCTCGGCTACA | 21 | 3p | 178 | 39.89 | −59.96 | 0 | 1.87 | 0 | 1.12 |
| novel_miR_28 | TCCATATTTCACTATCTCTTA | 21 | 3p | 105 | 32.38 | −48.6 | 11.25 | 16.01 | 72.54 | 22.66 |
| novel_miR_29 | TTGAACACCGAAGTAAAGCCAT | 22 | 5p | 209 | 41.15 | −82.8 | 0 | 0 | 0 | 1.78 |
| novel_miR_30 | TGCCAAATCAGGGAAGCGAA | 20 | 5p | 148 | 41.22 | −52.32 | 0 | 0 | 43.5 | 0 |
| novel_miR_31 | AGGCTGTGATGACGAGAAATTA | 22 | 3p | 237 | 30.38 | −47.71 | 32.94 | 24.1 | 16.92 | 0 |
| novel_miR_32 | TTTCCAATAGAAGAATGACAAAT | 23 | 5p | 140 | 27.14 | −54.8 | 1.36 | 1.09 | 1.03 | 0 |
| novel_miR_33 | AATCTCCCTCAAACGCTTCCAG | 22 | 5p | 118 | 45.76 | −47.84 | 0 | 0 | 9.66 | 0 |
| novel_miR_34 | TTTGGATTGAAGGGAGCTCTA | 21 | 3p | 203 | 42.36 | −92.45 | 0 | 0 | 141.2 | 0 |
| novel_miR_35 | TAGTGAGGATGGGAAATTTGT | 21 | 5p | 124 | 30.65 | −49.3 | 34.93 | 0 | 18.78 | 14 |
| novel_miR_36 | GAGCTTGGAAGTGCATCCGGC | 21 | 5p | 107 | 45.79 | −54.9 | 0 | 0 | 4.07 | 0 |
| novel_miR_37 | TCGCTTCCCTAATTTGGACGA | 21 | 3p | 148 | 41.22 | −52.32 | 36.33 | 31.09 | 0 | 85.09 |
| novel_miR_38 | TGACTCCTAGTACAACGGCCTC | 22 | 3p | 328 | 32.32 | −123.5 | 0 | 0 | 5.84 | 0 |
| novel_miR_39 | TTGAGCCGTGCCAATATCAATC | 22 | 3p | 108 | 47.22 | −49.81 | 0 | 0 | 226.44 | 0 |
| novel_miR_40 | CAAAGAGTAGAGGTATTGTGC | 21 | 5p | 272 | 41.54 | −112 | 0 | 13.89 | 1.23 | 0 |
| novel_miR_41 | ACAGGTTAGTAGAAATTAAGGTT | 23 | 5p | 115 | 41.74 | −22.6 | 0 | 1.09 | 0 | 0 |
| novel_miR_42 | CAGAATGACCAATTTACTCTTTA | 23 | 3p | 199 | 24.12 | −40.4 | 0 | 4.92 | 0 | 0 |
| novel_miR_43 | ACTCTCTTCCAAAGGCTTCAAG | 22 | 5p | 108 | 35.19 | −46 | 0 | 49.91 | 0 | 0 |
| novel_miR_44 | TGGTGCAGGTCGGGAACTGAT | 21 | 5p | 139 | 47.48 | −57.4 | 0 | 5.86 | 0 | 0 |
| novel_miR_45 | GTTCAATAAAGCTGTGGGAAG | 21 | 3p | 138 | 40.58 | −54.6 | 0 | 100.69 | 0 | 0 |
| novel_miR_46 | CAAAAGCAATGAAGAACTGGCCA | 23 | 5p | 331 | 34.14 | −78.9 | 0 | 1.97 | 0 | 0 |
| novel_miR_47 | ACAGTAGAAATGGATGGAATT | 21 | 3p | 153 | 30.07 | −48 | 0 | 3.99 | 0 | 0 |
| novel_miR_48 | TTGGATGGACGGTGCATTTATCT | 23 | 3p | 295 | 37.29 | −62.4 | 0 | 1.04 | 0 | 0 |
| novel_miR_49 | ATTATTGTTAATGTAGGAGGA | 21 | 5p | 371 | 31 | −61.26 | 0 | 1.92 | 0 | 0 |
| novel_miR_50 | TTGTACTAGGAGTCGGATTGC | 21 | 5p | 344 | 33.72 | −143.2 | 0 | 4.15 | 0 | 1.27 |
| novel_miR_51 | TTAGATCAAAGAGCAAACCGG | 21 | 5p | 210 | 36.67 | −95.5 | 1.32 | 0 | 0 | 0 |
| novel_miR_52 | CGAGACTTGCGGTAGAAACAAA | 22 | 3p | 149 | 40.27 | −35.2 | 6.74 | 0 | 0 | 0 |
| novel_miR_53 | TGGAGGCAGCGGTTCATCGATC | 22 | 5p | 110 | 42.73 | −40.1 | 0 | 0 | 0 | 12.37 |
| novel_miR_54 | GGGACTCTCCGGACTGTTTGGTT | 23 | 3p | 148 | 36.49 | −43.3 | 1.05 | 0 | 0 | 0 |
| novel_miR_55 | TGCCTGGCTCCCTGTATGCCA | 21 | 5p | 103 | 47.57 | −57.4 | 107.56 | 71.93 | 11.92 | 57.64 |
| novel_miR_56 | TTTTGCCAGCTCCGCCCATTCC | 22 | 3p | 122 | 40.98 | −45.01 | 66.24 | 87.89 | 0 | 1.73 |
| novel_miR_57 | CAGCCAAGGATGACTTGCCGG | 21 | 5p | 175 | 35.43 | −57.4 | 0.97 | 1.66 | 0 | 0.92 |
| novel_miR_58 | TTTTTTAGTAGAAGGAGCAAAAT | 23 | 5p | 254 | 32.68 | −56.8 | 0.35 | 0 | 0 | 0 |
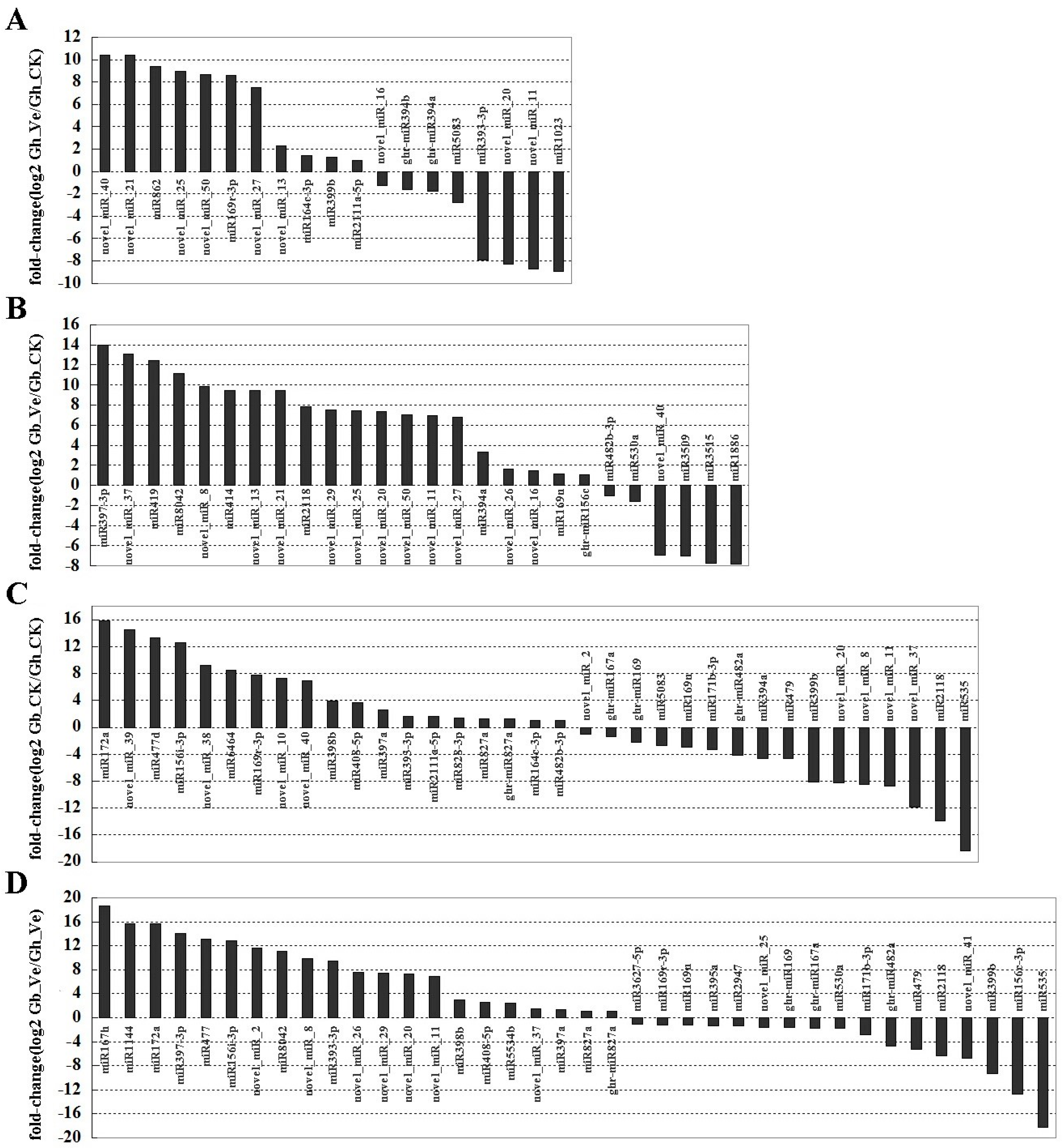
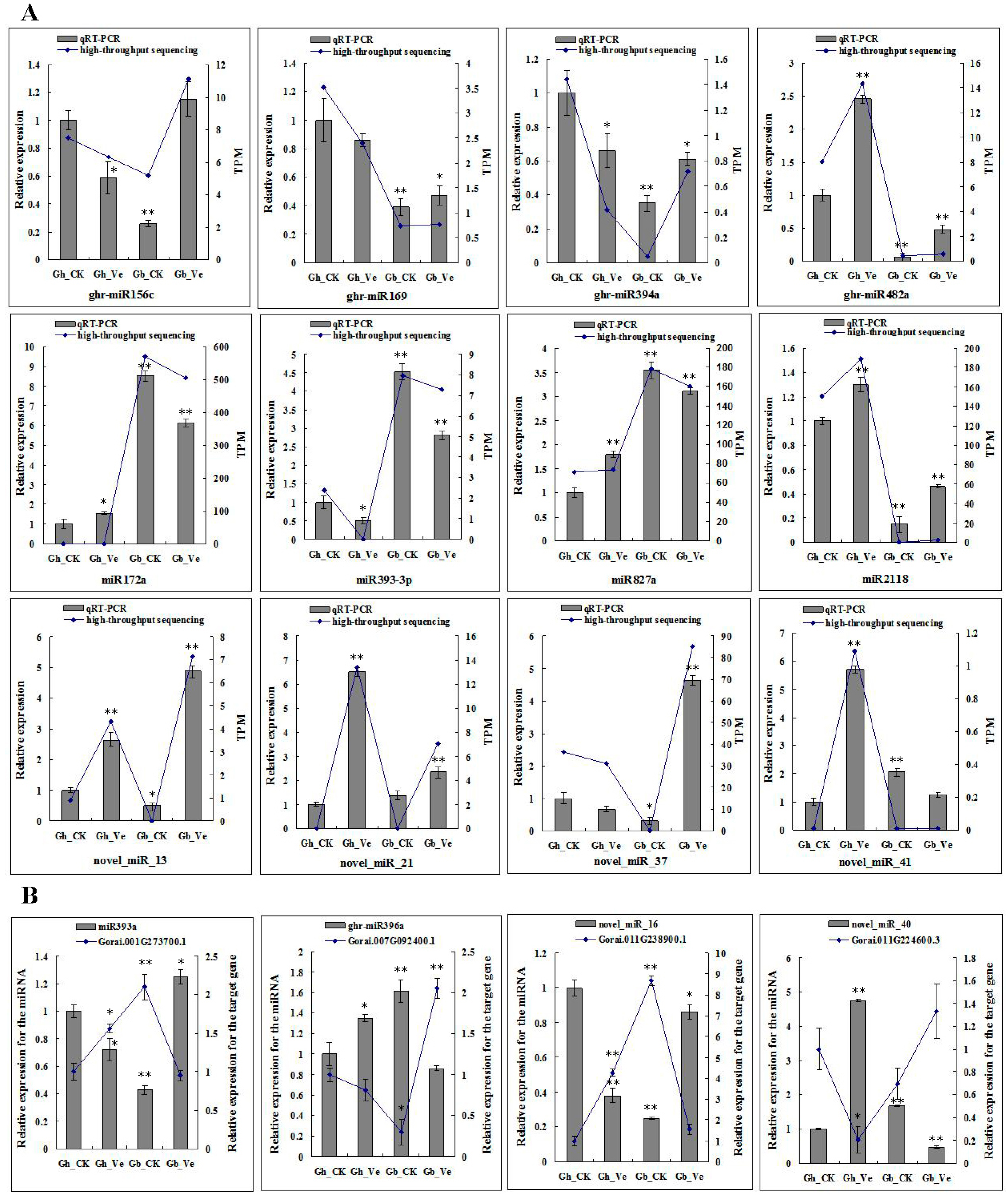
2.4. Expression Profiling of Differentially Expressed miRNAs in Response to V. dahliae Infection


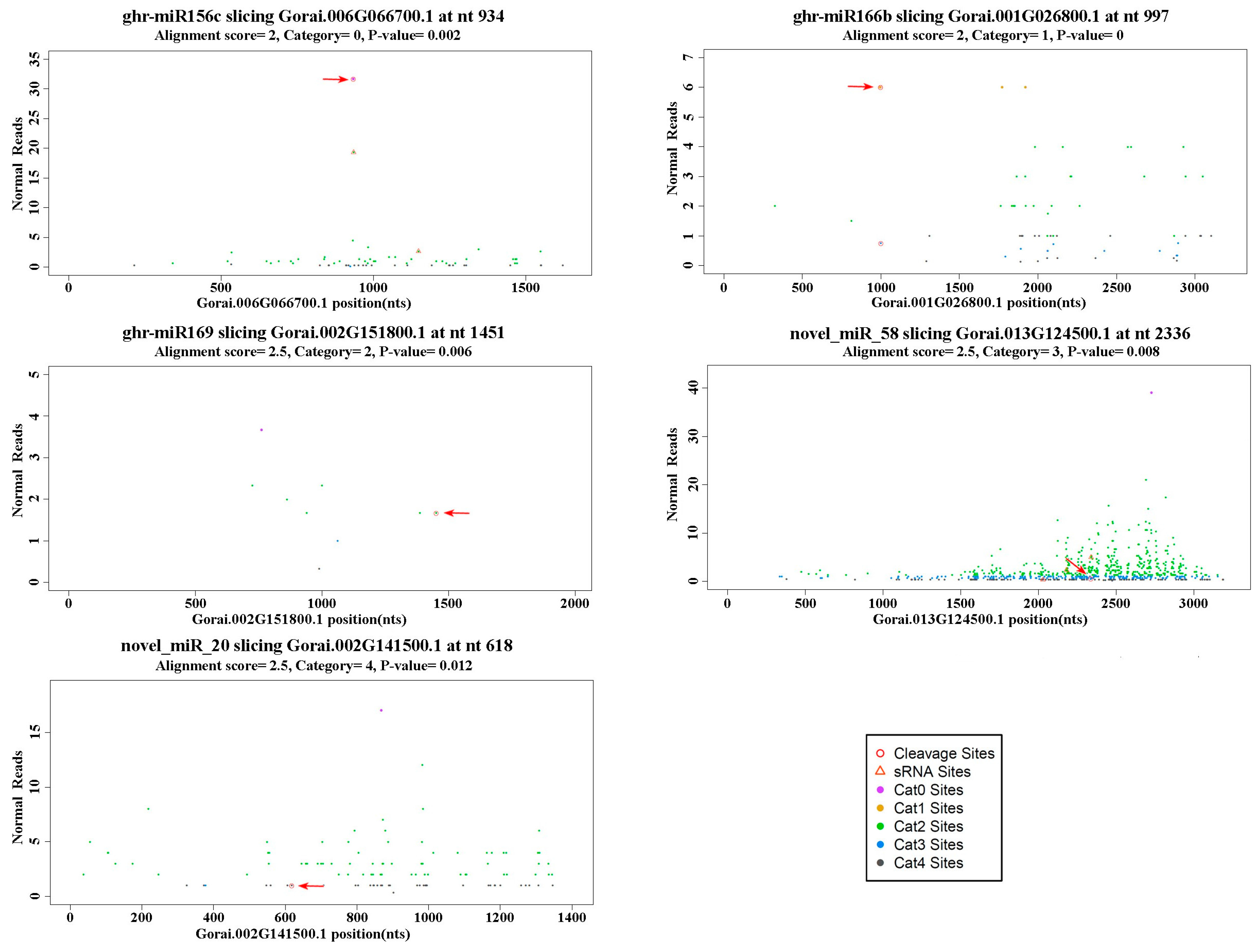
2.5. Target Genes of miRNAs Identified by Degradome Analysis

| miRNA | Target Gene | Cleavage Site | Categoty | Norm Reads | Target Description | |
|---|---|---|---|---|---|---|
| Gb | Gh | |||||
| novel_miR_12 | Gorai.005G020900.1 | 1117 | 1 | 0 | 3 | Ubiquitin carboxyl-terminal hydrolase family protein |
| novel_miR_16 | Gorai.003G142500.1 | 1315 | 0 | 58 | 54 | Auxin response factor 10 isoform 1 |
| Gorai.013G267100.1 | 1416 | 0 | 60 | 31 | Auxin response factor 10 isoform 1 | |
| Gorai.010G046000.1 | 1638 | 0 | 16 | 19 | Auxin response factor 17 | |
| Gorai.006G166400.1 | 1773 | 0 | 36 | 12 | Auxin response factor 19 | |
| Gorai.011G238900.1 | 1502 | 0 | 316 | 557 | Auxin response factor 19 | |
| Gorai.012G004800.1 | 1679 | 0 | 110 | 160 | Auxin response factor 19 | |
| novel_miR_20 | Gorai.002G141500.1 | 618 | 4 | 1 | 0 | Purine permease 3 |
| Gorai.003G066400.1 | 1545 | 4 | 1 | 0 | Ferrochelatase 2 isoform 1 | |
| Gorai.008G199200.1 | 1792 | 4 | 1 | 0 | Uncharacterized protein TCM_014264 | |
| novel_miR_21 | Gorai.009G146400.1 | 1251 | 0 | 7 | 0 | Uncharacterized protein TCM_029129 |
| novel_miR_24 | Gorai.006G008600.5 | 496 | 0 | 1 | 0 | TT12-2 MATE transporter |
| novel_miR_26 | Gorai.007G279100.4 | 319 | 2 | 0 | 2 | Intracellular protein transport protein USO1 |
| Gorai.012G074700.1 | 79 | 2 | 0 | 2 | Serine hydroxymethyltransferase 6 isoform 3 | |
| novel_miR_28 | Gorai.003G163700.1 | 1250 | 0 | 13 | 0 | Leucine-rich repeat containing protein |
| novel_miR_29 | Gorai.008G010200.1 | 855 | 2 | 7 | 5 | Glutathione S-transferase 7 isoform 1 |
| Gorai.008G120500.1 | 1587 | 3 | 0 | 2 | Fringe-related protein, putative | |
| novel_miR_30 | Gorai.005G163400.1 | 1744 | 0 | 28 | 0 | Uncharacterized protein TCM_010813 |
| novel_miR_40 | Gorai.011G224600.3 | 1555 | 1 | 0 | 1 | Inositol 1,3,4-trisphosphate 5/6-kinase 4 |
| novel_miR_48 | Gorai.007G279100.4 | 319 | 2 | 0 | 2 | Intracellular protein transport protein USO1 |
| Gorai.012G074700.1 | 79 | 2 | 0 | 2 | Serine hydroxymethyltransferase 6 isoform 3 | |
| novel_miR_56 | Gorai.007G319800.1 | 613 | 0 | 10 | 1 | LRR and NB-ARC domains-containing disease resistance-like protein |
| novel_miR_57 | Gorai.004G172100.1 | 1219 | 0 | 19 | 26 | Nuclear transcription factor Y subunit A-1, putative isoform 1 |
| novel_miR_58 | Gorai.013G124500.1 | 2336 | 3 | 1 | 1 | Bacterial-induced lipoxygenase |
3. Discussion
4. Experimental Section
4.1. Plant Material and Total RNA Isolation
4.2. sRNA Library and Degradome Library Construction and Sequencing
4.3. Analysis of Sequencing Data
4.4. qRT-PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Katiyar-Agarwal, S.; Morgan, R.; Dahlbeck, D.; Borsani, O.; Villegas, A.; Zhu, J.-K.; Staskawicz, B.J.; Jin, H. A pathogen-inducible endogenous siRNA in plant immunity. Proc. Natl. Acad. Sci. USA 2006, 103, 18002–18007. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, C.; Zhang, X.; Jin, H. Host small RNAs are big contributors to plant innate immunity. Curr. Opin. Plant Biol. 2009, 12, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, H.; Gao, S.; Wang, W.-C.; Katiyar-Agarwal, S.; Huang, H.-D.; Raikhel, N.; Jin, H. Arabidopsis argonaute 2 regulates innate immunity via miRNA393*-mediated silencing of a golgi-localized SNARE gene, MEMB12. Mol. Cell 2011, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Ellendorff, U.; Fradin, E.F.; De Jonge, R.; Thomma, B.P. H.J. RNA silencing is required for Arabidopsis defence against Verticillium wilt disease. J. Exp. Bot. 2009, 60, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ferrer, V.; Voinnet, O. Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 2009, 60, 485–510. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.G.; Subbarao, K.V. Host range specificity in Verticillium dahliae. Phytopathology 1999, 89, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Sink, K.C.; Grey, W.E. A root-injection method to assess verticillium wilt resistance of peppermint (Mentha × piperita L.) and its use in identifying resistant somaclones of cv. Black Mitcham. Euphytica 1999, 106, 223–230. [Google Scholar] [CrossRef]
- Hampton, R.E.; Wullschleger, S.D.; Oosterhuis, D.M. Impact of verticillium wilt on net photosynthesis, respiration and photorespiration in field-grown cotton ( Gossypium hirsutum L.). Physiol. Mol. Plant Pathol. 1990, 37, 271–280. [Google Scholar] [CrossRef]
- Cai, Y.; Xiaohong, H.; Mo, J.; Sun, Q.; Yang, J.; Liu, J. Molecular research and genetic engineering of resistance to Verticillium wilt in cotton: A review. Afr. J. Biotechnol. 2009, 8, 7363–7372. [Google Scholar]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhu, L.; Tu, L.; Liu, L.; Yuan, D.; Jin, L.; Long, L.; Zhang, X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J. Exp. Bot. 2011, 62, 5607–5621. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genomics 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Guo, W.; Li, G.; Gao, F.; Lin, S.; Zhang, T. QTLs mapping for Verticillium wilt resistance at seedling and maturity stages in Gossypium barbadense L. Plant Sci. 2008, 174, 290–298. [Google Scholar] [CrossRef]
- Yin, Z.; Li, Y.; Han, X.; Shen, F. Genome-wide profiling of miRNAs and other small non-coding RNAs in the Verticillium dahliae-inoculated cotton roots. PLoS ONE 2012, 7, e35765. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Woodward, A.W.; Agarwal, V.; Guan, X.; Ha, M.; Ramachandran, V.; Chen, X.; Triplett, B.A.; Stelly, D.M.; Chen, Z.J. Genome-wide analysis reveals rapid and dynamic changes in miRNA and siRNA sequence and expression during ovule and fiber development in allotetraploid cotton (Gossypium hirsutum L.). Genome Biol. 2009, 10, R122. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Bowman, J.L. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008, 13, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant microRNAs. Plant Cell Online 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Zheng, Y.; Addo-Quaye, C.; Zhang, L.; Saini, A.; Jagadeeswaran, G.; Axtell, M.J.; Zhang, W.; Sunkar, R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010, 62, 742–759. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar] [PubMed]
- Zhang, Y.; Wang, X.F.; Ding, Z.G.; Ma, Q.; Zhang, G.R.; Zhang, S.L.; Li, Z.K.; Wu, L.Q.; Zhang, G.Y.; Ma, Z.Y. Transcriptome profiling of Gossypium barbadense inoculated with Verticillium dahliae provides a resource for cotton improvement. BMC Genomics 2013, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jiang, H.; Zhu, X.; Wang, W.; He, X.; Shi, Y.; Yuan, Y.; Du, X.; Cai, Y. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genomics 2013, 14, 852. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liang, Z.; Yang, B.; Tian, H.; Ma, J.; Zhang, H. Derepression of microRNA-mediated protein translation inhibition by apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G (APOBEC3G) and its family members. J. Biol. Chem. 2007, 282, 33632–33640. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jue, D.; Li, W.; Zhang, R.; Chen, M.; Yang, Q. Identification of miRNA from eggplant (Solanum melongena L.) by small RNA deep sequencing and their response to Verticillium dahliae infection. PLoS ONE 2013, 8, e72840. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, Y.-H.; Shi, R.; Clark, C.; Li, L.; Chiang, V.L. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell Online 2005, 17, 2186–2203. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, Q.; Jiang, H.; Zhu, X.; Mo, J.; Long, L.; Xiang, L.; Xie, Y.; Shi, Y.; Yuan, Y.; Cai, Y. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. SpringerPlus 2014, 3, 564. [Google Scholar] [CrossRef] [PubMed]
- Llorente, F.; Muskett, P.; Sánchez-Vallet, A.; López, G.; Ramos, B.; Sánchez-Rodríguez, C.; Jordá, L.; Parker, J.; Molina, A. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol. Plant 2008, 1, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Carra, A.; Gambino, G.; Schubert, A. A cetyltrimethylammonium bromide-based method to extract low-molecular-weight RNA from polysaccharide-rich plant tissues. Anal. Biochem. 2007, 360, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- The mfold Web Server. Available online: http://mfold.rna.albany.edu/?q=mfold/download-mfold (accessed on 1 April 2014).
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, W.; Chen, J.; Liu, J.; Xia, M.; Shen, F. Identification of miRNAs and Their Targets in Cotton Inoculated with Verticillium dahliae by High-Throughput Sequencing and Degradome Analysis. Int. J. Mol. Sci. 2015, 16, 14749-14768. https://doi.org/10.3390/ijms160714749
Zhang Y, Wang W, Chen J, Liu J, Xia M, Shen F. Identification of miRNAs and Their Targets in Cotton Inoculated with Verticillium dahliae by High-Throughput Sequencing and Degradome Analysis. International Journal of Molecular Sciences. 2015; 16(7):14749-14768. https://doi.org/10.3390/ijms160714749
Chicago/Turabian StyleZhang, Yujuan, Wei Wang, Jie Chen, Jubo Liu, Minxuan Xia, and Fafu Shen. 2015. "Identification of miRNAs and Their Targets in Cotton Inoculated with Verticillium dahliae by High-Throughput Sequencing and Degradome Analysis" International Journal of Molecular Sciences 16, no. 7: 14749-14768. https://doi.org/10.3390/ijms160714749