
Salvianolic Acid B (Sal B) Protects Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death by Activating Glutaredoxin 1 (Grx1)


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
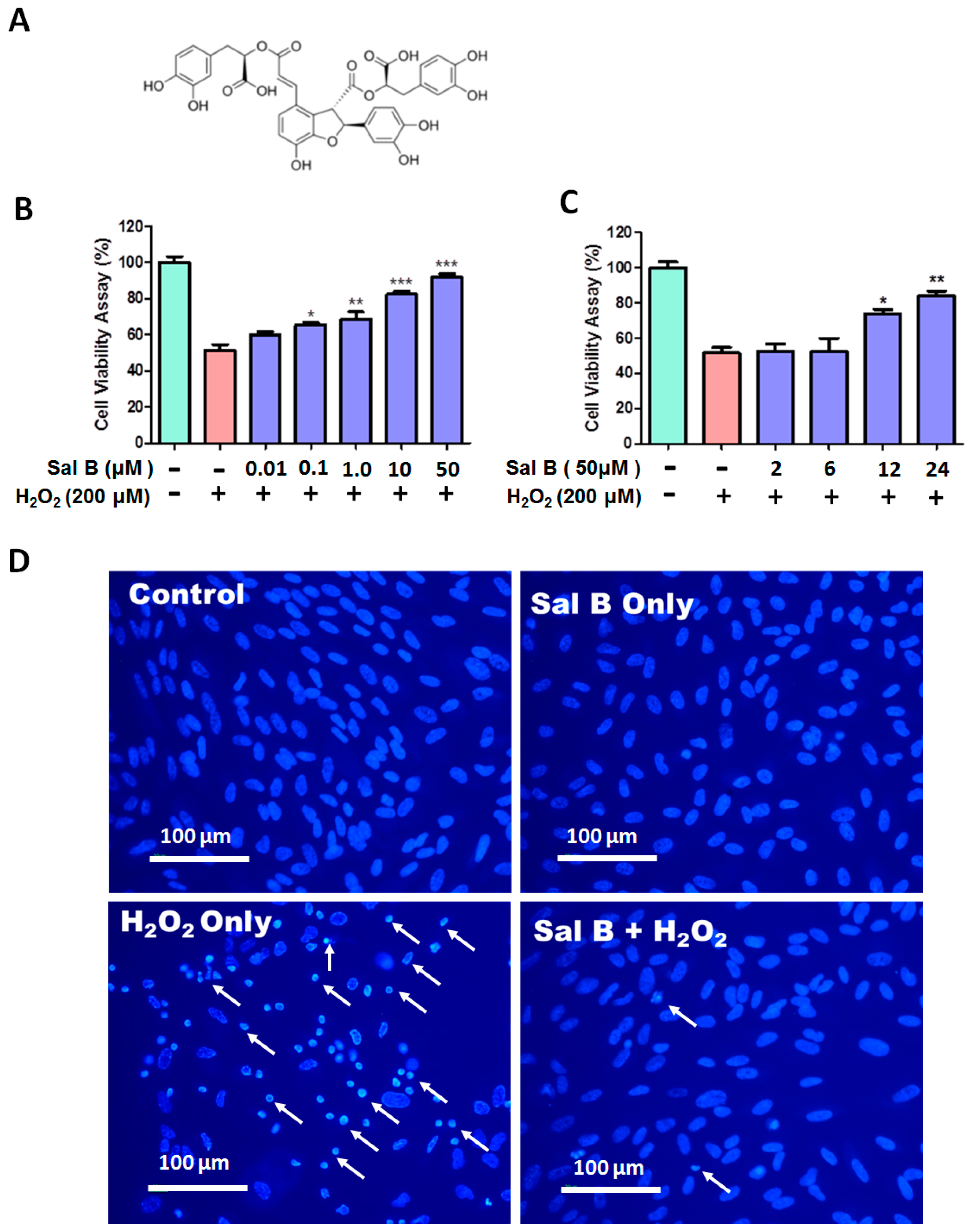
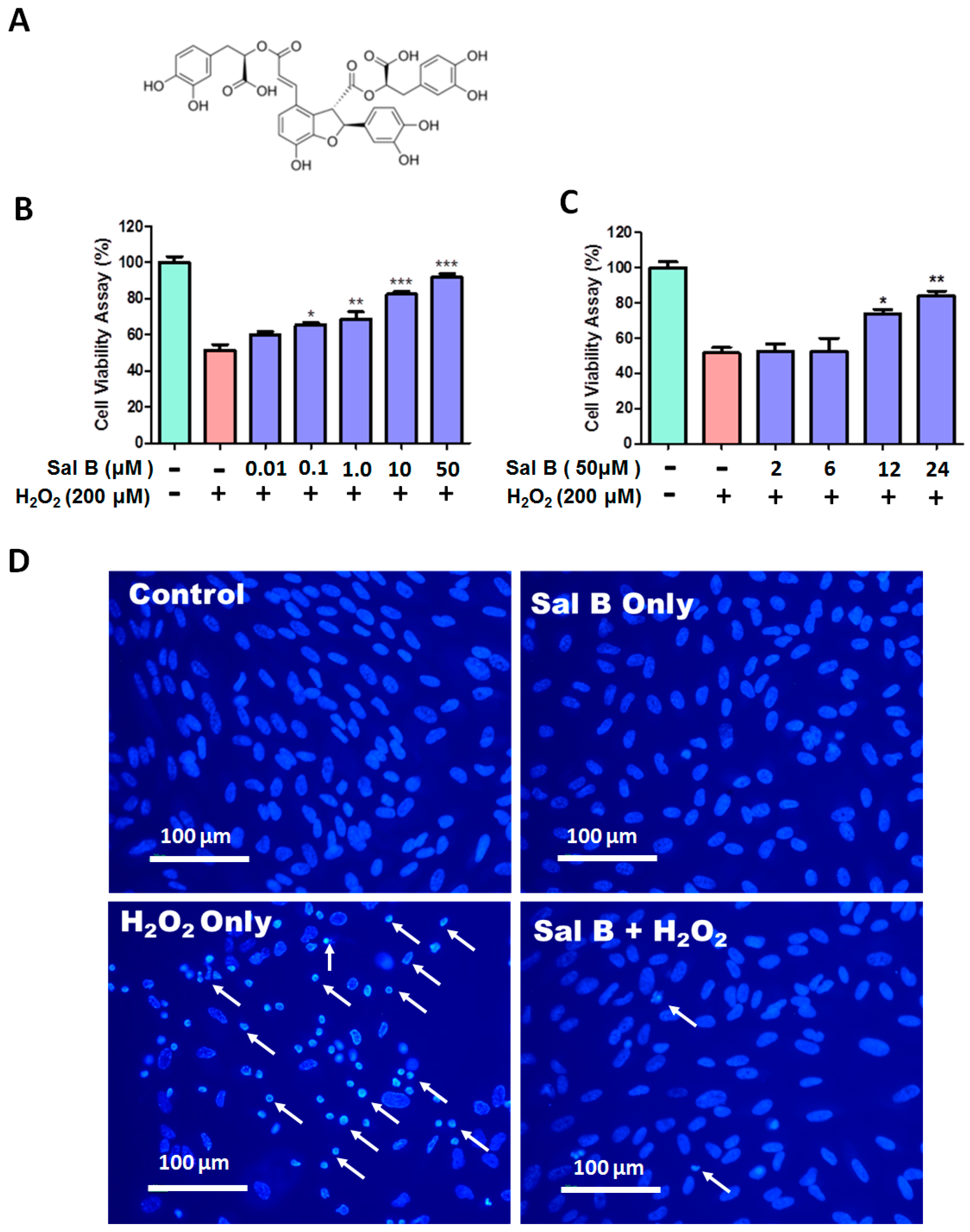
2.1. Salvianolic Acid B (Sal B) Prevents H2O2-Induced Cell Damage and Death in a Dose- and Time-Dependent Assay
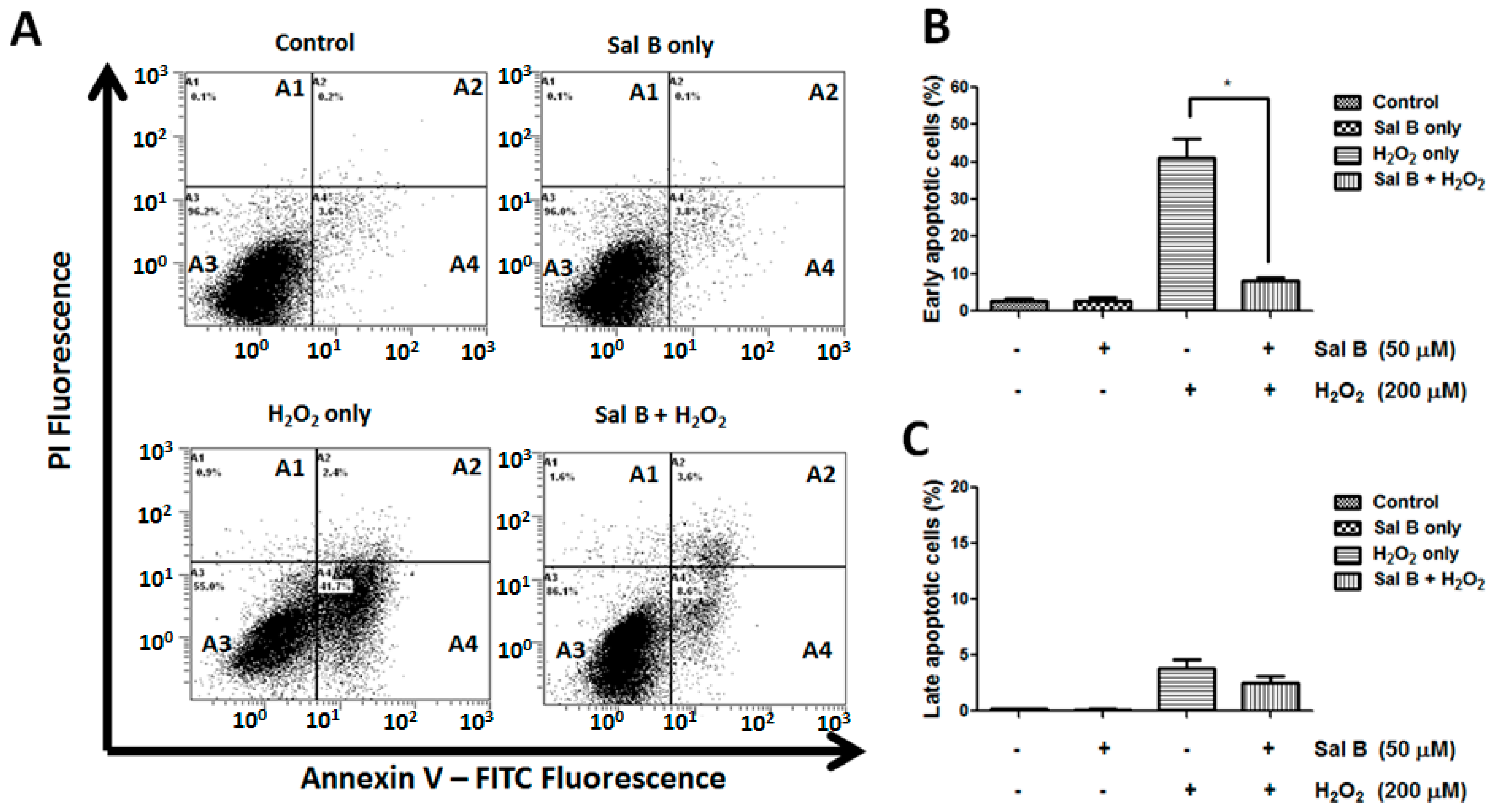
2.2. Sal B Treatment Reduces Apoptosis in H2O2-Treated Retinal Pigment Epithelial (RPE) Cells
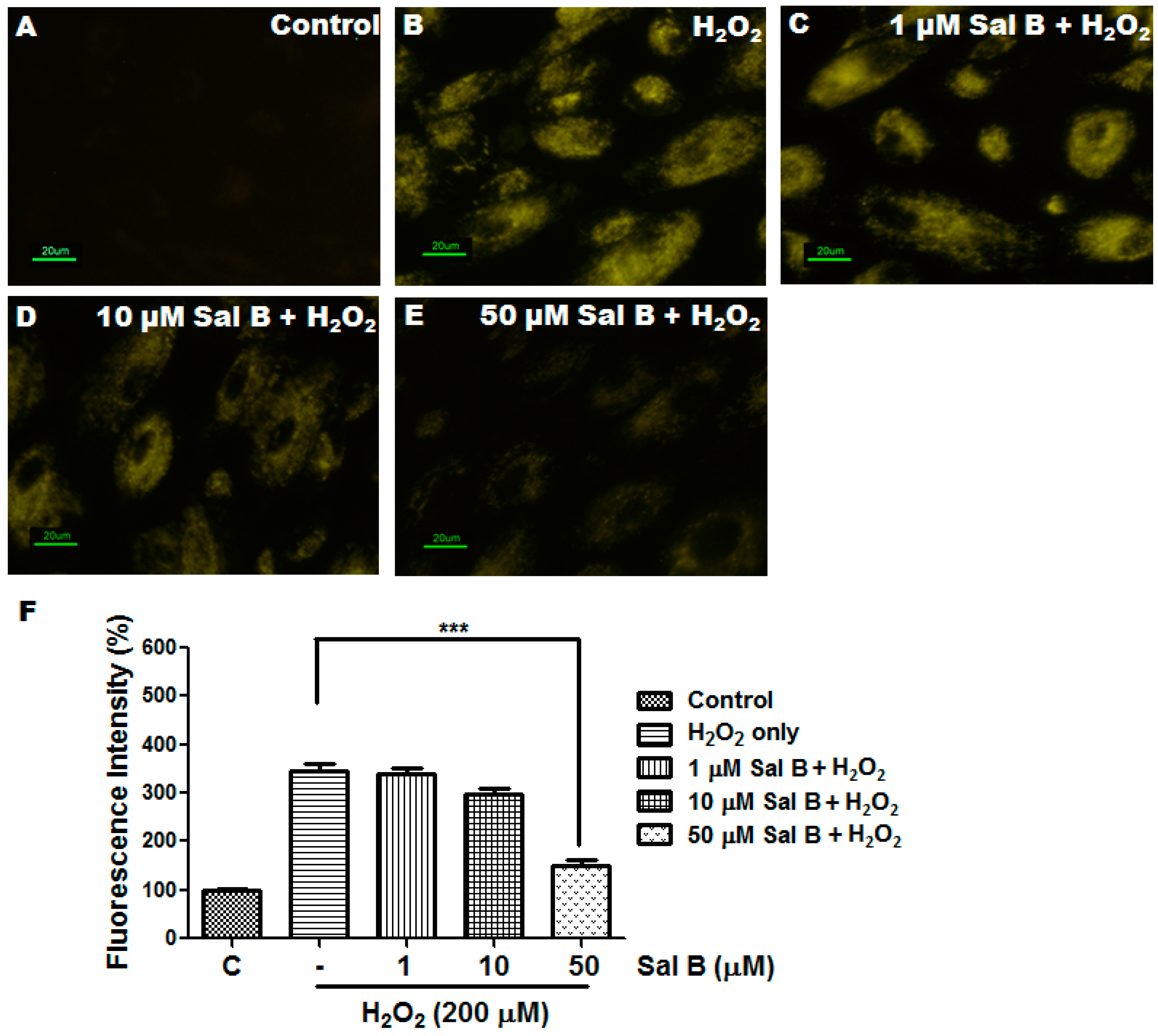
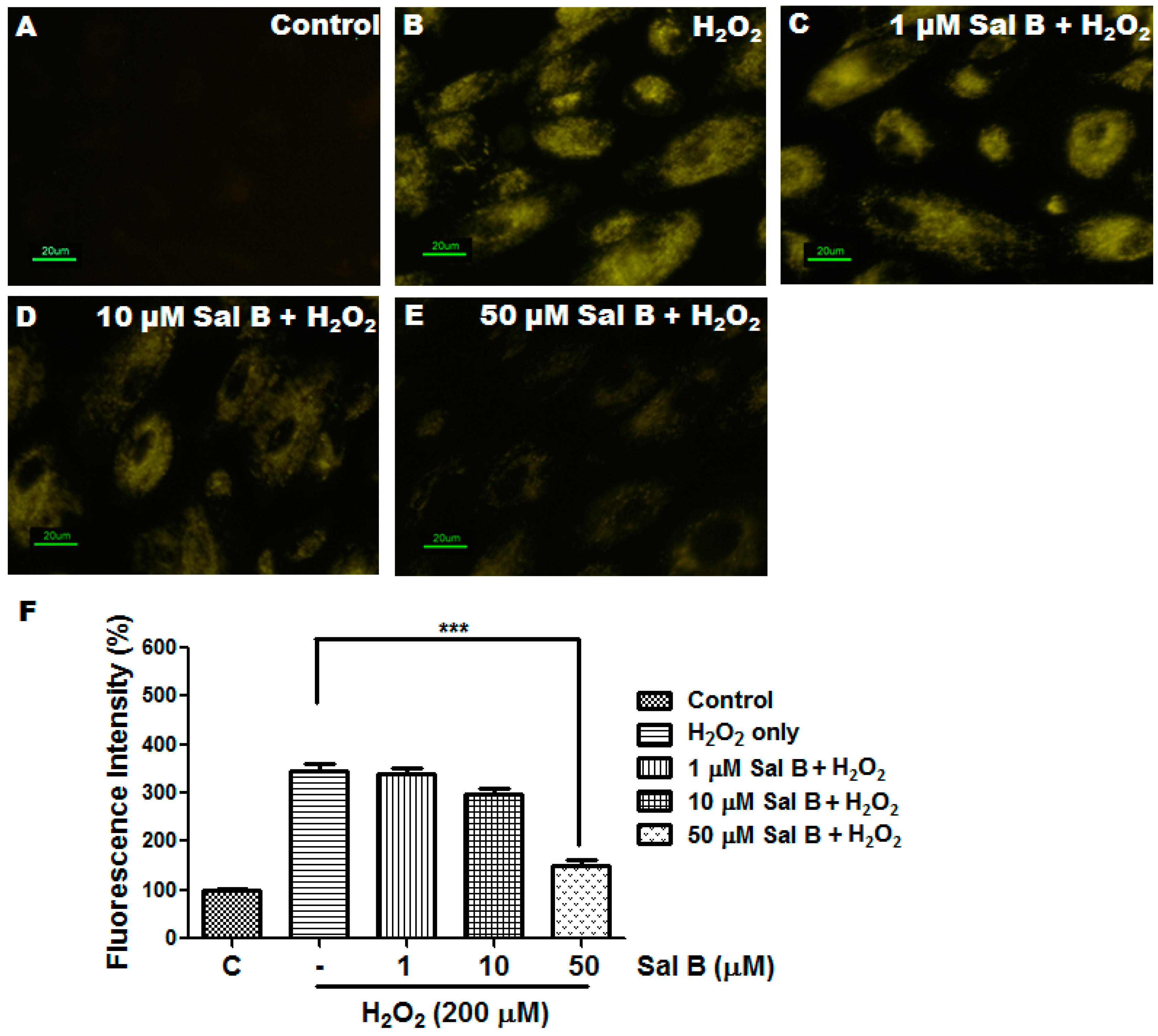
2.3. Sal B Has Strong Reactive Oxygen Species (ROS) Scavenging Activity
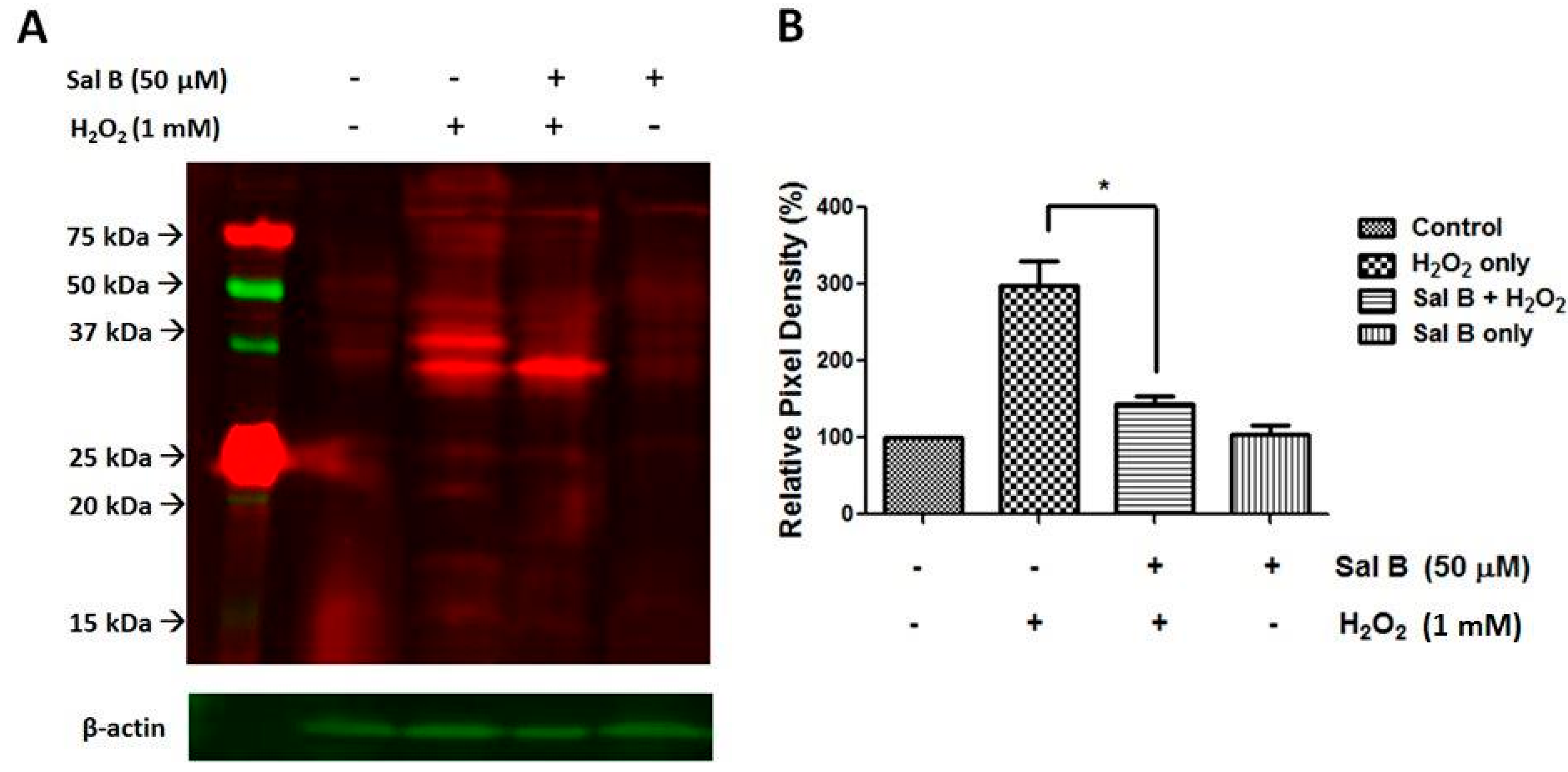
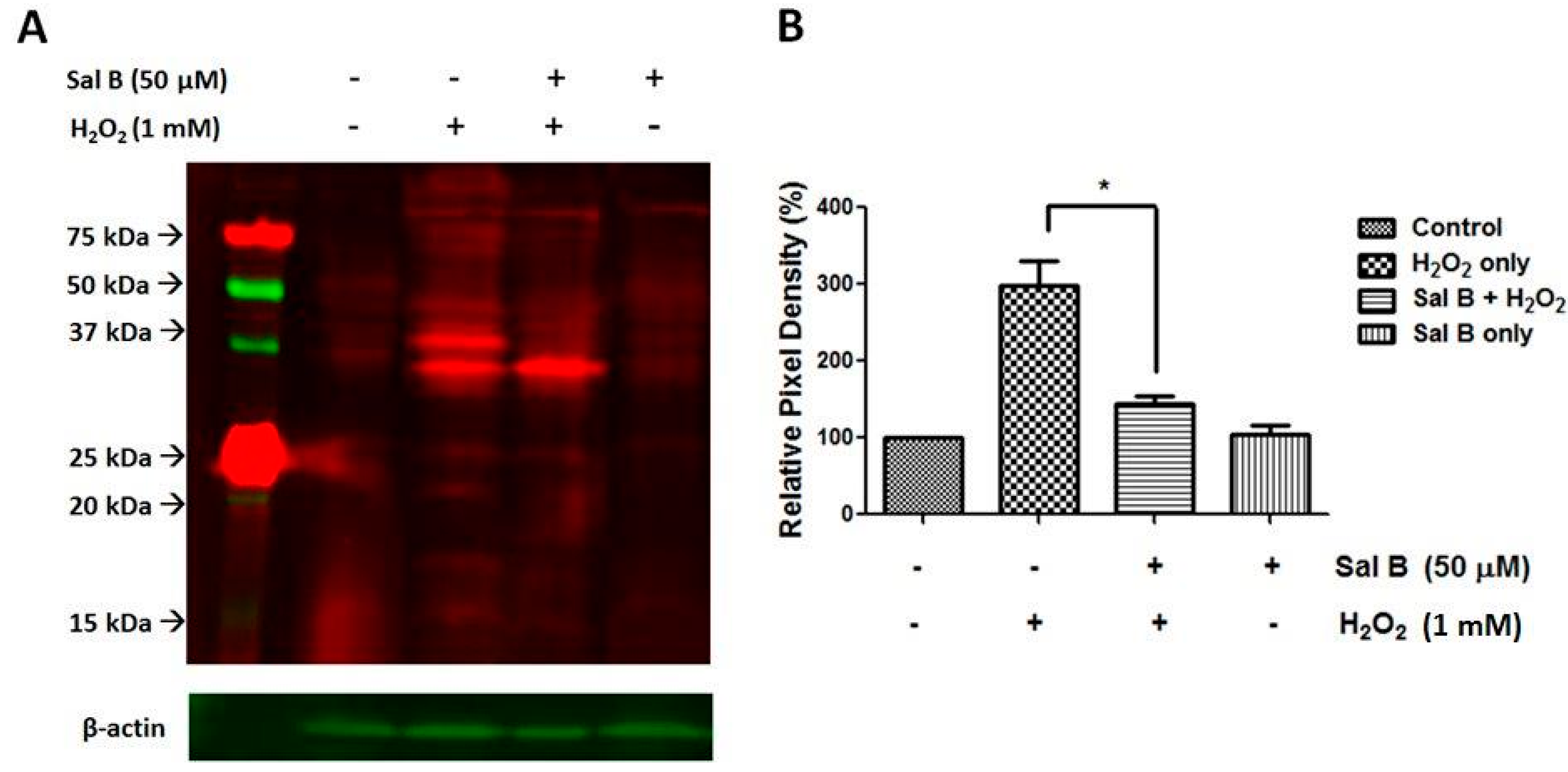
2.4. Sal B Decreases Protein Glutathionylation in RPE Cells
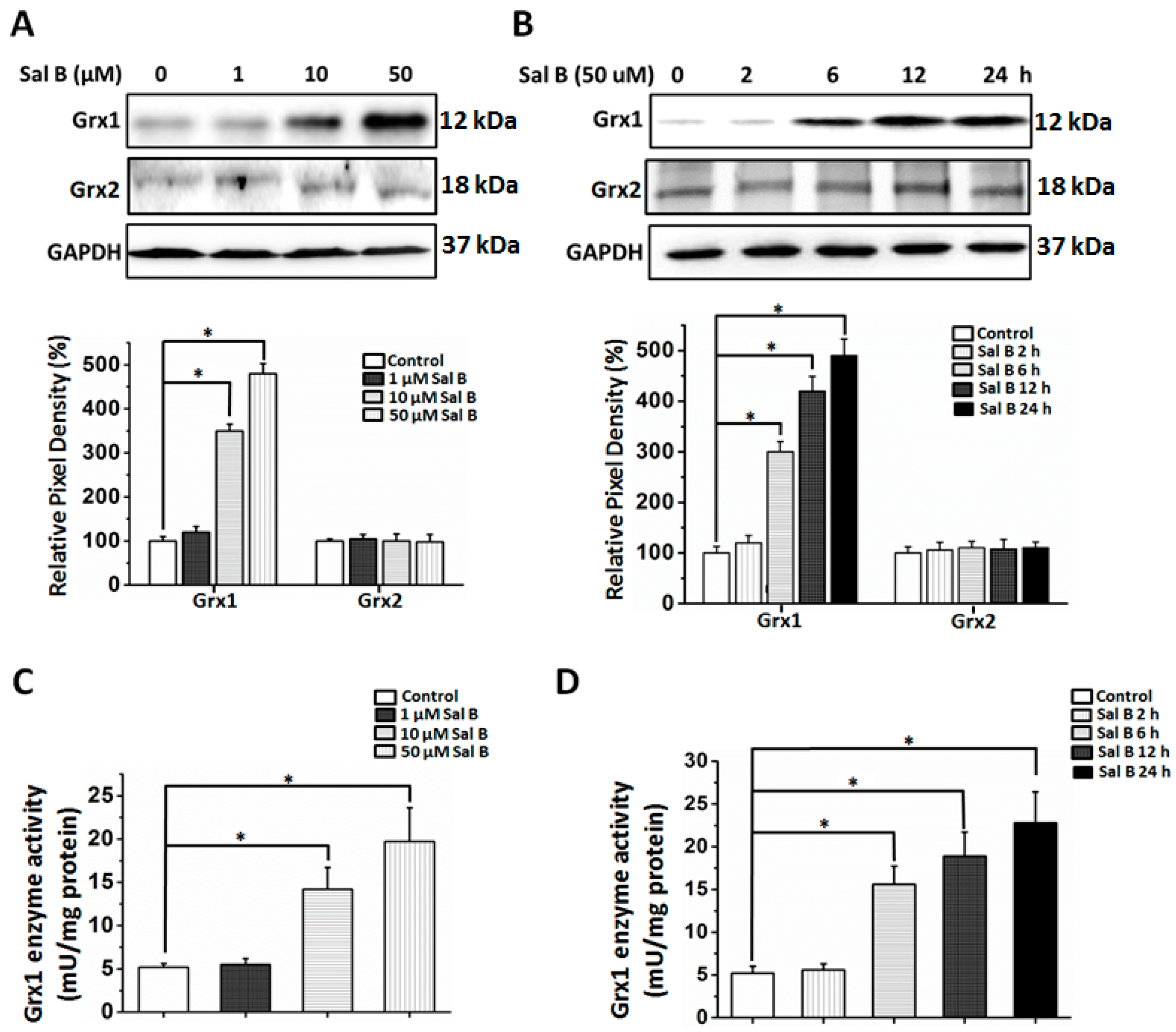
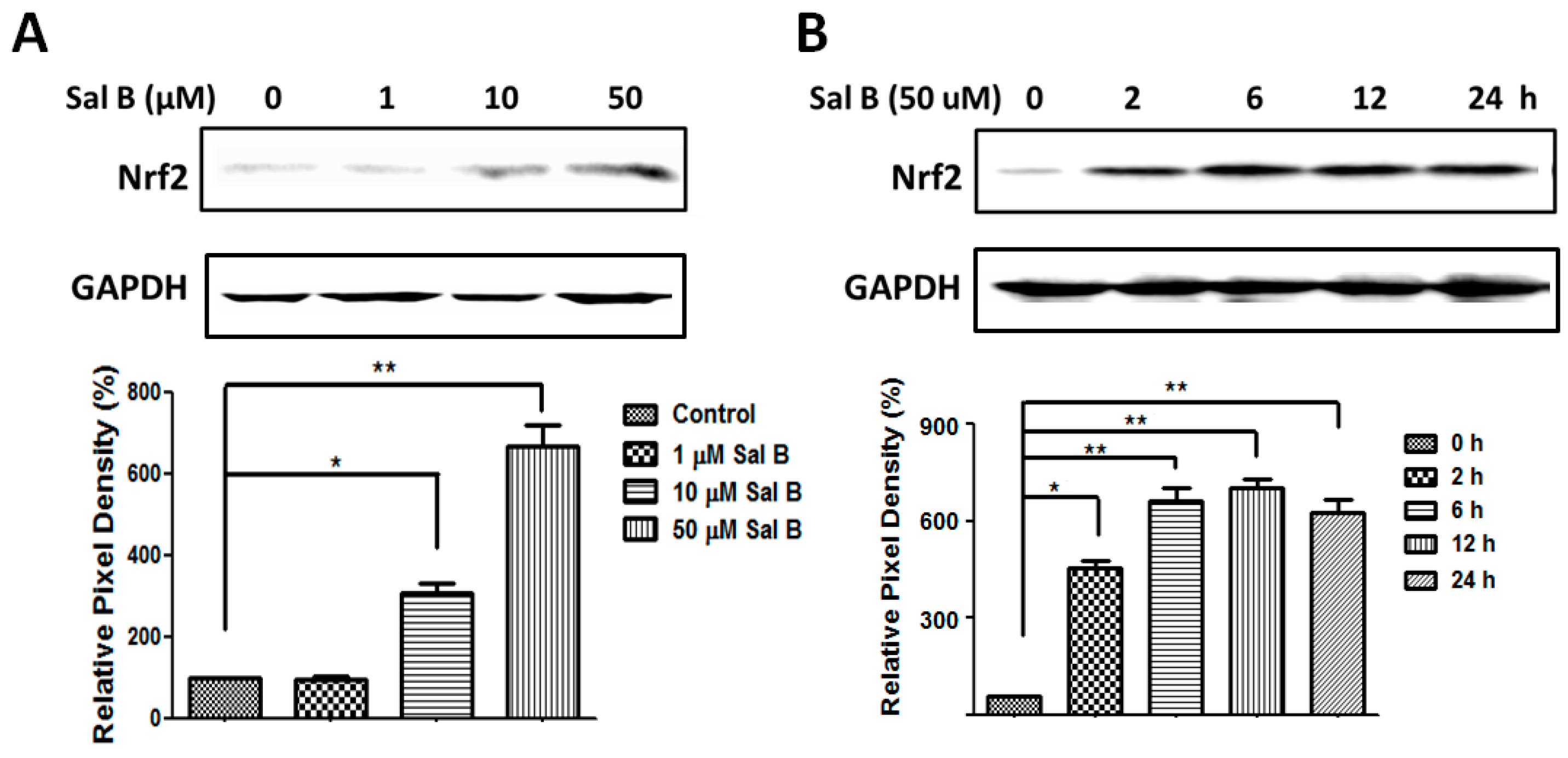
2.5. Sal B Time- and Dose-Dependently Up-Regulates Glutaredoxin 1 (Grx1)
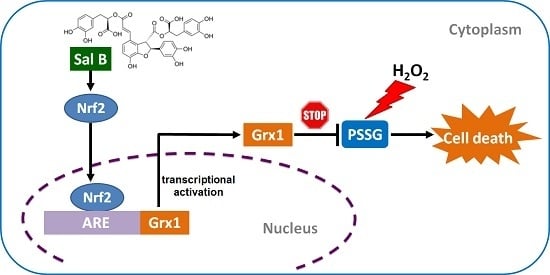
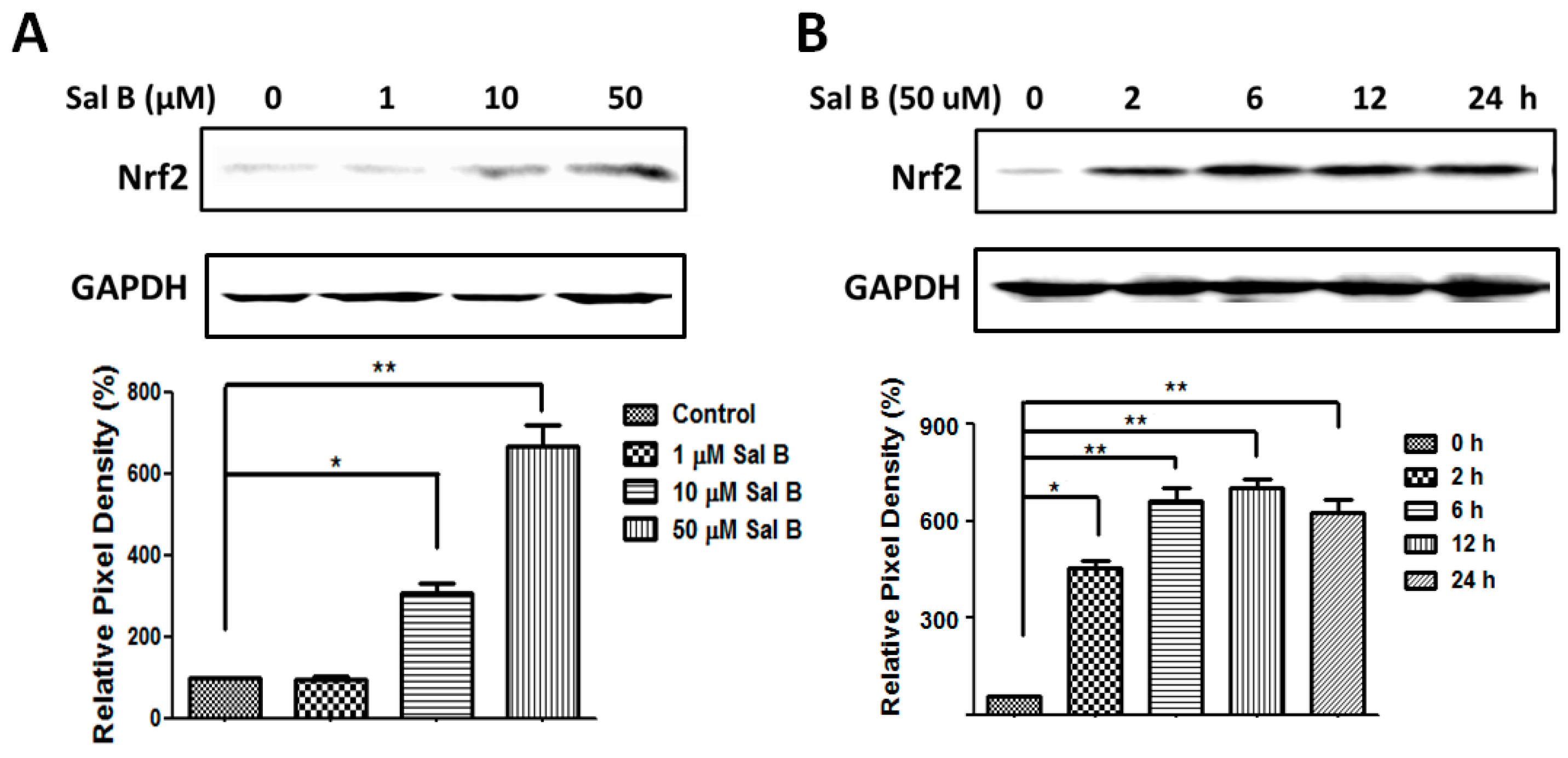
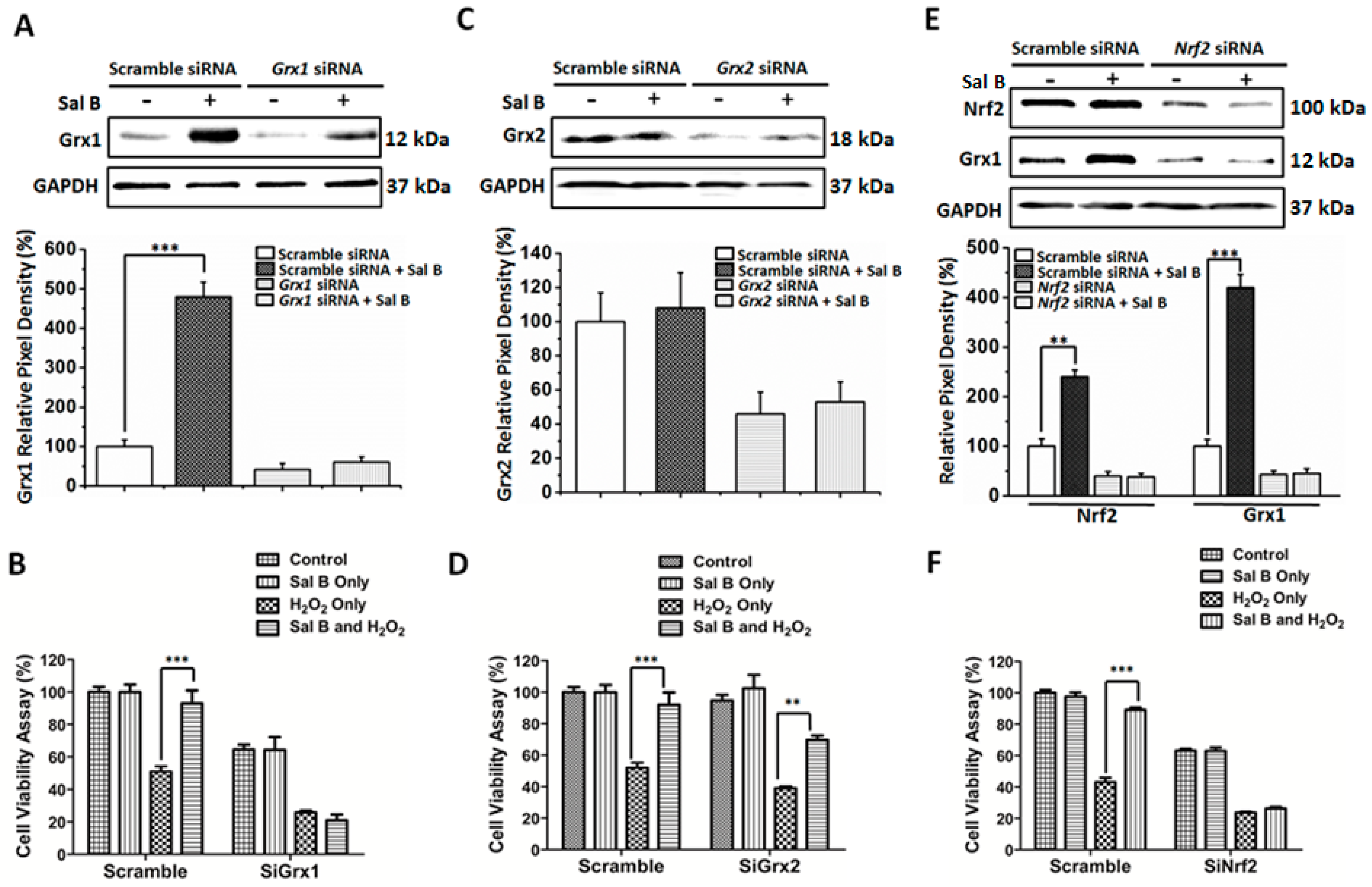
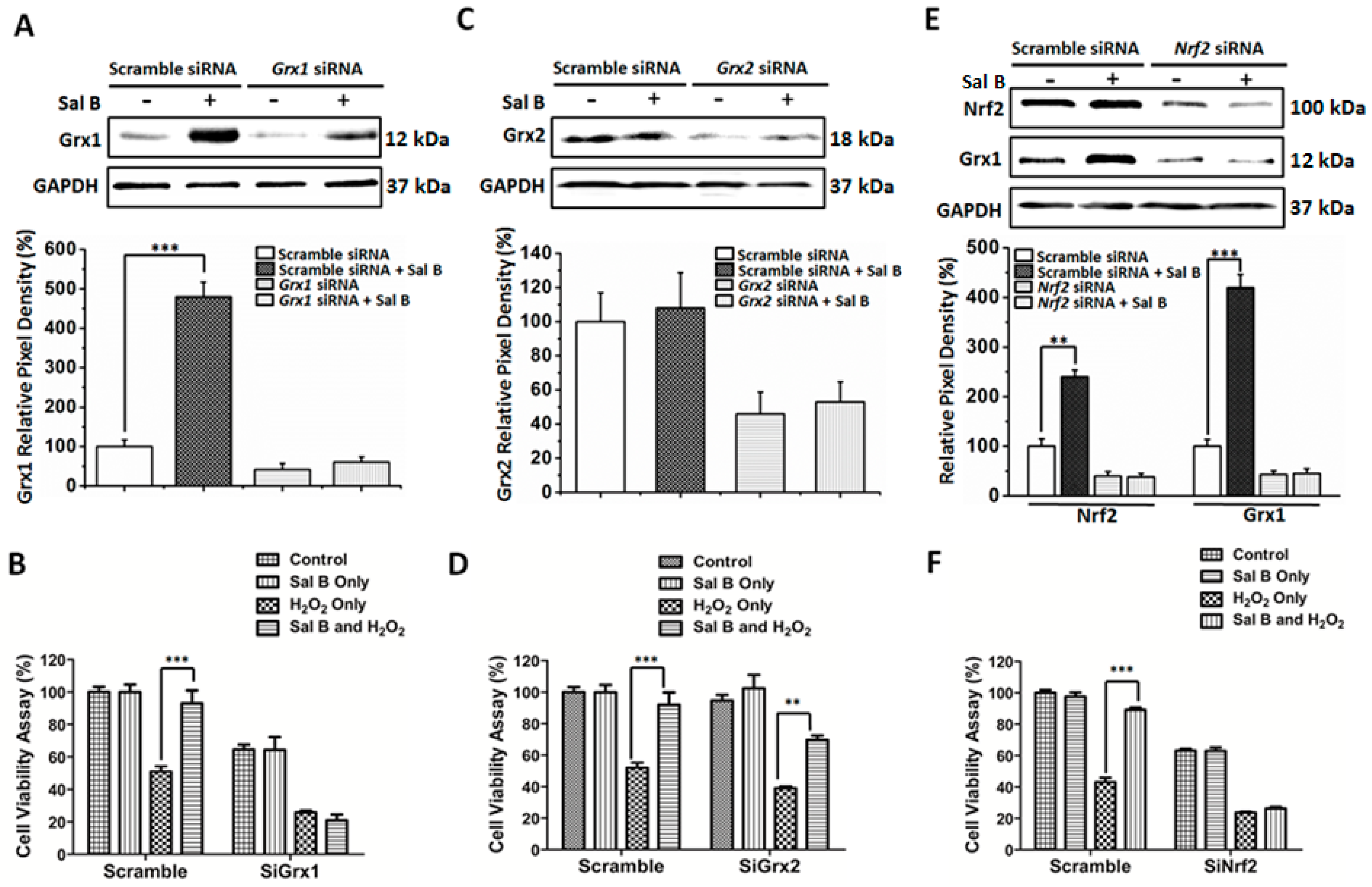
2.6. Sal B Up-Rregulates Grx1 through the NF-E2-Related Factor 2 (Nrf2) Transcriptional Pathway
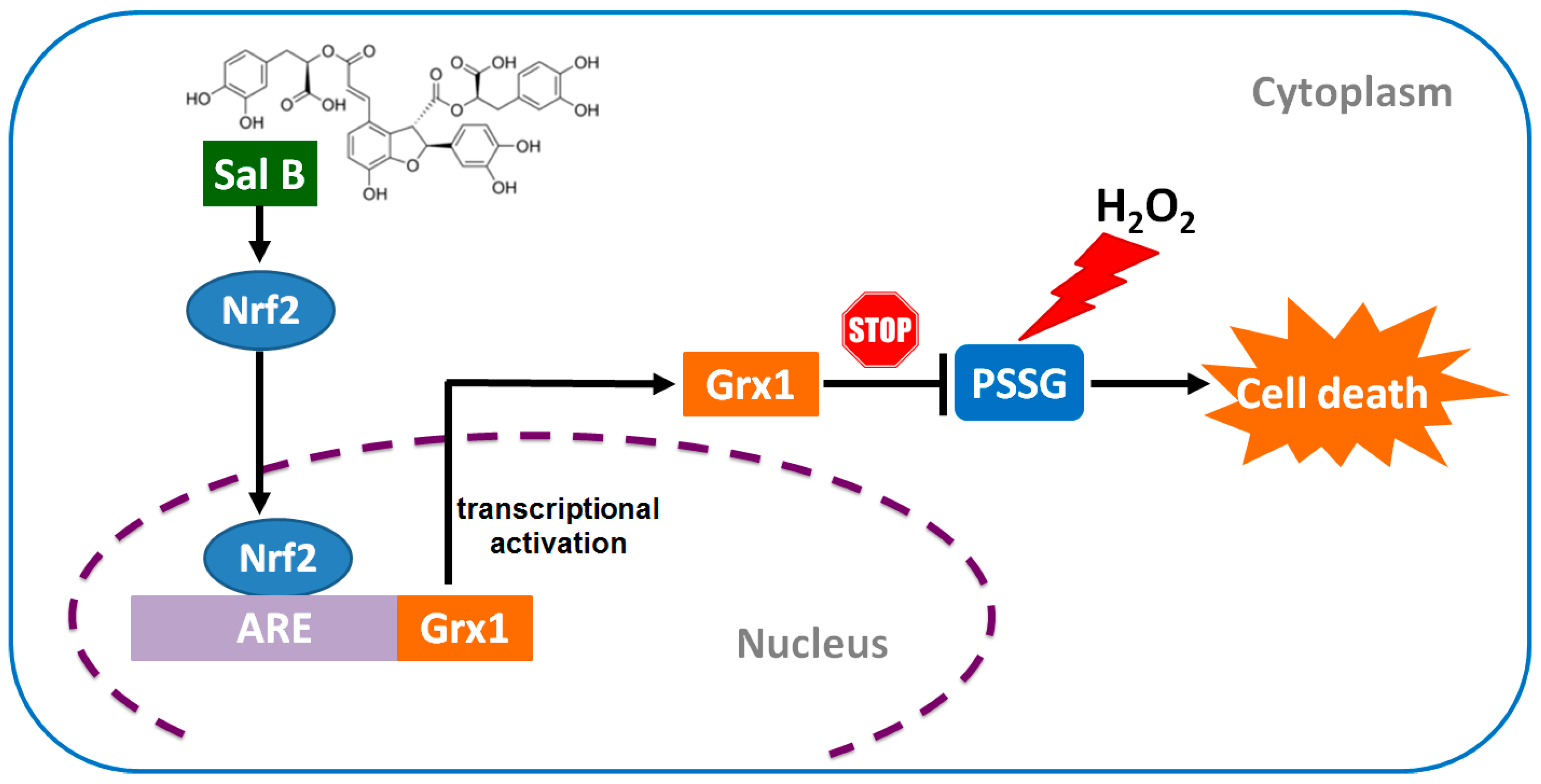
2.7. Sal B Protects Cells from Oxidative Stress-Induced Cell Injury by Promoting Grx1 Expression through Nrf2 Activation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Human Retinal Pigment Epithelial (RPE) Cell Culture
4.3. Cell Viability Assay
4.4. Hoechst 33342 Fluorescent Staining
4.5. Flow Cytometry Analysis of Cell Apoptosis
4.6. ROS Detection
4.7. Protein Glutathionylation Detection
4.8. Western Blot Analysis
4.9. Enzyme Activity Assays
4.10. siRNA-Mediated Knockdown of Grx1, Grx2, and Nrf2 and Cell Viability Testing
4.11. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Nelson, K.C.; Wu, M.; Sternberg, P., Jr.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- De Jong, P.T. Age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, G.; Chai, Y.C.; Crabb, J.W.; Sears, J. Protein S-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp. Eye Res. 2004, 78, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005, 7, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, G.M.; Raghavachari, N.; Lou, M.F. Distribution of thioltransferase (glutaredoxin) in ocular tissues. Investig. Ophthalmol. Vis. Sci. 1998, 39, 476–480. [Google Scholar]
- Lofgren, S.; Fernando, M.R.; Xing, K.Y.; Wang, Y.; Kuszynski, C.A.; Ho, Y.S.; Lou, M.F. Effect of thioltransferase (glutaredoxin) deletion on cellular sensitivity to oxidative stress and cell proliferation in lens epithelial cells of thioltransferase knockout mouse. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4497–4505. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) protects human retinal pigment epithelial cells from oxidative damage by preventing AKT glutathionylation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zuo, Z.; Chow, M.S. Danshen: An overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J. Clin. Pharmacol. 2005, 45, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.O. Cardiovascular effects of danshen. Int. J. Cardiol. 2007, 121, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Xie, L.X.; Li, M.; Durairajan, S.S.; Goto, S.; Huang, J.D. Salvianolic acid B inhibits hydrogen peroxide-induced endothelial cell apoptosis through regulating PI3K/Akt signaling. PLoS ONE 2007, 2, e1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.R.; Zhang, H.M.; Ye, T.X.; Xiang, Z.J.; Yuan, Y.J.; Guo, Z.X.; Zhao, L.B. Characterization of the radical scavenging and antioxidant activities of danshensu and salvianolic acid B. Food Chem. Toxicol. 2008, 46, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Liu, A.H.; Wu, H.L.; Westenbroek, C.; Song, Q.L.; Yu, H.M.; Ter Horst, G.J.; Li, X.J. Salvianolic acid B, an antioxidant from salvia miltiorrhiza, prevents Aβ(25–35)-induced reduction in BPRP in PC12 cells. Biochem. Biophys. Res. Commun. 2006, 348, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.L.; Li, Y.H.; Lin, Y.H.; Wang, R.; Li, Y.B.; Tie, L.; Song, Q.L.; Guo, D.A.; Yu, H.M.; Li, X.J. Salvianolic acid B protects human endothelial cells from oxidative stress damage: A possible protective role of glucose-regulated protein 78 induction. Cardiovasc. Res. 2009, 81, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar] [PubMed]
- Chen, Y.L.; Hu, C.S.; Lin, F.Y.; Chen, Y.H.; Sheu, L.M.; Ku, H.H.; Shiao, M.S.; Chen, J.W.; Lin, S.J. Salvianolic acid B attenuates cyclooxygenase-2 expression in vitro in LPS-treated human aortic smooth muscle cells and in vivo in the apolipoprotein-E-deficient mouse aorta. J. Cell. Biochem. 2006, 98, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Lee, I.T.; Chen, Y.H.; Lin, F.Y.; Sheu, L.M.; Ku, H.H.; Shiao, M.S.; Chen, J.W.; Chen, Y.L. Salvianolic acid B attenuates MMP-2 and MMP-9 expression in vivo in apolipoprotein-E-deficient mouse aorta and in vitro in LPS-treated human aortic smooth muscle cells. J. Cell. Biochem. 2007, 100, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, L.; Shen, J.; Hao, S.; Ming, A.; Wang, X.; Su, F.; Zhang, Z. Salvianolic acid B attenuates apoptosis and inflammation via SIRT1 activation in experimental stroke rats. Brain Res. Bull. 2015, 115, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Fu, F.; Li, G.; Wang, Y.; Gao, Y.; Liu, Z.; Zhang, S. SMND-309, a novel derivate of salvianolic acid B, ameliorates cerebral infarction in rats: Characterization and role. Brain Res. 2009, 1263, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.F. Redox regulation in the lens. Prog. Retin. Eye Res. 2003, 22, 657–682. [Google Scholar] [CrossRef]
- Xing, K.Y.; Lou, M.F. Effect of H2O2 on human lens epithelial cells and the possible mechanism for oxidative damage repair by thioltransferase. Exp. Eye Res. 2002, 74, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Lou, M.F. The possible physiological function of thioltransferase in cells. FASEB J. 2003, 17, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Yeager, R.L.; Yamamoto, M.; Klaassen, C.D. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol. Sci. 2009, 108, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Yin, W.; Shan, W.; Dai, J.; Yang, Y.; Li, L. Combination of chlorogenic acid and salvianolic acid B protects against polychlorinated biphenyls-induced oxidative stress through Nrf2. Environ. Toxicol. Pharmacol. 2016, 46, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhai, X.; Wang, G.; Tian, X.; Gao, D.; Shi, L.; Wu, H.; Fan, Q.; Peng, J.; Liu, K.; et al. Salvianolic acid B protects against acetaminophen hepatotoxicity by inducing Nrf2 and phase II detoxification gene expression via activation of the PI3K and PKC signaling pathways. J. Pharmacol. Sci. 2015, 127, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cao, B.; Zhang, D.; Xiao, N.; Chen, H.; Li, G.Q.; Peng, S.C.; Wei, L.Q. Salvianolic acid B protects against paraquat-induced pulmonary injury by mediating Nrf2/Nox4 redox balance and TGF-β1/Smad3 signaling. Toxicol. Appl. Pharmacol. 2016, 309, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qu, X.D.; Li, Z.Y.; Wei, J.; Liu, Q.; Ma, Y.H.; He, J.J. Salvianolic acid B attenuates toxin-induced neuronal damage via Nrf2-dependent glial cells-mediated protective activity in Parkinson’s disease models. PLoS ONE 2014, 9, e101668. [Google Scholar] [CrossRef] [PubMed]
- Tongqiang, L.; Shaopeng, L.; Xiaofang, Y.; Nana, S.; Xialian, X.; Jiachang, H.; Ting, Z.; Xiaoqiang, D. Salvianolic acid B prevents iodinated contrast media-induced acute renal injury in rats via the PI3K/Akt/Nrf2 pathway. Oxid. Med. Cell. Longev. 2016, 2016, 7079487. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Xavier, C.; Jann, J.; Wu, H. Salvianolic Acid B (Sal B) Protects Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death by Activating Glutaredoxin 1 (Grx1). Int. J. Mol. Sci. 2016, 17, 1835. https://doi.org/10.3390/ijms17111835
Liu X, Xavier C, Jann J, Wu H. Salvianolic Acid B (Sal B) Protects Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death by Activating Glutaredoxin 1 (Grx1). International Journal of Molecular Sciences. 2016; 17(11):1835. https://doi.org/10.3390/ijms17111835
Chicago/Turabian StyleLiu, Xiaobin, Christy Xavier, Jamieson Jann, and Hongli Wu. 2016. "Salvianolic Acid B (Sal B) Protects Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death by Activating Glutaredoxin 1 (Grx1)" International Journal of Molecular Sciences 17, no. 11: 1835. https://doi.org/10.3390/ijms17111835
APA StyleLiu, X., Xavier, C., Jann, J., & Wu, H. (2016). Salvianolic Acid B (Sal B) Protects Retinal Pigment Epithelial Cells from Oxidative Stress-Induced Cell Death by Activating Glutaredoxin 1 (Grx1). International Journal of Molecular Sciences, 17(11), 1835. https://doi.org/10.3390/ijms17111835