
The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae


Abstract
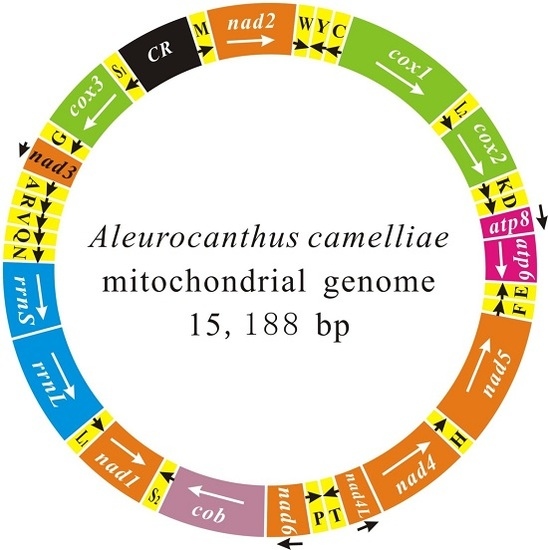
:
1. Introduction
2. Results and Discussion
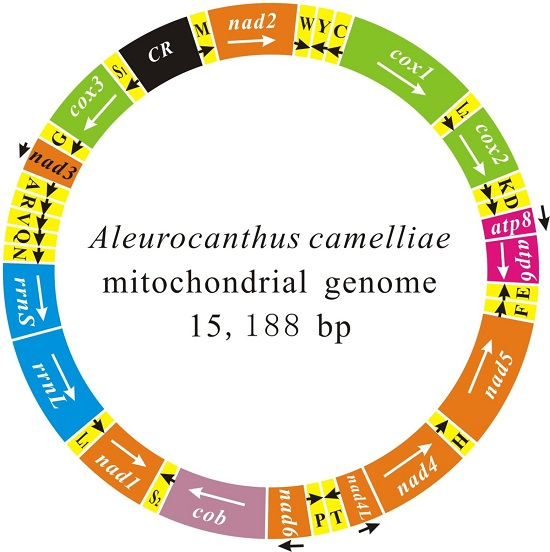
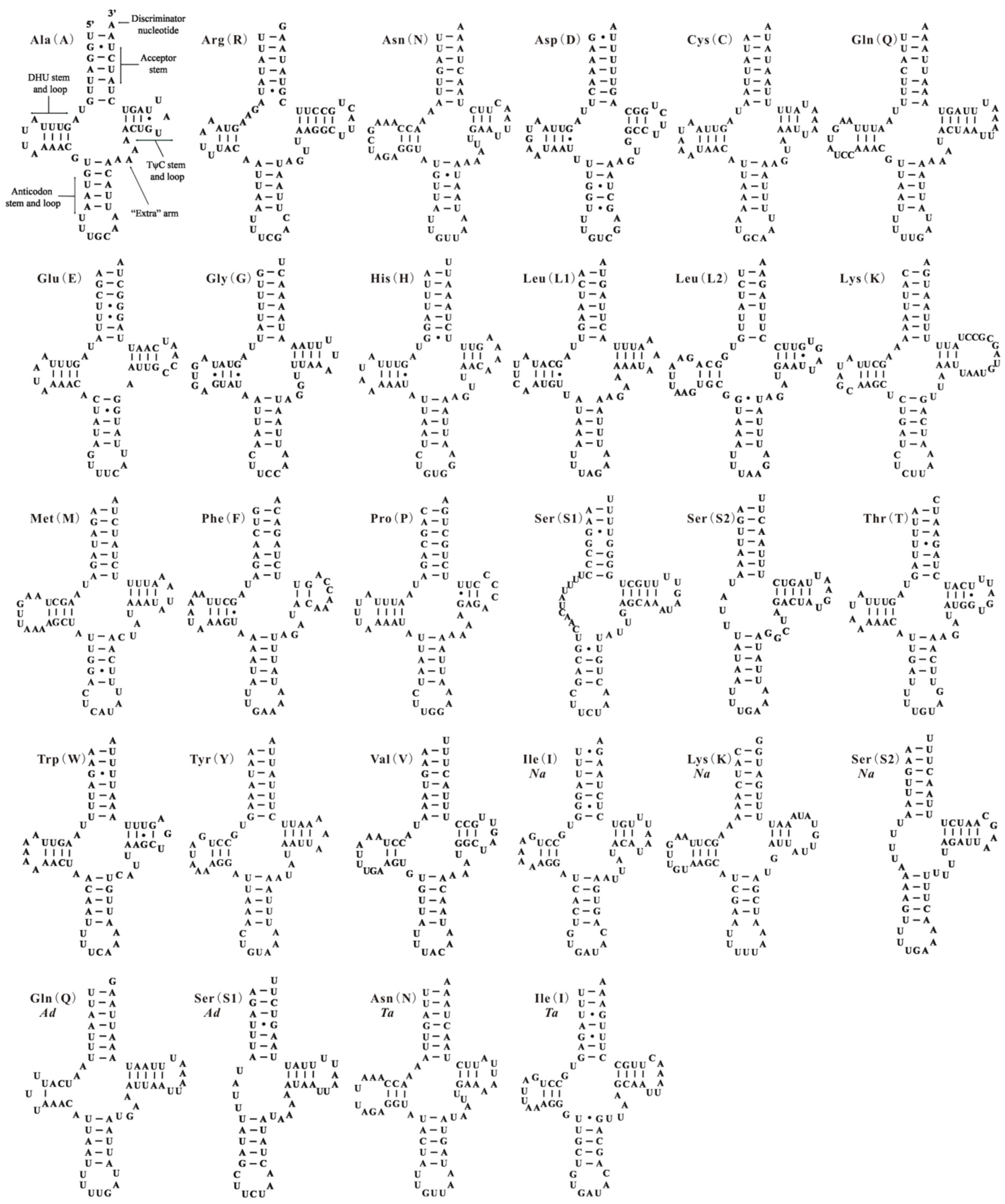
2.1. Genome Content and Nucleotide Composition
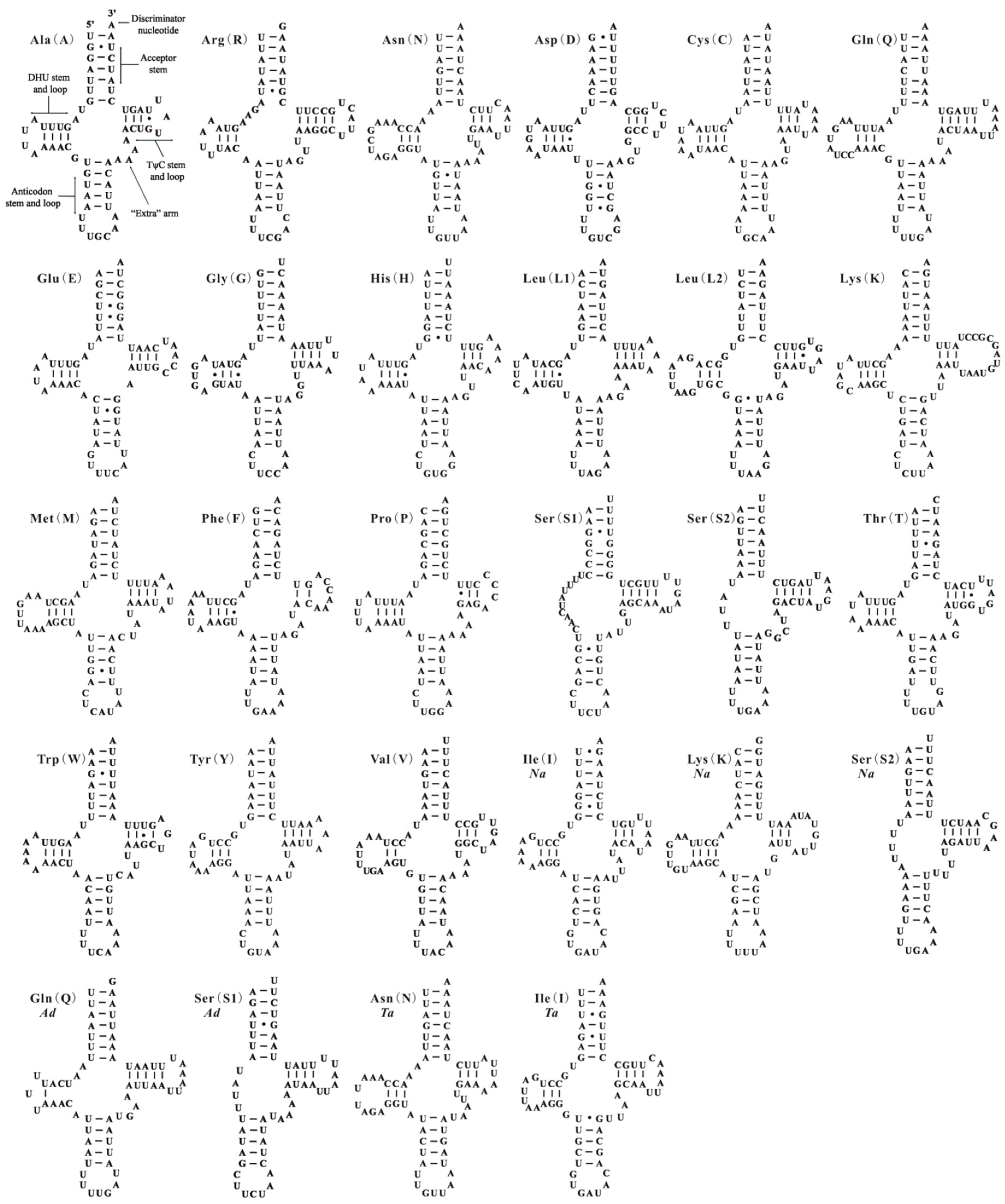
2.2. Re-Annotation for Whitefly Mitochondrial Genomes
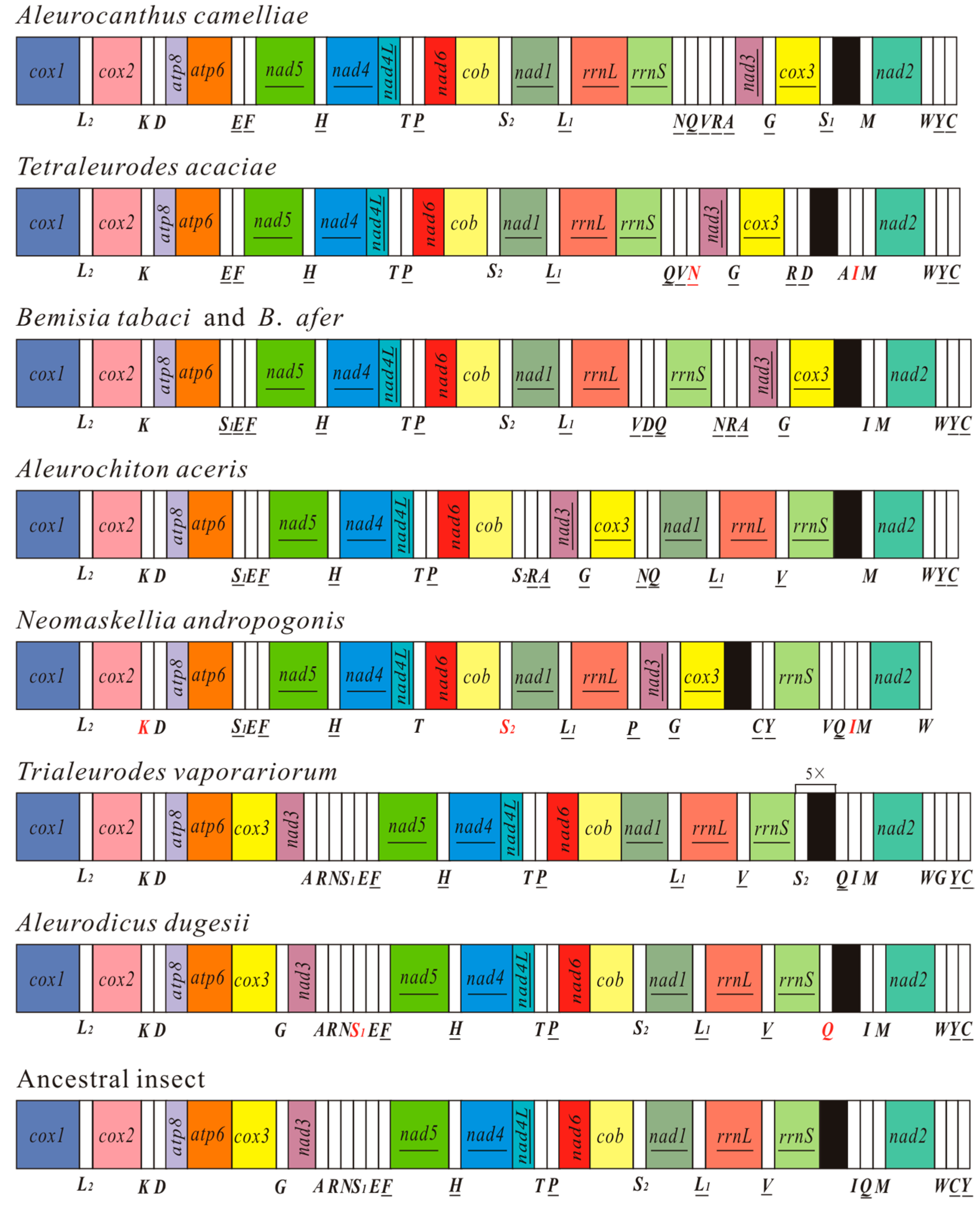
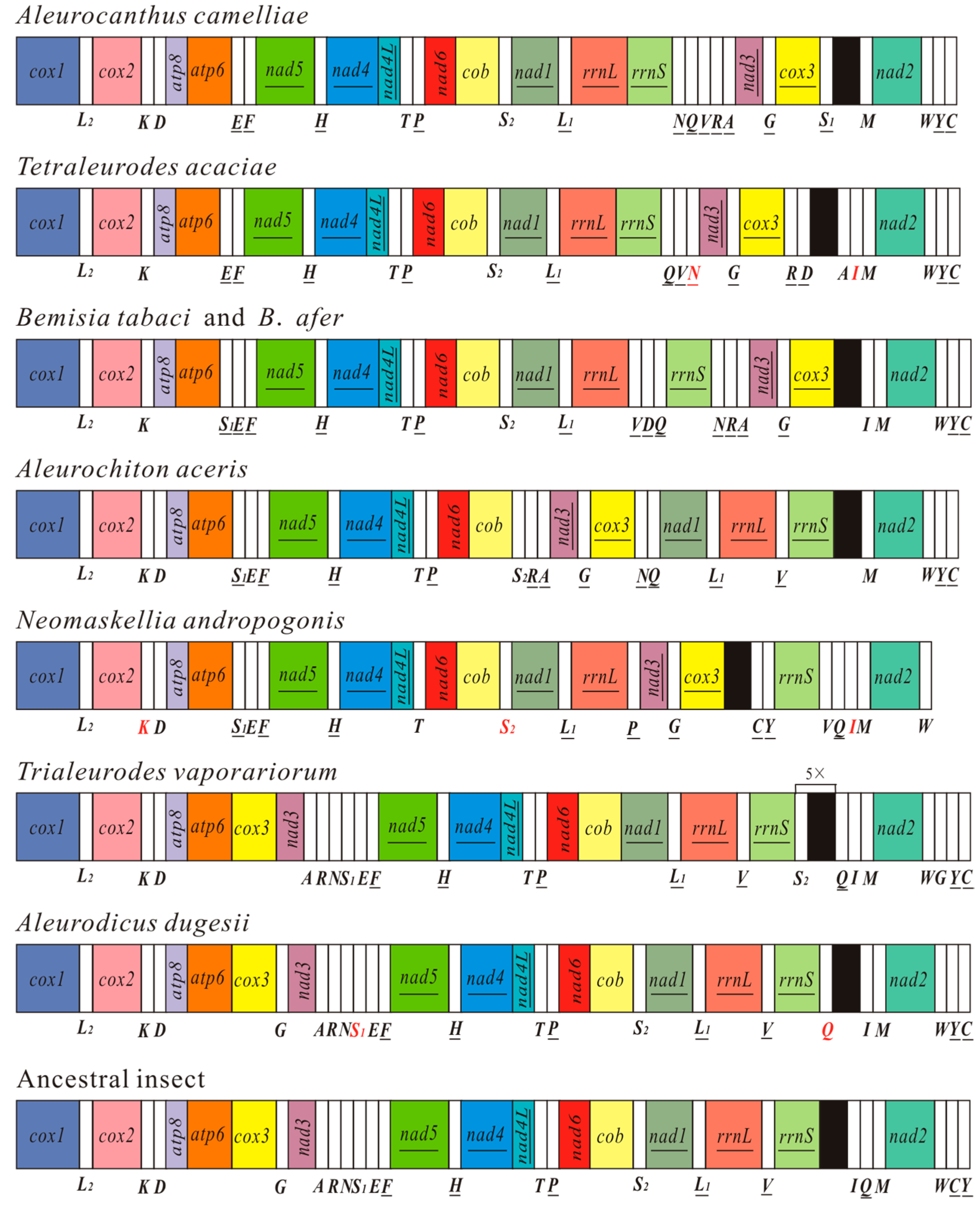
2.3. Gene Arrangement
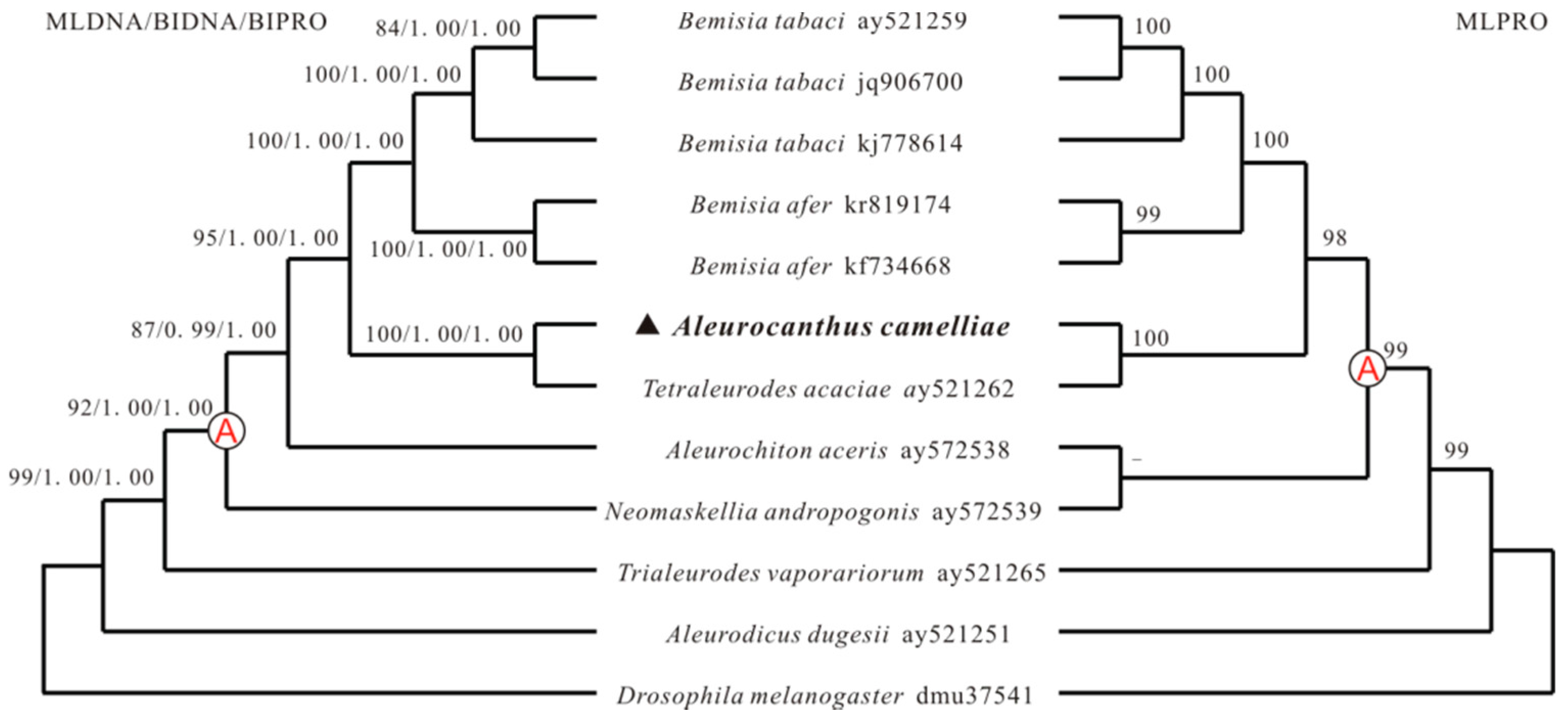
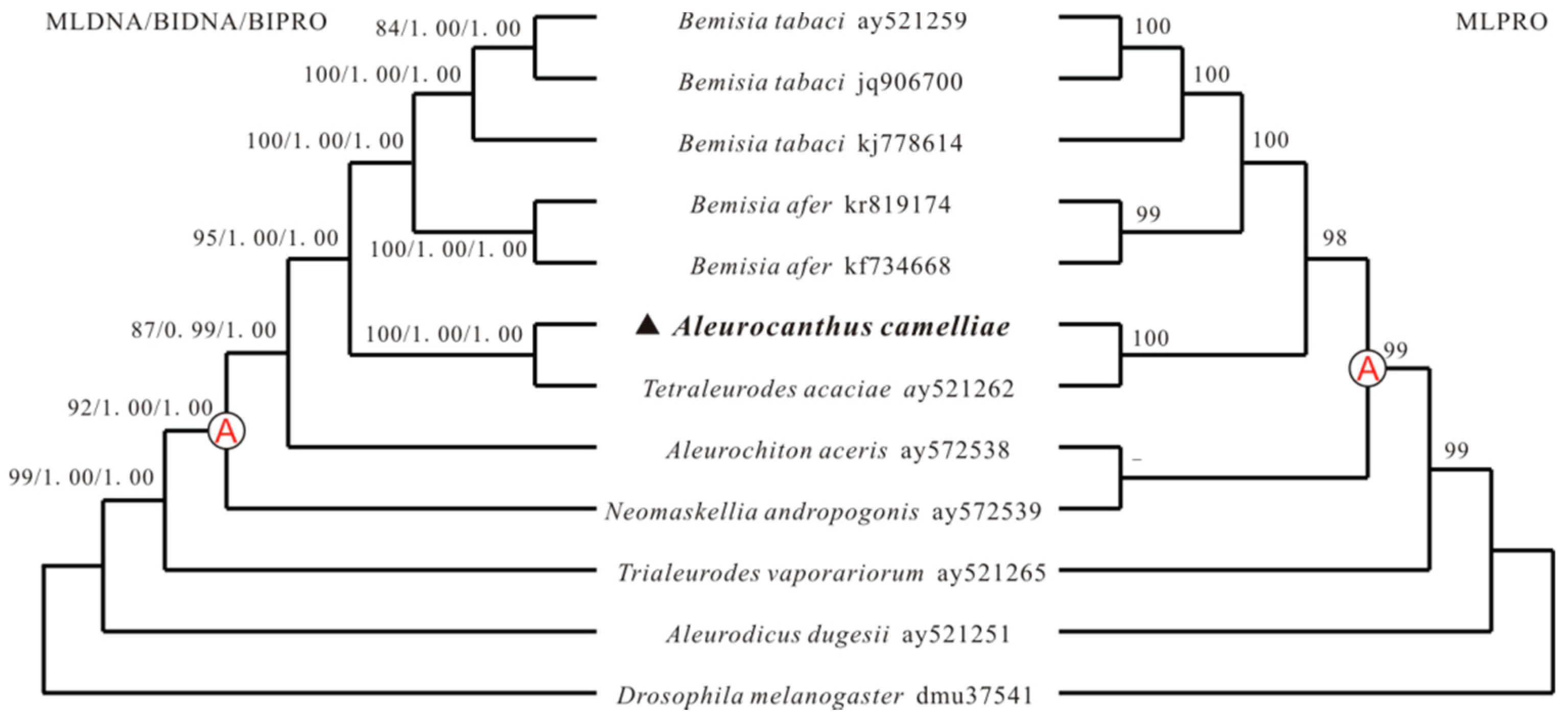
2.4. Phylogenetic Analyses
3. Materials and Methods
3.1. Ethics Statement and Sample Collection
3.2. DNA Extraction and mt Genome Amplification
3.3. Sequence Annotation and Analysis
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fu, J.Y.; Han, B.Y.; Xiao, Q. Mitochondrial COI and 16sRNA evidence for a single species hypothesis of E. vitis, J. formosana and E. onukii in East Asia. PLoS ONE 2014, 9, e115259. [Google Scholar] [CrossRef] [PubMed]
- Kanmiya, K.; Ueda, S.; Kasai, A.; Yamashita, K.; Sato, Y.; Yoshiyasu, Y. Proposal of new specific status for tea-infesting populations of the nominal citrus spiny whitefly Aleurocanthus spiniferus (Homoptera: Aleyrodidae). Zootaxa 2011, 2797, 25–44. [Google Scholar]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.C.; Wei, D.D.; Shao, R.; Dou, W.; Wang, J.J. The complete mitochondrial genome of the booklouse, Liposcelis decolor: Insights into gene arrangement and genome organization within the genus Liposcelis. PLoS ONE 2014, 9, e91902. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Barker, S.C.; Li, H.; Song, S.; Poudel, S.; Su, Y. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Zhu, X.Q.; Barker, S.C.; Herd, K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol. Evol. 2012, 4, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Yoshizawa, K.; Mizukoshi, A.; Whiting, M.F.; Johnson, K.P. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.C.; Wei, D.D.; Shao, R.; Shi, J.X.; Dou, W.; Wang, J.J. Evolution of multipartite mitochondrial genomes in the booklice of the genus Liposcelis (Psocoptera). BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Yuan, M.L.; Wang, B.J.; Zhou, A.W.; Dou, W.; Wang, J.J. Population genetics of two asexually and sexually reproducing psocids species inferred by the analysis of mitochondrial and nuclear DNA sequences. PLoS ONE 2012, 7, e33883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Yuan, Z.J.; Zhang, C.X.; Yin, K.S.; Tang, M.J.; Guo, H.W.; Fu, J.Y.; Xiao, Q. Detecting deep divergence in seventeen populations of tea geometrid (Ectropis obliqua Prout) in China by COI mtDNA and cross-breeding. PLoS ONE 2014, 9, e99373. [Google Scholar] [CrossRef] [PubMed]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Dickey, A.M.; Kumar, V.; Morgan, J.K.; Jara-Cavieres, A.; Shatters, R.G., Jr.; McKenzie, C.L.; Osborne, L.S. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): Extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Shao, R.; Yuan, M.L.; Dou, W.; Barker, S.C.; Wang, J.J. The multipartite mitochondrial genome of Liposcelis bostrychophila: Insights into the evolution of mitochondrial genomes in bilateral animals. PLoS ONE 2012, 7, e33973. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.; Mifsud, D.; Rapisarda, C. The whiteflies (Hemiptera: Aleyrodidae) of Europe and the Mediterranean Basin. Bull. Entomol. Res. 2000, 90, 407–448. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.L.; Xiao, N.; Yang, J.; Wang, X.W.; Colvin, J.; Liu, S.S. The complete mitochondrial genome of Bemisia afer (Hemiptera: Aleyrodidae). Mitochondr. DNA A 2016, 27, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Yang, J.; Boykin, L.M.; Zhao, Q.Y.; Li, Q.; Wang, X.W.; Liu, S.S. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Zhang, Z.; Bing, X.L.; Liu, Y.Q.; Liu, S.S.; Wang, X.W. A complete mitochondrial DNA genome derived from a Chinese population of the Bemisia afer species complex (Hemiptera: Aleyrodidae). Mitochondr. DNA A 2015, 27, 3500–35011. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Complete mitochondrial genome sequence of the polychaete annelid Platynereis dumerilii. Mol. Biol. Evol. 2001, 18, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Elfekih, S.; Court, L.; Gordon, K.H.; de Barro, P.J. Complete mitochondrial DNA genome of Bemisia tabaci cryptic pest species complex Asia I (Hemiptera: Aleyrodidae). Mitochondr. DNA 2016, 27, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Helfenbein, K.G.; Fourcade, H.M.; Vanjani, R.G.; Boore, J.L. The mitochondrial genome of Paraspadella gotoi is highly reduced and reveals that chaetognaths are a sister group to protostomes. Proc. Natl. Acad. Sci. USA 2004, 101, 10639–10643. [Google Scholar] [CrossRef] [PubMed]
- Kayal, E.; Lavrov, D.V. The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene 2008, 410, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Duchene, A.M.; Pujol, C.; Marechal-Drouard, L. Import of tRNAs and aminoacyl-tRNA synthetases into mitochondria. Curr. Genet. 2009, 55, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.S.; Suga, K.; Sakakura, Y.; Park, H.G.; Hagiwara, A.; Rhee, J.S.; Lee, J.S. Complete mitochondrial genome of the monogonont rotifer, Brachionus koreanus (Rotifera, Brachionidae). Mitochondr. DNA 2014, 25, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Suga, K.; Welch, D.B.M.; Tanaka, Y.; Sakakura, Y.; Hagiwarak, A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 2008, 25, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Jeyaprakash, A.; Hoy, M.A. The mitochondrial genome of the predatory mite Metaseiulus occidentalis (Arthropoda:Chelicerata:Acari:Phytoseiidae) is unexpectedly large and contains several novel features. Gene 2007, 391, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Dermauw, W.; Vanholme, B.; Tirry, L.; van Leeuwen, T. Mitochondrial genome analysis of the predatory mite Phytoseiulus persimilis and a revisit of the Metaseiulus occidentalis mitochondrial genome. Genome 2010, 53, 285–301. [Google Scholar] [PubMed]
- Stewart, J.B.; Beckenbach, A.T. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 2009, 445, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. Comp. Genom. 2000, 1, 133–147. [Google Scholar]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the evaniomorpha (hymenoptera)—A group with an intermediate rate of gene rearrangement. Genome Biol. Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Dowton, M. Complete mitochondrial genomes of Ceratobaeus sp. and Idris sp. (Hymenoptera: Scelionidae): Shared gene rearrangements as potential phylogenetic markers at the tribal level. Mol. Biol. Rep. 2014, 41, 6419–6427. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Campbell, N.J. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.L.; Farr, C.L.; Farquhar, A.L.; Kaguni, L.S. Sequence, organization, and evolution of the A + T region of Drosophila melanogaster mitochondrial DNA. Mol. Biol. Evol. 1994, 11, 523–538. [Google Scholar] [PubMed]
- Dowton, M.; Castro, L.R.; Austin, A.D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome “morphology“. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Uesugi, K.; Sato, Y.; Han, B.Y.; Huang, Z.-D.; Yara, K.; Furuhashi, K. Molecular evidence for multiple phylogenetic groups within two species of invasive spiny whiteflies and their parasitoid wasp. Bull. Entomol. Res. 2016, 106, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, D.R.; Torres-Jerez, I.; Bedford, I.D.; Markham, P.G.; Brown, J.K. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene-sequences and a compilation of conserved polymerase chain-reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene a | Region | Size | Inc b | AT% | AT-Skew c | GC-Skew | Start Codon | Stop Codon | Anticodon |
|---|---|---|---|---|---|---|---|---|---|
| cox1 | 1–1539 | 1539 | 1 | 64.39 | −0.322 | 0.263 | ATG | TAA | - |
| trnL2 | 1542–1606 | 65 | 2 | 70.77 | −0.087 | 0.474 | - | - | TAA |
| cox2 | 1607–2270 | 664 | 0 | 68.67 | −0.197 | 0.202 | ATT | T- e | - |
| trnK | 2271–2340 | 70 | 0 | 71.43 | 0.000 | −0.100 | - | - | CTT |
| trnD | 2347–2408 | 62 | 6 | 66.13 | −0.171 | 0.333 | - | - | GTC |
| atp8 | 2418–2564 | 147 | 9 | 62.59 | −0.217 | 0.273 | ATA | TAA | - |
| atp6 | 2558–3208 | 651 | −7 | 71.27 | −0.388 | 0.241 | ATT | TAG | - |
| trnE | 3233–3293 | 61 | 24 | 72.13 | 0.000 | 0.059 | - | - | TTC |
| trnF | 3301–3363 | 63 | 7 | 76.19 | 0.292 | 0.067 | - | - | GAA |
| nad5 | 3364–5023 | 1660 | 0 | 73.13 | 0.137 | −0.166 | ATA | T- | - |
| trnH | 5024–5084 | 61 | 0 | 81.97 | 0.040 | 0.455 | - | - | GTG |
| nad4 | 5086–6366 | 1281 | 1 | 69.95 | −0.007 | −0.179 | ATA | TAG | - |
| nad4L | 6363–6659 | 297 | −4 | 76.77 | −0.018 | −0.246 | ATG | TAA | - |
| trnT | 6661–6724 | 64 | 1 | 76.56 | −0.102 | 0.333 | - | - | TGT |
| trnP | 6723–6784 | 62 | −2 | 72.58 | 0.111 | −0.059 | - | - | TGG |
| nad6 | 6816–7250 | 435 | 31 | 70.57 | −0.270 | 0.328 | ATA | TAA | - |
| cob | 7250–8383 | 1134 | −1 | 64.90 | −0.351 | 0.186 | ATG | TAA | - |
| trnS2 | 8384–8439 | 56 | 0 | 80.36 | −0.067 | 0.273 | - | - | TGA |
| nad1 | 8450–9359 | 910 | 10 | 72.86 | 0.005 | −0.198 | ATT | T- | - |
| trnL1 | 9360–9422 | 63 | 0 | 84.13 | 0.132 | 0.200 | - | - | TAG |
| rrnL | 9423–10612 | 1190 | 0 | 77.23 | 0.134 | −0.055 | - | - | - |
| rrnS | 10707–11435 | 729 | 94 | 75.99 | 0.022 | 0.074 | - | - | - |
| trnN | 11483–11548 | 66 | 47 | 78.79 | 0.115 | 0.143 | - | - | ATT |
| trnQ | 11553–11623 | 71 | 4 | 87.32 | 0.000 | −0.111 | - | - | TTG |
| trnV | 11617–11680 | 64 | −7 | 73.44 | −0.021 | 0.059 | - | - | TAC |
| trnR | 11678–11742 | 65 | −3 | 73.85 | −0.042 | 0.059 | - | - | TCG |
| trnA | 11744–11805 | 62 | 1 | 75.81 | −0.021 | 0.200 | - | - | TGC |
| nad3 | 11806–12159 | 354 | 0 | 72.60 | −0.276 | 0.052 | ATT | TAA | - |
| trnG | 12161–12222 | 63 | 1 | 82.54 | −0.038 | 0.273 | - | - | TCC |
| cox3 | 12223–13009 | 786 | 0 | 66.92 | −0.198 | 0.115 | ATG | TAA | - |
| trnS1 | 13025–13086 | 62 | 15 | 61.29 | −0.263 | 0.083 | - | - | TCT |
| CR d | 13087–13961 | 875 | 0 | 66.86 | −0.050 | 0.117 | - | - | - |
| trnM | 13962–14028 | 67 | 0 | 77.61 | 0.077 | −0.067 | - | - | CAT |
| nad2 | 14029–15003 | 975 | 0 | 68.72 | −0.287 | 0.279 | ATA | TAA | - |
| trnW | 15002–15064 | 63 | −2 | 82.54 | 0.077 | 0.091 | - | - | TCA |
| trnY | 15063–15124 | 60 | −2 | 86.67 | 0.154 | 0.500 | - | - | GTA |
| trnC | 15125–15187 | 63 | 0 | 92.06 | 0.138 | 0.200 | - | - | GCA |
| Region | A% | C% | G% | T% | AT% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|
| Full length | 31.02 | 12.38 | 16.72 | 39.87 | 70.90 | −0.125 | 0.149 |
| PCGs | 29.27 | 14.12 | 16.64 | 39.96 | 69.49 | −0.154 | 0.082 |
| 1st codon | 35.12 | 13.20 | 20.62 | 31.05 | 66.37 | 0.061 | 0.219 |
| 2nd codon | 18.81 | 17.70 | 15.12 | 48.37 | 68.08 | −0.440 | −0.078 |
| 3rd codon | 33.88 | 11.47 | 14.18 | 40.47 | 74.02 | −0.089 | 0.106 |
| tRNA genes | 39.51 | 9.75 | 13.04 | 37.71 | 77.19 | 0.023 | 0.145 |
| rRNA genes | 41.90 | 11.67 | 11.57 | 34.86 | 76.61 | 0.092 | −0.004 |
| Codon | N | % | RSCU | Codon | N | % | RSCU | Codon | N | % | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UAA (*) | 8 | 0.22 | 1.60 | AAA (K) | 79 | 2.19 | 1.30 | AGA (S) | 76 | 2.11 | 1.63 |
| UAG (*) | 2 | 0.06 | 0.40 | AAG (K) | 43 | 1.19 | 0.71 | AGC (S) | 23 | 0.64 | 0.49 |
| GCA (A) | 62 | 1.72 | 1.56 | CUA (L) | 91 | 2.52 | 1.54 | AGG (S) | 22 | 0.61 | 0.47 |
| GCC (A) | 23 | 0.64 | 0.58 | CUC (L) | 16 | 0.44 | 0.27 | AGU (S) | 55 | 1.52 | 1.18 |
| GCG (A) | 14 | 0.39 | 0.35 | CUG (L) | 42 | 1.16 | 0.71 | UCA (S) | 83 | 2.30 | 1.78 |
| GCU (A) | 60 | 1.66 | 1.51 | CUU (L) | 87 | 2.41 | 1.48 | UCC (S) | 18 | 0.50 | 0.39 |
| UGC (C) | 17 | 0.47 | 0.74 | UUA (L) | 178 | 4.93 | 1.33 | UCG (S) | 17 | 0.47 | 0.36 |
| UGU (C) | 29 | 0.80 | 1.26 | UUG (L) | 90 | 2.49 | 0.67 | UCU (S) | 80 | 2.22 | 1.71 |
| GAC (D) | 22 | 0.61 | 0.70 | AUA (M) | 225 | 6.23 | 1.54 | ACA (T) | 76 | 2.11 | 1.62 |
| GAU (D) | 41 | 1.14 | 1.30 | AUG (M) | 67 | 1.86 | 0.46 | ACC (T) | 19 | 0.53 | 0.40 |
| GAA (E) | 57 | 1.58 | 1.37 | AAC (N) | 48 | 1.33 | 0.63 | ACG (T) | 6 | 0.17 | 0.13 |
| GAG (E) | 26 | 0.72 | 0.63 | AAU (N) | 104 | 2.88 | 1.37 | ACU (T) | 87 | 2.41 | 1.85 |
| UUC (F) | 46 | 1.27 | 0.26 | CCA (P) | 26 | 0.72 | 1.11 | GUA (V) | 85 | 2.35 | 1.30 |
| UUU (F) | 304 | 8.42 | 1.74 | CCC (P) | 18 | 0.50 | 0.77 | GUC (V) | 21 | 0.58 | 0.32 |
| GGA (G) | 62 | 1.72 | 1.39 | CCG (P) | 13 | 0.36 | 0.55 | GUG (V) | 49 | 1.36 | 0.75 |
| GGC (G) | 20 | 0.55 | 0.45 | CCU (P) | 37 | 1.02 | 1.57 | GUU (V) | 106 | 2.94 | 1.63 |
| GGG (G) | 55 | 1.52 | 1.23 | CAA (Q) | 29 | 0.80 | 1.14 | UGA (W) | 68 | 1.88 | 1.36 |
| GGU (G) | 42 | 1.16 | 0.94 | CAG (Q) | 22 | 0.61 | 0.86 | UGG (W) | 32 | 0.89 | 0.64 |
| CAC (H) | 22 | 0.61 | 0.86 | CGA (R) | 18 | 0.50 | 1.60 | UAC (Y) | 48 | 1.33 | 0.65 |
| CAU (H) | 29 | 0.80 | 1.14 | CGC (R) | 5 | 0.14 | 0.44 | UAU (Y) | 99 | 2.74 | 1.35 |
| AUC (I) | 48 | 1.33 | 0.28 | CGG (R) | 12 | 0.33 | 1.07 | - | - | - | - |
| AUU (I) | 291 | 8.06 | 1.72 | CGU (R) | 10 | 0.28 | 0.89 | - | - | - | - |
| Region | Primer Name | Primer Sequence (5′–3′) | References |
|---|---|---|---|
| cox1-nad5 | C1-J-2195 | TTGATTTTTTGGTCATCCAGAAGT | [45] |
| N5-N7793 | TTAGGTTGRGATGGNYTAGG | [12] | |
| nad5-cob | AC-N5J | CAGCAGTTACTAGAGTTGATGAGT | In this study |
| AC-CBN | TCTCCACAAACAATAAAACAAGC | In this study | |
| cob-rrnL | CBF1 | TATGTACTACCATGAGGACAAATATC | [46] |
| AC-16SN | GTCTAAGTCTTAAATAAACTCTGC | In this study | |
| rrnL | 16Sar | CGCCTGTTTAACAAAAACAT | [46] |
| 16Sbr | CCGGTCTGAACTCAGATCACGT | [46] | |
| rrnL-rrnS | AC-16SJ | TTAGAGGAACCTGCTTAGTAATC | In this study |
| AC-12SN | GCAGAGTTTATTTAAGACTTAGAC | In this study | |
| rrnS | 12SRNA-F | AAACTAGGATTAGATACCCTATTAT | [46] |
| 12SRNA-R | AAGAGCGACGGGCGATGTGT | [46] | |
| rrnS-cox1 | AC-12SJ | CCCTGATACAAAAGGTACAAGTCT | In this study |
| AC-C1N | CCCATAATAGCAAATACAATCCCT | In this study |
| Species | Genome Size | Acc. Number | References |
|---|---|---|---|
| Bemisia tabaci complex sp. Asia I | 15,210 bp | KJ778614 | [27] |
| Bemisia tabaci | 15,632 bp | JQ906700 | [23] |
| Bemisia tabaci | 15,322 bp | AY521259 | [21] |
| Bemisia afer | 15,300 bp | KR819174 | [24] |
| Bemisia afer | 14,968 bp | KF734668 | [22] |
| Tetraleurodes acaciae | 15,080 bp | AY521262 | [21] |
| Neomaskellia andropogonis | 14,496 bp | AY572539 | [21] |
| Aleurochiton aceris | 15,388 bp | AY572538 | [21] |
| Aleurodicus dugesii | 15,723 bp | AY521251 | [21] |
| Trialeurodes vaporariorum | 18,414 bp | AY521265 | [21] |
| Drosophila melanogaster | 19,517 bp | DMU37541 | [42] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-C.; Wang, X.-Q.; Li, P.-W.; Hu, X.; Wang, J.-J.; Peng, P. The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae. Int. J. Mol. Sci. 2016, 17, 1843. https://doi.org/10.3390/ijms17111843
Chen S-C, Wang X-Q, Li P-W, Hu X, Wang J-J, Peng P. The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae. International Journal of Molecular Sciences. 2016; 17(11):1843. https://doi.org/10.3390/ijms17111843
Chicago/Turabian StyleChen, Shi-Chun, Xiao-Qing Wang, Pin-Wu Li, Xiang Hu, Jin-Jun Wang, and Ping Peng. 2016. "The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae" International Journal of Molecular Sciences 17, no. 11: 1843. https://doi.org/10.3390/ijms17111843
APA StyleChen, S. -C., Wang, X. -Q., Li, P. -W., Hu, X., Wang, J. -J., & Peng, P. (2016). The Complete Mitochondrial Genome of Aleurocanthus camelliae: Insights into Gene Arrangement and Genome Organization within the Family Aleyrodidae. International Journal of Molecular Sciences, 17(11), 1843. https://doi.org/10.3390/ijms17111843