
CPP-Assisted Intracellular Drug Delivery, What Is Next?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
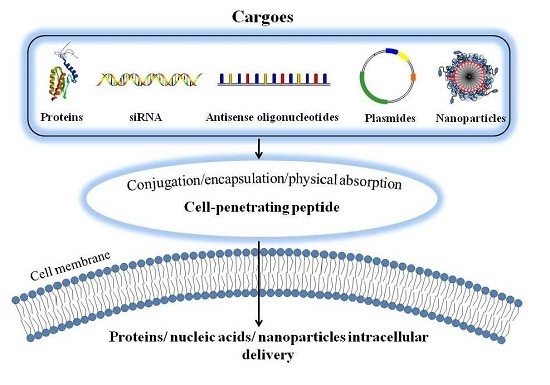
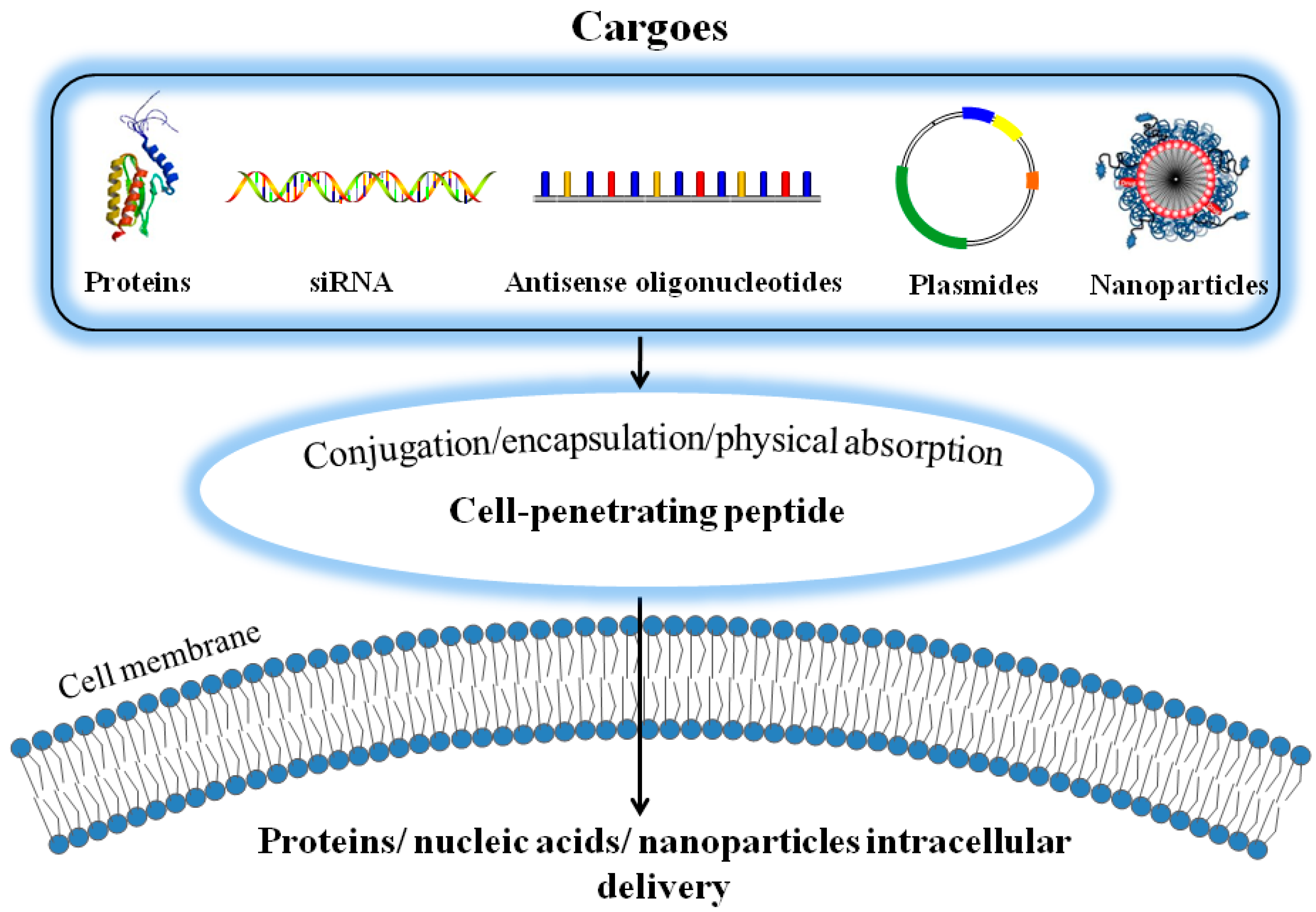
2. Cell Penetrating Peptides (CPPs)-Assisted Strategies for Intracellular Drug Delivery
2.1. Covalent Conjugation of Therapeutic Agents with CPP
2.2. Encapsulation of Therapeutic Agents into CPP-Linked Nano-Carriers
2.3. Physical Adsorption of Therapeutic Agents with CPPs via Electrostatic Complexation
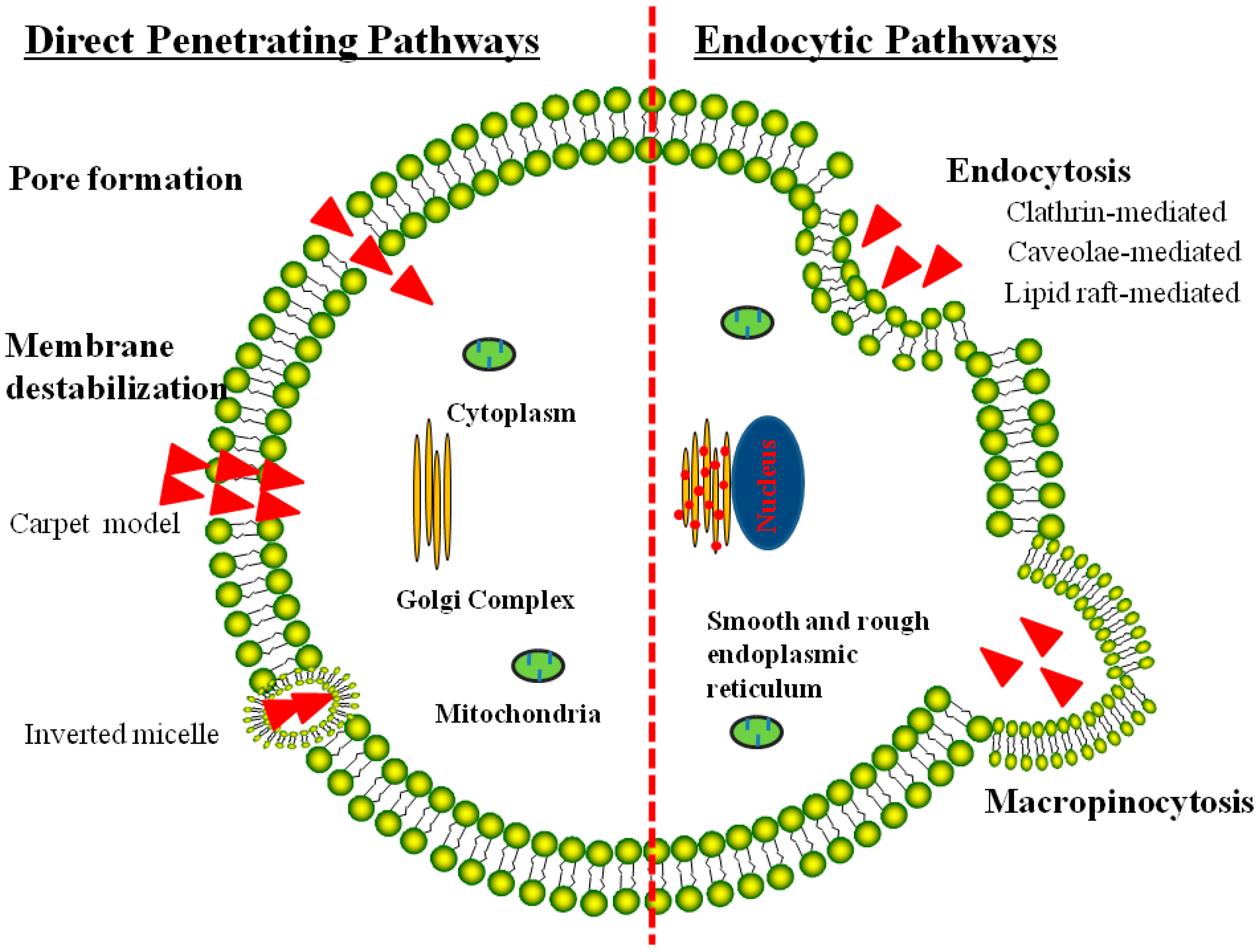
3. Mechanisms of the CPP-Mediated Intracellular Uptake
3.1. Direct Translocation of CPPs across Biological Membranes
3.2. Endocytosis Pathway for CPP-Mediated Cell Internalization
4. Technical Limitations Concerning CPPs-Assisted Intracellular Delivery
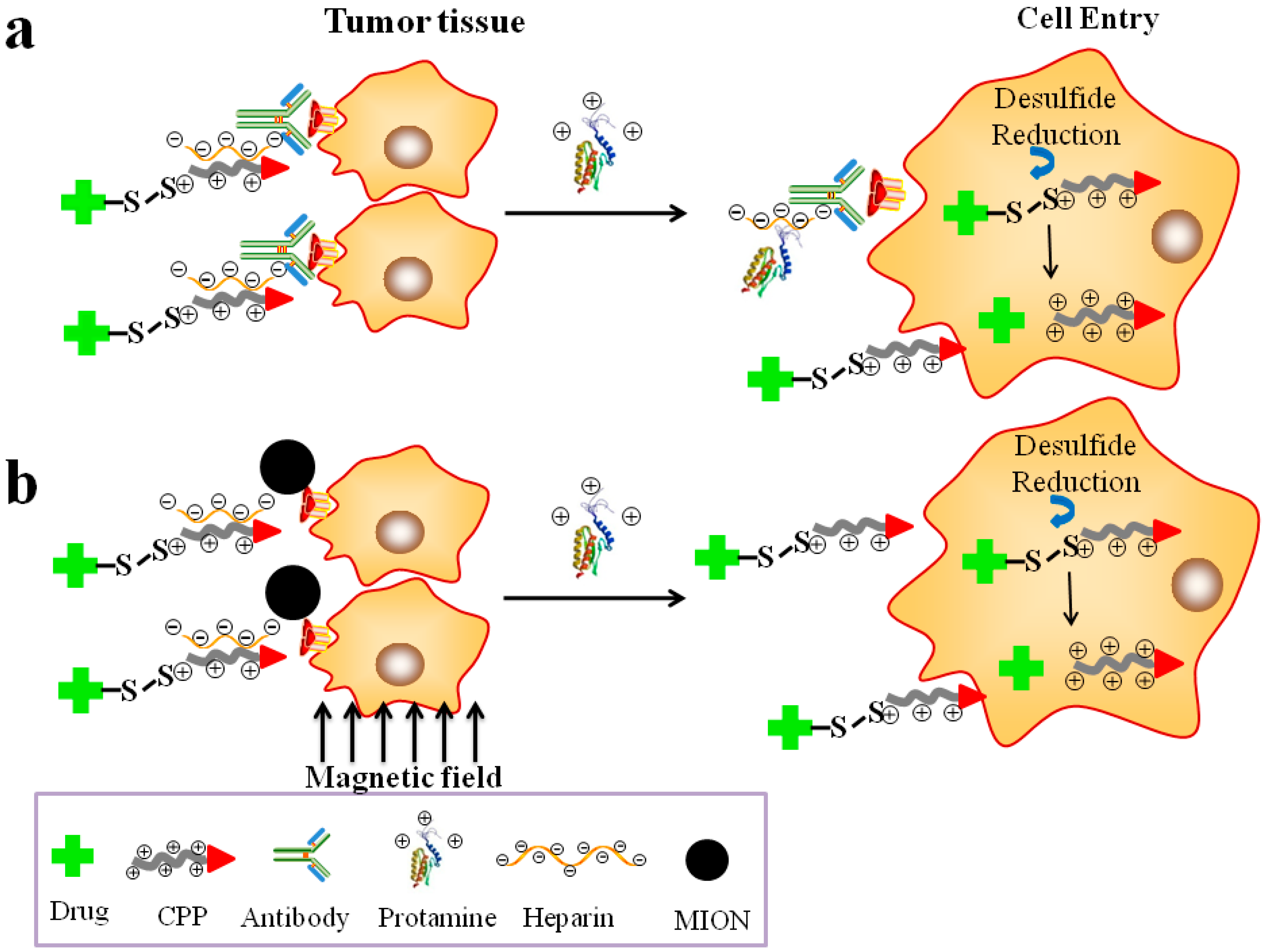
5. Stimulus-Responsive “Smart” Systems for CPP-Mediated Cancer Therapy
5.1. Prodrug-Based, CPP-Mediated Smart Drug Delivery Systems
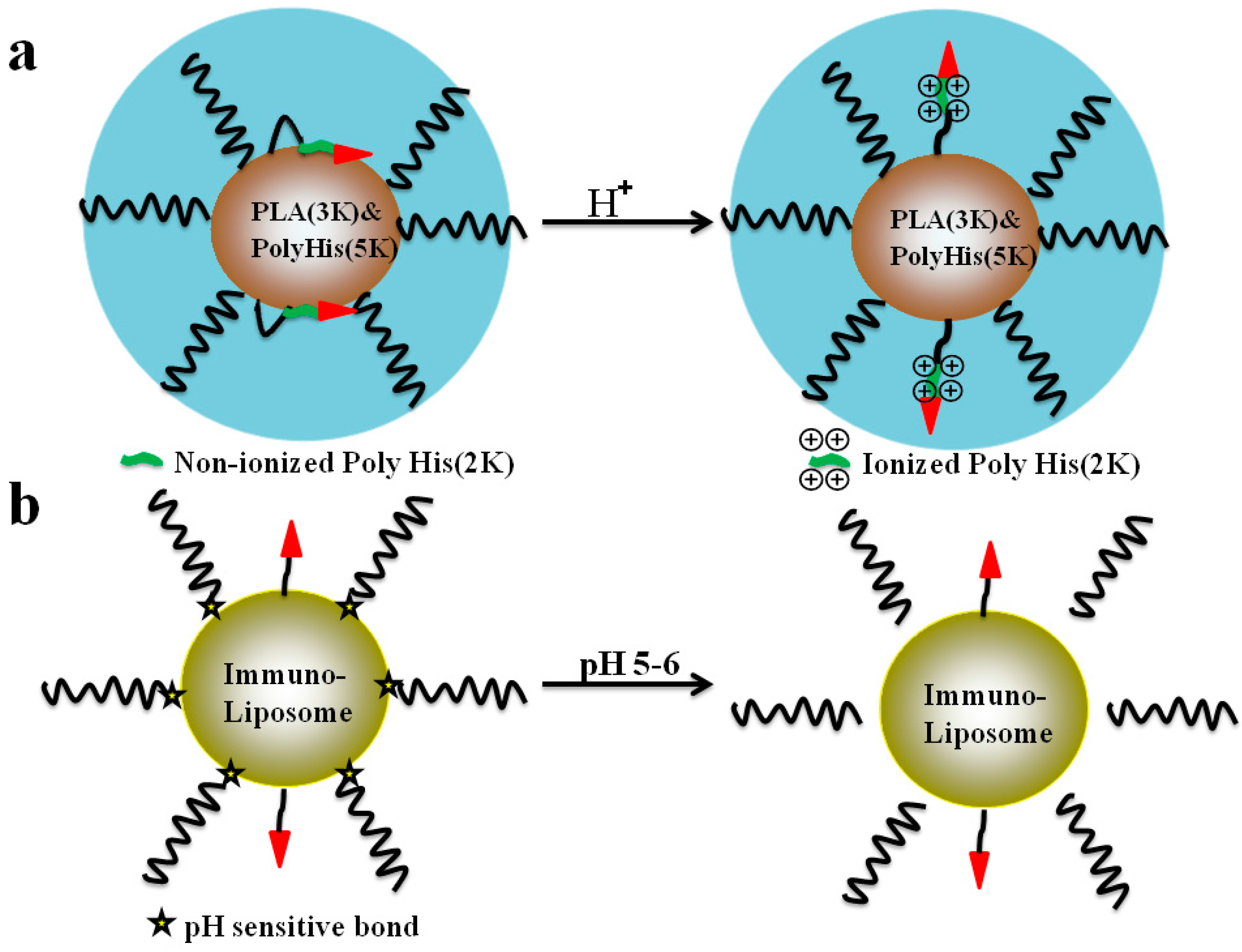
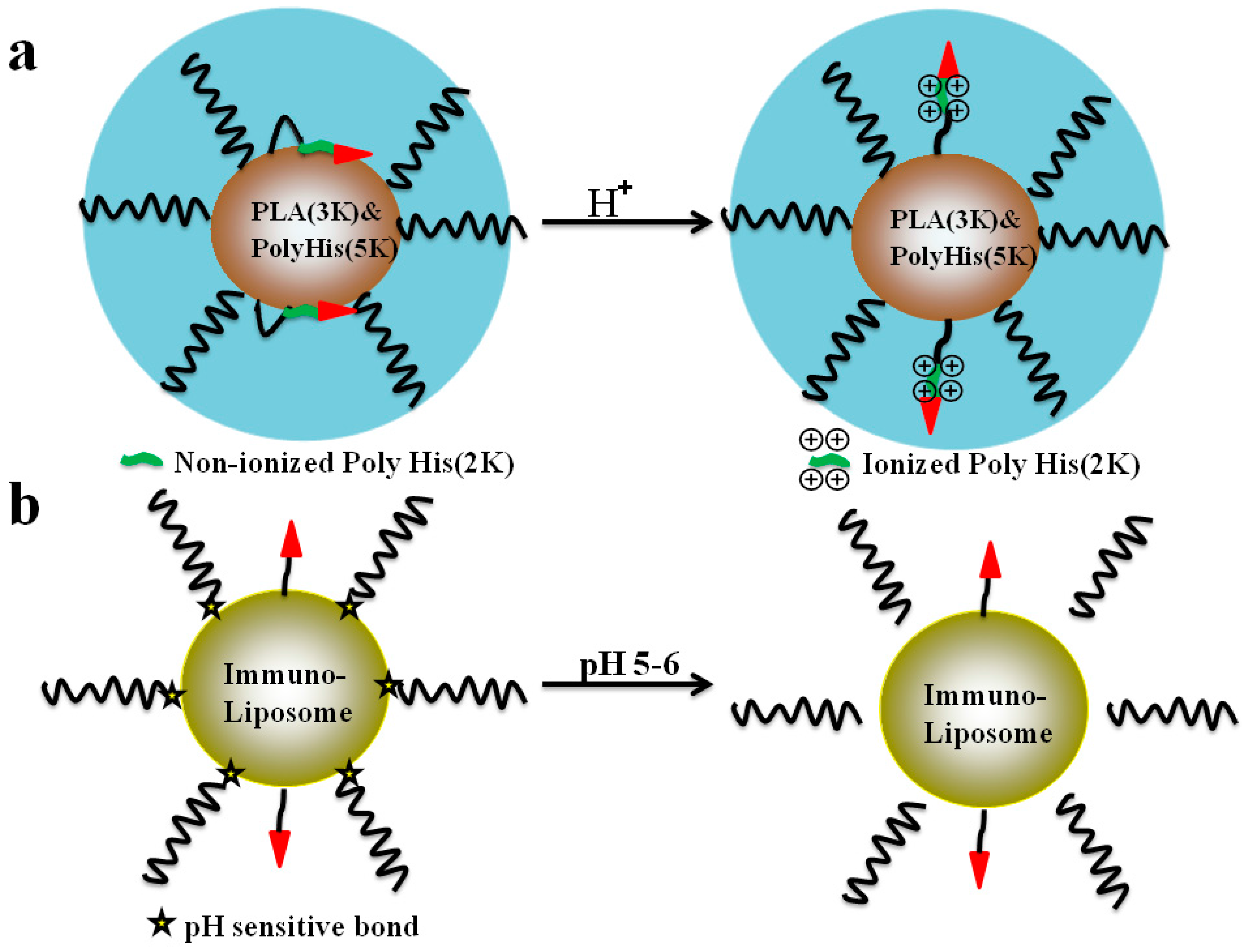
5.2. The pH-Triggered, CPP-Mediated, “Smart” Intracellular Drug Delivery Systems
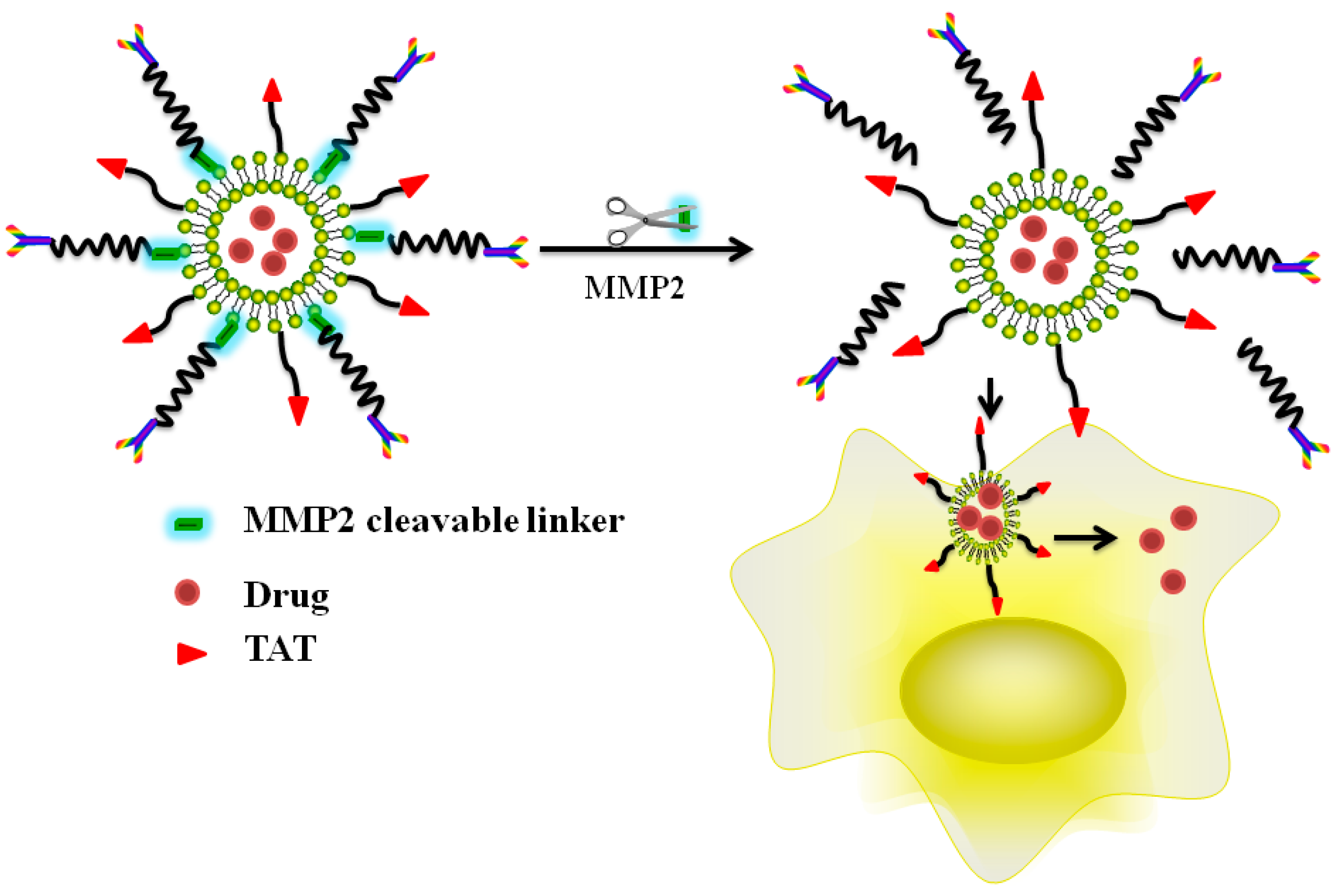
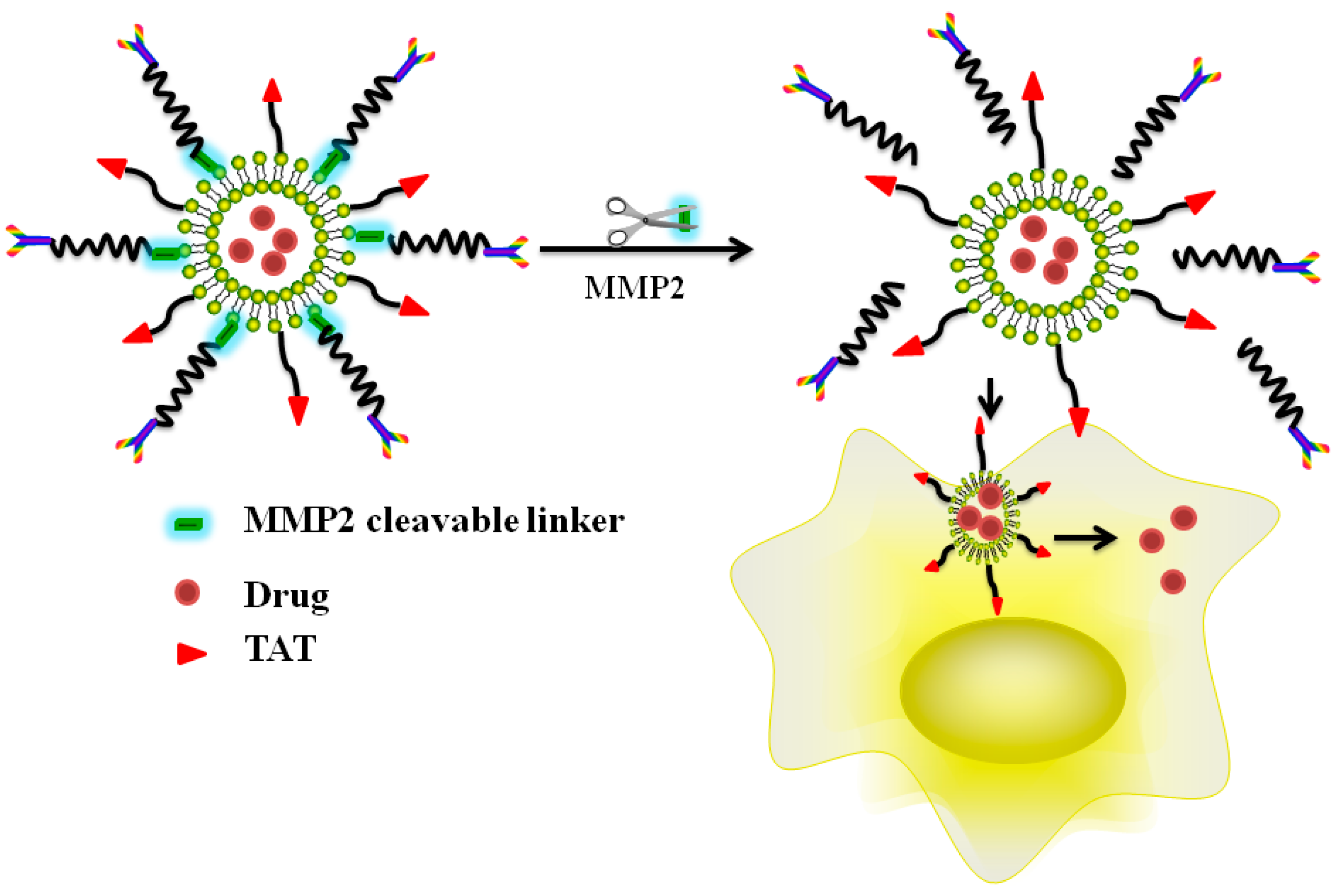
5.3. Enzyme Triggered, CPP-Mediated “Smart” Drug Delivery Systems
6. Conclusions and Outlooks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Intracellular delivery of protein and peptide therapeutics. Drug Discov. Today Technol. 2008, 5, e95–e103. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006, 8, 343–375. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.; Lebleu, B.; Vives, E. Tat peptide-mediated cellular delivery: Back to basics. Adv. Drug Deliv. Rev. 2005, 57, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, R.; Divita, G. Happy birthday cell penetrating peptides: Already 20 years. Biochim. Biophys. Acta 2010, 1798, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Hallbrink, M.; Floren, A.; Elmquist, A.; Pooga, M.; Bartfai, T.; Langel, U. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta 2001, 1515, 101–109. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Cell-penetrating peptides: Tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Dietz, G.P.; Kilic, E.; Bahr, M. Inhibition of neuronal apoptosis in vitro and in vivo using TAT-mediated protein transduction. Mol. Cell. Neurosci. 2002, 21, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.M.; Li, Y.T.; Liang, J.F.; Park, Y.J.; Chang, L.C.; Yang, V.C. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: A proof-of-concept investigation. J. Control. Release 2008, 130, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Weissleder, R. Intracellular cargo delivery using tat peptide and derivatives. Med. Res. Rev. 2004, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vives, E. Present and future of cell-penetrating peptide mediated delivery systems: Is the Trojan horse too wild to go only to Troy? J. Control. Release 2005, 109, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Park, Y.S.; Moon, C.; David, A.E.; Chung, H.S.; Yang, V.C. Synthetic skin-permeable proteins enabling needleless immunization. Angew. Chem. Int. Ed. Eng. 2010, 49, 2724–2727. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ye, J.; Wang, Y.; Liu, Q.; Chung, H.S.; Kwon, Y.M.; Shin, M.C.; Lee, K.; Yang, V.C. Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. J. Control. Release 2014, 176, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Dietz, G.P.; Bahr, M. Peptide-enhanced cellular internalization of proteins in neuroscience. Brain Res. Bull. 2005, 68, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Morishita, M.; Eda, Y.; Ida, N.; Nishio, R.; Takayama, K. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J. Control. Release 2008, 132, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Morishita, M.; Takayama, K. Importance of intermolecular interaction on the improvement of intestinal therapeutic peptide/protein absorption using cell-penetrating peptides. J. Control. Release 2009, 136, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Barnett, E.M.; Elangovan, B.; Bullok, K.E.; Piwnica-Worms, D. Selective cell uptake of modified TAT peptide-fluorophore conjugates in rat retina in ex vivo and in vivo models. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2589–2595. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.B.; Brophy, C.M.; Furnish, E.; Flynn, C.R.; Sparks, O.; Komalavilas, P.; Joshi, L.; Panitch, A.; Bentley, M.V. Comparative study of the skin penetration of protein transduction domains and a conjugated peptide. Pharm. Res. 2005, 22, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus TAT trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- He, H.; Ye, J.; Liu, E.; Liang, Q.; Liu, Q.; Yang, V.C. Low molecular weight protamine (LMWP): A nontoxic protamine substitute and an effective cell-penetrating peptide. J. Control. Release 2014, 193, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Munyendo, W.L.; Lv, H.; Benza-Ingoula, H.; Baraza, L.D.; Zhou, J. Cell penetrating peptides in the delivery of biopharmaceuticals. Biomolecules 2012, 2, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Kurrikoff, K.; Langel, U. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Dong, W.; Gong, J.; Wang, J.; Yang, V.C. Developing macromolecular therapeutics: The future drug-of-choice. Front. Chem. Eng. China 2010, 4, 10–17. [Google Scholar] [CrossRef]
- Jarver, P.; Langel, K.; El-Andaloussi, S.; Langel, U. Applications of cell-penetrating peptides in regulation of gene expression. Biochem. Soc. Trans. 2007, 35, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Liu, B.R.; Dai, Y.H.; Lee, C.Y.; Chan, M.H.; Chen, H.H.; Chiang, H.J.; Lee, H.J. A gene delivery system for insect cells mediated by arginine-rich cell-penetrating peptides. Gene 2011, 493, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Chugh, A.; Eudes, F. Cellular uptake of cell-penetrating peptides pVEC and transportan in plants. J. Pept. Sci. 2008, 14, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Dietz, G.P.; Bahr, M. Synthesis of cell-penetrating peptides and their application in neurobiology. Methods Mol. Biol. 2007, 399, 181–198. [Google Scholar] [PubMed]
- Henriques, S.T.; Costa, J.; Castanho, M.A. Translocation of β-galactosidase mediated by the cell-penetrating peptide PEP-1 into lipid vesicles and human HeLa cells is driven by membrane electrostatic potential. Biochemistry 2005, 44, 10189–10198. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [PubMed]
- He, H.N.; Ye, J.X.; Sheng, J.Y.; Wang, J.X.; Huang, Y.Z.; Chen, G.Y. Overcoming oral insulin delivery barriers: Application of cell penetrating peptide and silica-based nanoporous composites. Front. Chem. Sci. Eng. 2013, 7, 9–19. [Google Scholar] [CrossRef]
- Dohmen, C.; Wagner, E. Multifunctional CPP polymer system for tumor-targeted pDNA and siRNA delivery. Methods Mol. Biol. 2011, 683, 453–463. [Google Scholar] [PubMed]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.W.; Liu, B.R.; Wu, C.Y.; Lu, S.W.; Lee, H.J. Protein transport in human cells mediated by covalently and noncovalently conjugated arginine-rich intracellular delivery peptides. Peptides 2009, 30, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Harenberg, J.; Jeschek, M.; Acker, M.; Malsch, R.; Huhle, G.; Heene, D.L. Effects of low-molecular-weight dermatan sulfate on coagulation, fibrinolysis and tissue factor pathway inhibitor in healthy volunteers. Blood Coagul. Fibrinolysis 1996, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Dowdy, S.F. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Deliv. Rev. 2005, 57, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Dietz, G.P.; Bahr, M. Delivery of bioactive molecules into the cell: The Trojan horse approach. Mol. Cell. Neurosci. 2004, 27, 85–131. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Chou, J.C.; Chen, C.P.; Liu, B.R.; Lee, H.J. Noncovalent protein transduction in plant cells by macropinocytosis. New Phytol. 2007, 174, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.W.; Chan, M.H.; Hsu, H.R.; Liu, B.R.; Chen, C.P.; Chen, H.H.; Lee, H.J. Transdermal delivery of proteins mediated by non-covalently associated arginine-rich intracellular delivery peptides. Exp. Dermatol. 2007, 16, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides—An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shin, M.C.; Liang, Q.; He, H.; Yang, V.C. 15 years of ATTEMPTS: A macromolecular drug delivery system based on the CPP-mediated intracellular drug delivery and antibody targeting. J. Control. Release 2015, 10, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.R.; Xu, G.; Becker-Hapak, M.; Dowdy, S.F.; McLeod, H.L. The kinetics and tissue distribution of protein transduction in mice. Eur. J. Pharm. Sci. 2006, 27, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, S.; Ziegler, E.; Rolver, L.; Linkermann, A.; Keyser, K.A.; Steen, P.; Wollert, K.C.; Korf-Klingebiel, M.; Kunzendorf, U. Effective blockage of both the extrinsic and intrinsic pathways of apoptosis in mice by TAT-crmA. J. Biol. Chem. 2010, 285, 19997–20005. [Google Scholar] [CrossRef] [PubMed]
- Essafi, M.; Baudot, A.D.; Mouska, X.; Cassuto, J.P.; Ticchioni, M.; Deckert, M. Cell-penetrating TAT-FOXO3 fusion proteins induce apoptotic cell death in leukemic cells. Mol. Cancer Ther. 2011, 10, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Dowdy, S.F. In vivo protein transduction: Intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharm. Sci. 2000, 21, 45–48. [Google Scholar] [CrossRef]
- Zanta, M.A.; Belguise-Valladier, P.; Behr, J.P. Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. USA 1999, 96, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Liang, J.F.; Ko, K.S.; Kim, S.W.; Yang, V.C. Low molecular weight protamine as an efficient and nontoxic gene carrier: In vitro study. J. Gene Med. 2003, 5, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Turner, J.J.; Oretskaya, T.S.; Gait, M.J. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr. Pharm. Des. 2005, 11, 3639–3654. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.; Schillinger, U.; Ortiz, A.; Tabatt, K.; Plank, C.; Muller, R.H.; Rosenecker, J. Application of novel solid lipid nanoparticle (SLN)-gene vector formulations based on a dimeric HIV-1 TAT-peptide in vitro and in vivo. Pharm. Res. 2004, 21, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers 2008, 90, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. TATP-mediated intracellular delivery of pharmaceutical nanocarriers. Biochem. Soc. Trans. 2007, 35, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Fuente, J.M.; Berry, C.C. TAT peptide as an efficient molecule to translocate gold nanoparticles into the cell nucleus. Bioconjug. Chem. 2005, 16, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Meyer, K.; Rennert, R.; Neundorf, I. Quantum dot-carrier peptide conjugates suitable for imaging and delivery applications. Bioconjug. Chem. 2008, 19, 2346–2356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shin, M.C.; David, A.E.; Zhou, J.; Lee, K.; He, H.; Yang, V.C. Long-circulating heparin-functionalized magnetic nanoparticles for potential application as a protein drug delivery platform. Mol. Pharm. 2013, 10, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; David, A.E.; Yang, V.C. Magnetic tumor targeting of β-glucosidase immobilized iron oxide nanoparticles. Nanotechnology 2011, 24, 375102. [Google Scholar] [CrossRef] [PubMed]
- Weisz, P.B.; Joullie, M.M.; Hunter, C.M.; Kumor, K.M.; Zhang, Z.; Levine, E.; Macarak, E.; Weiner, D.; Barnathan, E.S. A basic compositional requirement of agents having heparin-like cell-modulating activities. Biochem. Pharmacol. 1997, 54, 149–157. [Google Scholar] [CrossRef]
- Sheng, J.; He, H.; Han, L.; Qin, J.; Chen, S.; Ru, G.; Li, R.; Yang, P.; Wang, J.; Yang, V.C. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J. Control. Release 2016, 233, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Sheng, J.; Ye, J.; Wang, Y.; Gong, J.; Yang, V.C.; Wang, J.; He, H. CPP mediated insulin delivery: Current status and promising future. Curr. Pharm. Biotechnol. 2014, 15, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Stroh, M.; Zimmer, J.P.; Duda, D.G.; Levchenko, T.S.; Cohen, K.S.; Brown, E.B.; Scadden, D.T.; Torchilin, V.P.; Bawendi, M.G.; Fukumura, D.; et al. Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo. Nat. Med. 2005, 11, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Lee, J.Y.; Suh, J.S.; Kwon, Y.M.; Lee, S.J.; Chung, J.K.; Lee, D.S.; Yang, V.C.; Chung, C.P.; Park, Y.J. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials 2010, 31, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Herbig, M.E.; Weller, K.; Krauss, U.; Beck-Sickinger, A.G.; Merkle, H.P.; Zerbe, O. Membrane surface-associated helices promote lipid interactions and cellular uptake of human calcitonin-derived cell penetrating peptides. Biophys. J. 2005, 89, 4056–4066. [Google Scholar] [CrossRef] [PubMed]
- Zaro, J.L.; Shen, W.C. Cationic and amphipathic cell-penetrating peptides (CPPs): Their structures and studies in drug delivery. Front. Chem. Sci. Eng. 2015, 9, 407–427. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Tunnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Richard, J.P.; Rispal, C.; Lebleu, B. TAT peptide internalization: Seeking the mechanism of entry. Curr. Protein. Pept. Sci. 2003, 4, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Sugishita, K.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with lipopolysaccharide-containing liposomes as a model for outer membranes of gram-negative bacteria. FEBS Lett. 1999, 449, 221–224. [Google Scholar] [CrossRef]
- Jarver, P.; Langel, U. Cell-penetrating peptides—A brief introduction. Biochim. Biophys. Acta 2006, 1758, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Xu, W.; Ding, Y.; Lu, S.; Chen, P. Uptake Mechanism and Direct Translocation of a New CPP for siRNA Delivery. Mol. Pharm. 2016, 13, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.E.; Nguyen, C.M.; Zelphati, O.; Tsai, Y.; Felgner, P.L. Analytical methods for the characterization of cationic lipid-nucleic acid complexes. Hum. Gene Ther. 1998, 9, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppe, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [PubMed]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. TAT peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar] [PubMed]
- Grunwald, J.; Rejtar, T.; Sawant, R.; Wang, Z.; Torchilin, V.P. TAT peptide and its conjugates: Proteolytic stability. Bioconjug. Chem. 2009, 20, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Apte, A.; Sawant, R.R.; Grunwald, J.; Torchilin, V.P. Cell-penetrating TAT peptide in drug delivery systems: Proteolytic stability requirements. Drug Deliv. 2011, 18, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Verdurmen, W.P.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hallbrink, M.; van Kuppevelt, T.H.; Brock, R. Preferential uptake of l-versus d-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.M.; Hurley, J.P.; Salmaso, S.; Kale, A.; Tolcheva, E.; Levchenko, T.S.; Torchilin, V.P. “SMART” drug delivery systems: Double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug. Chem. 2006, 17, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Omata, D.; Negishi, Y.; Hagiwara, S.; Yamamura, S.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K.; Nomizu, M.; Aramaki, Y. Bubble liposomes and ultrasound promoted endosomal escape of TAT-PEG liposomes as gene delivery carriers. Mol. Pharm. 2011, 8, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Yuan, W.; Li, W.; Qin, Y.; Tang, J.; Yuan, M.; Fu, L.; Ran, R.; Zhang, Z.; He, Q. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol. Pharm. 2011, 8, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- He, H.; David, A.; Chertok, B.; Cole, A.; Lee, K.; Zhang, J.; Wang, J.; Huang, Y.; Yang, V.C. Magnetic nanoparticles for tumor imaging and therapy: A so-called theranostic system. Pharm. Res. 2013, 30, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhang, L.; Huang, Y.; Sun, K.; David, A.E.; Yang, V.C. The magnetophoretic mobility and superparamagnetism of core-shell iron oxide nanoparticles with dual targeting and imaging functionality. Biomaterials 2010, 31, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Olsnes, S.; Pihl, A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J. Biol. Chem. 1980, 255, 6947–6953. [Google Scholar] [PubMed]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Moon, C.; Balthasar, J.P.; Yang, V.C. Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. J. Control. Release 2014, 194, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Park, Y.S.; Wang, J.; Moon, C.; Kwon, Y.M.; Chung, H.S.; Park, Y.J.; Yang, V.C. ATTEMPTS system: A macromolecular prodrug strategy for cancer drug delivery. Curr. Pharm. Des. 2010, 16, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Kwon, Y.M.; Spangrude, G.J.; Liang, J.F.; Chung, H.S.; Park, Y.J.; Yang, V.C. Preliminary in vivo evaluation of the protein transduction domain-modified ATTEMPTS approach in enhancing asparaginase therapy. J. Biomed. Mater. Res. A 2009, 91, 209–220. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liang, Q.; Shin, M.C.; Lee, K.; Gong, J.; Ye, J.; Liu, Q.; Wang, J.; Yang, V. Significance and strategies in developing delivery systems for bio-macromolecular drugs. Front. Chem. Sci. Eng. 2013, 7, 496–507. [Google Scholar] [CrossRef]
- Lee, E.S.; Gao, Z.; Kim, D.; Park, K.; Kwon, I.C.; Bae, Y.H. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J. Control. Release 2008, 129, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Apte, A.; Jani, A.; Torchilin, V.P. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J. Control. Release 2012, 160, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.Y.; Chen, J.X.; Wang, H.Y.; Li, C.; Chang, C.; Zhang, X.Z.; Zhuo, R.X. Core-shell nanosized assemblies mediated by the alpha-beta cyclodextrin dimer with a tumor-triggered targeting property. ACS Nano 2010, 4, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Karve, S.; Bandekar, A.; Ali, M.R.; Sofou, S. The pH-dependent association with cancer cells of tunable functionalized lipid vesicles with encapsulated doxorubicin for high cell-kill selectivity. Biomaterials 2010, 31, 4409–4416. [Google Scholar] [CrossRef] [PubMed]
- Du, J.Z.; Du, X.J.; Mao, C.Q.; Wang, J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Sun, L.; Ye, J.; Liu, E.; Chen, S.; Liang, Q.; Shin, M.C.; Yang, V.C. Enzyme-triggered, cell penetrating peptide-mediated delivery of anti-tumor agents. J. Control. Release 2016, 240, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kate, P.; Torchilin, V.P. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano 2012, 6, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; von Maltzahn, G.; Lord, M.E.; Park, J.H.; Agrawal, A.; Min, D.H.; Sailor, M.J.; Bhatia, S.N. Protease-triggered unveiling of bioactive nanoparticles. Small 2008, 4, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Ito, E.; Hayashi, Y.; Oishi, M.; Nagasaki, Y.; Danev, R.; Nagayama, K.; Kaji, N.; Kikuchi, H.; et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 2011, 32, 4306–4316. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Liu, E.; Yu, Z.; Pei, X.; Chen, S.; Zhang, P.; Shin, M.-C.; Gong, J.; He, H.; Yang, V.C. CPP-Assisted Intracellular Drug Delivery, What Is Next? Int. J. Mol. Sci. 2016, 17, 1892. https://doi.org/10.3390/ijms17111892
Ye J, Liu E, Yu Z, Pei X, Chen S, Zhang P, Shin M-C, Gong J, He H, Yang VC. CPP-Assisted Intracellular Drug Delivery, What Is Next? International Journal of Molecular Sciences. 2016; 17(11):1892. https://doi.org/10.3390/ijms17111892
Chicago/Turabian StyleYe, Junxiao, Ergang Liu, Zhili Yu, Xing Pei, Sunhui Chen, Pengwei Zhang, Meong-Cheol Shin, Junbo Gong, Huining He, and Victor C. Yang. 2016. "CPP-Assisted Intracellular Drug Delivery, What Is Next?" International Journal of Molecular Sciences 17, no. 11: 1892. https://doi.org/10.3390/ijms17111892