
Mechanisms of p53 Functional De-Regulation: Role of the IκB-α/p53 Complex



Abstract
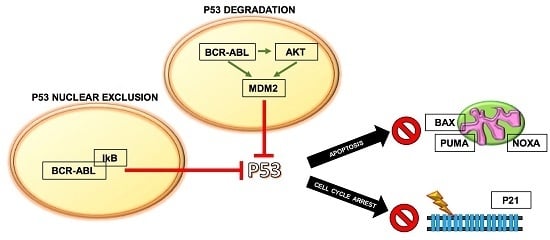
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
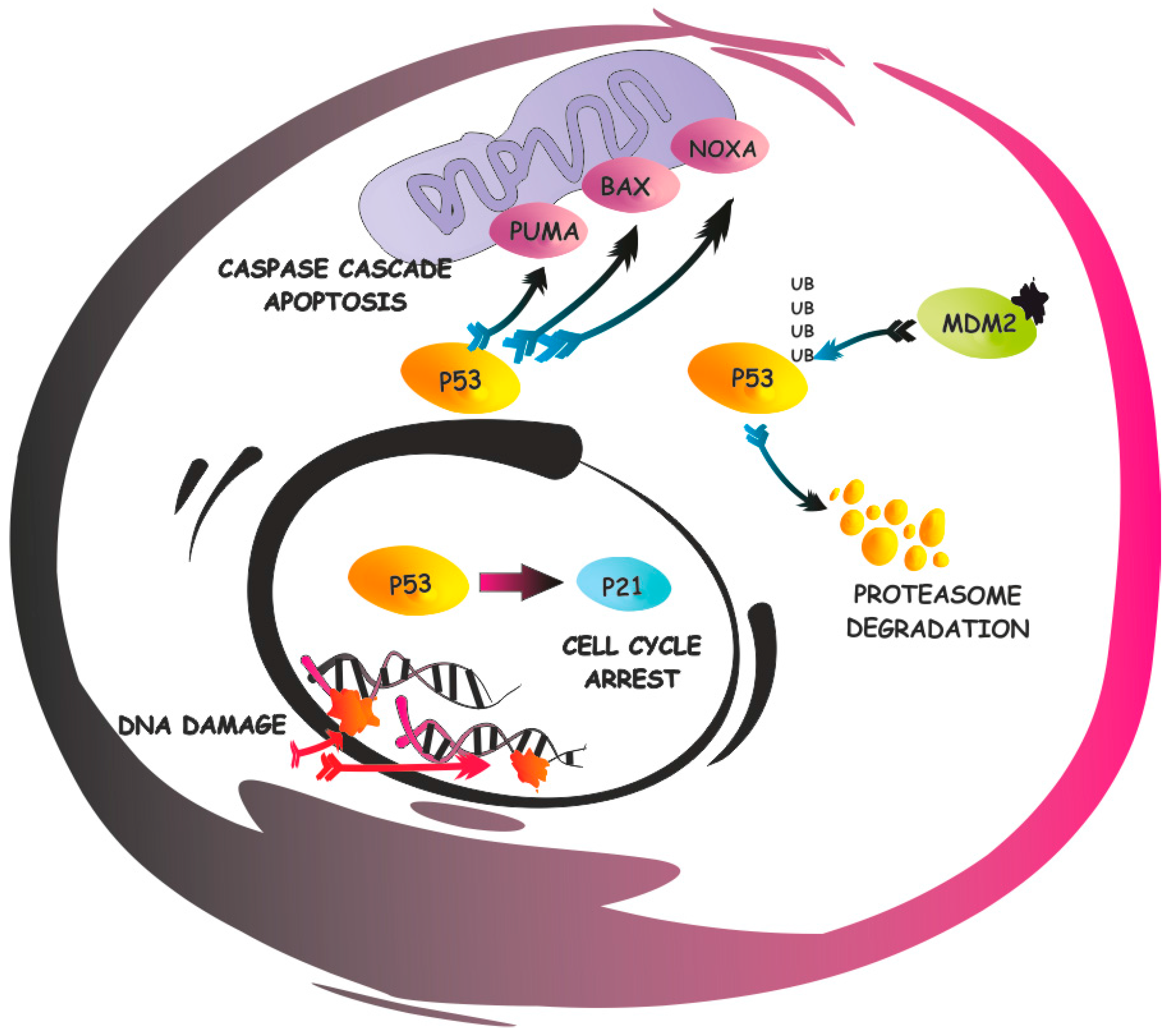
2. p53 Functional Inactivation
2.1. Mechanisms of p53 Shuttling
2.2. p53 Regulatory Proteins
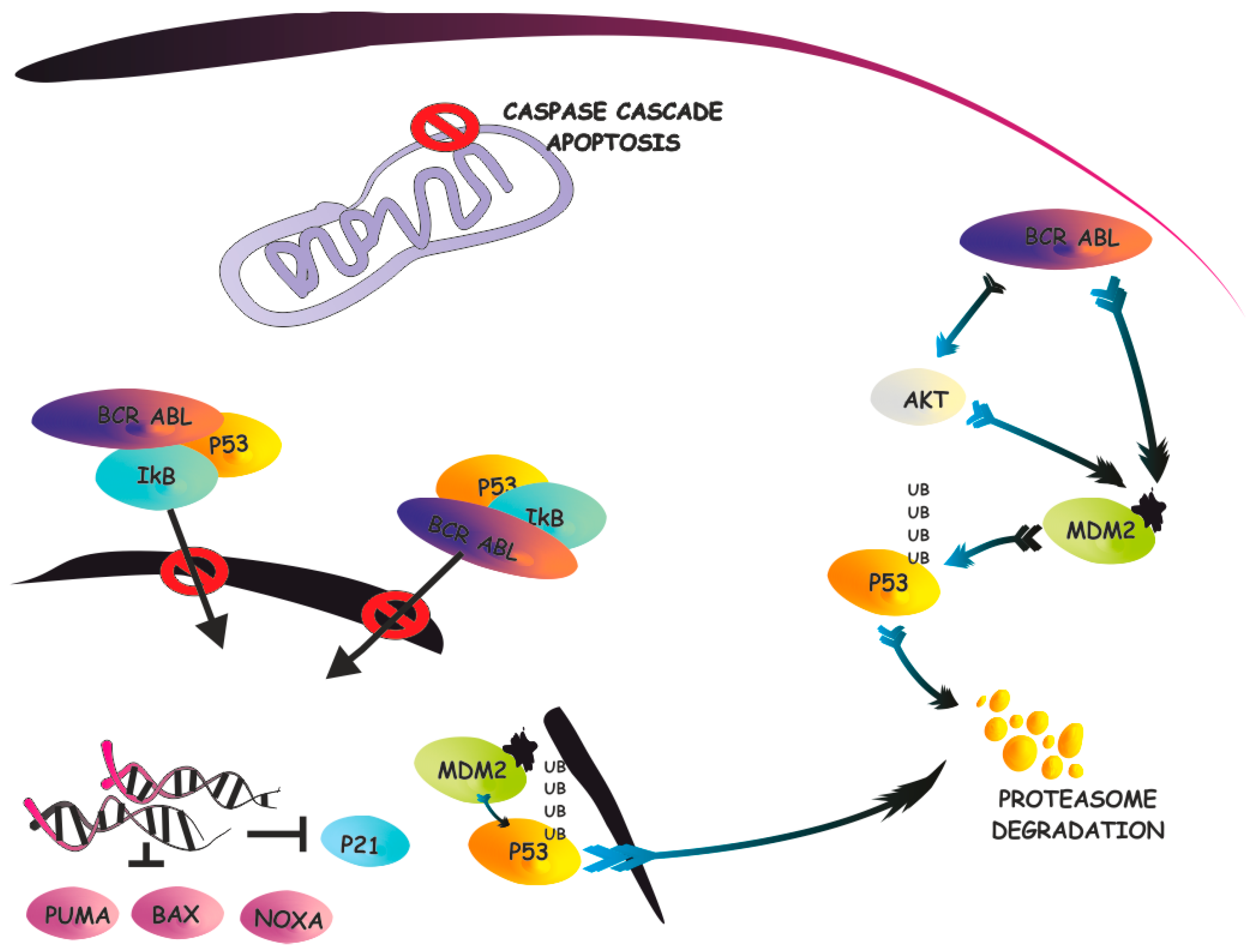
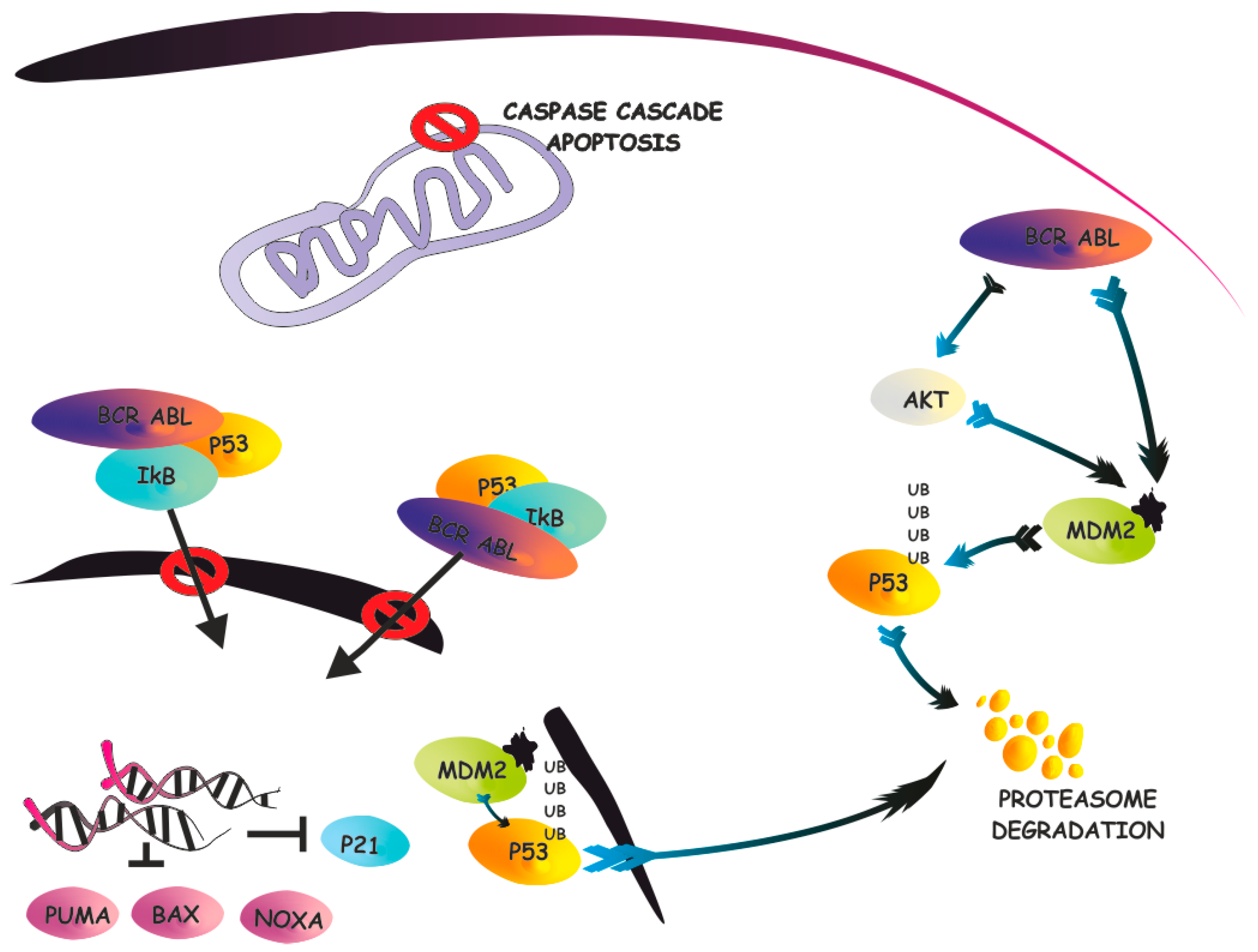
3. The IκB-α/p53 Connection
4. Targeting the IκB-α/p53 Network
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K.G. Shaping genetic alterations in human cancer: The p53 mutation paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Stracquadanio, G.; Wang, X.; Wallace, M.D.; Grawenda, A.M.; Zhang, P.; Hewitt, J.; Zeron-Medina, J.; Castro-Giner, F.; Tomlinson, I.P.; Goding, C.R.; et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat. Rev. Cancer 2016, 16, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Lowe, S.W. Cancer: Mutant p53 and chromatin regulation. Nature 2015, 525, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Hoe, K.K.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Attardi, L.D. Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 2013, 20, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.G.; Espinosa, J.M. The crusade against mutant p53: Does the COMPASS point to the Holy Grail? Cancer Cell 2015, 28, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Feng, X.; Huang, J. Genome-wide studies of the transcriptional regulation by p53. Biochim. Biophys. Acta 2012, 1819, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012, 22, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Kamp, W.M.; Wang, P.Y.; Hwang, P.M. TP53 mutation, mitochondria and cancer. Curr. Opin. Genet. Dev. 2016, 38, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. Tumor suppression by p53: Fall of the triumvirate? Cell 2012, 149, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Mandinova, A.; Lee, S.W. The p53 pathway as a target in cancer therapeutics: Obstacles and promise. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Hupp, T.R.; Hayward, R.L.; Vojtesek, B. Strategies for p53 reactivation in human sarcoma. Cancer Cell 2012, 22, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, M.; Henderson, B.R. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp. Cell Res. 2003, 282, 59–69. [Google Scholar] [CrossRef]
- Salmena, L.; Pandolfi, P.P. Changing venues for tumour suppression: Balancing destruction and localization by monoubiquitylation. Nat. Rev. Cancer 2007, 7, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.M.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.P.; et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Morotti, A.; Panuzzo, C.; Crivellaro, S.; Pergolizzi, B.; Familiari, U.; Berger, A.H.; Saglio, G.; Pandolfi, P.P. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 2014, 28, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Mackenzie, R.J.; Besson, A.; Jeng, S.; Carey, A.; LaTocha, D.H.; Fleischman, A.G.; Duquesnes, N.; Eide, C.A.; Vasudevan, K.B.; et al. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood 2014, 124, 3260–3273. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, K.; Li, H.; Zhang, Y. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol. Cell. Biol. 2003, 23, 6396–6405. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Dynamics in the p53-Mdm2 ubiquitination pathway. Cell Cycle 2004, 3, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Li, M.; Gu, W. Monoubiquitination: The signal for p53 nuclear export? Cell Cycle 2004, 3, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. p53 regulation by ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 2009, 284, 3250–3263. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007, 26, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Li, M.; Hu, M.; Shi, Y.; Gu, W. The p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007, 26, 7262–7266. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, A.W.; del Sal, G.; Lu, X. Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 2006, 13, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Tabakin-Fix, Y.; Azran, I.; Schavinky-Khrapunsky, Y.; Levy, O.; Aboud, M. Functional inactivation of p53 by human T-cell leukemia virus type 1 Tax protein: Mechanisms and clinical implications. Carcinogenesis 2006, 27, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Pise-Masison, C.A.; Mahieux, R.; Radonovich, M.; Jiang, H.; Duvall, J.; Guillerm, C.; Brady, J.N. Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res. Hum. Retrovir. 2000, 16, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Kurki, S.; Peltonen, K.; Latonen, L.; Kiviharju, T.M.; Ojala, P.M.; Meek, D.; Laiho, M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475. [Google Scholar] [CrossRef]
- Haupt, S.; Raghu, D.; Haupt, Y. Mutant p53 drives cancer by subverting multiple tumor suppression pathways. Front. Oncol. 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple mechanisms define biologic activity in cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. 2014, 46, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Krämer, O.H. NFκB/p53 crosstalk—A promising new therapeutic target. Biochim. Biophys. Acta 2011, 1815, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V.; Perkins, N.D. p53 and NF-κB crosstalk: IKKα tips the balance. Mol. Cell 2007, 26, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Panwalkar, A.; Verstovsek, S.; Giles, F. Nuclear factor-κB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004, 100, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S. The non-ankyrin C terminus of IκB-α physically interacts with p53 in vivo and dissociates in response to apoptotic stress, hypoxia, DNA damage, and transforming growth factor-β 1-mediated growth suppression. J. Biol. Chem. 2002, 277, 10323–10331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Gu, L.; Zhu, N.; Woods, W.G.; Findley, H.W. Transfection of a dominant-negative mutant NF-κB inhibitor (IκBm) represses p53-dependent apoptosis in acute lymphoblastic leukemia cells: Interaction of IκBm and p53. Oncogene 2003, 22, 8137–8144. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, D.H.; Nagasawa, M.; Gelfand, E.W.; Ghoda, L.Y. Modulation of p53 activity by IκB-α: Evidence suggesting a common phylogeny between NF-κB and p53 transcription factors. BMC Immunol. 2005, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xing, D.; Wang, J.; Zhu, D.B.; Zhang, L.; Chen, X.J.; Sun, F.Y.; Hong, A. Effects of IκB-α and its mutants on NF-κB and p53 signaling pathways. World J. Gastroenterol. 2006, 12, 6658–6664. [Google Scholar] [CrossRef] [PubMed]
- Morotti, A.; Fava, C.; Saglio, G. Milestones and monitoring. Curr. Hematol. Malig. Rep. 2015, 10, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Saglio, G.; Morotti, A.; Mattioli, G.; Messa, E.; Giugliano, E.; Volpe, G.; Rege-Cambrin, G.; Cilloni, D. Rational approaches to the design of therapeutics targeting molecular markers: The case of chronic myelogenous leukemia. Ann. N. Y. Acad. Sci. 2004, 1028, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Crivellaro, S.; Panuzzo, C.; Carrà, G.; Volpengo, A.; Crasto, F.; Gottardi, E.; Familiari, U.; Papotti, M.; Torti, D.; Piazza, R.; et al. Non genomic loss of function of tumor suppressors in CML: BCR-ABL promotes IκB-α mediated p53 nuclear exclusion. Oncotarget 2015, 6, 25217–25225. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrà, G.; Crivellaro, S.; Taulli, R.; Guerrasio, A.; Saglio, G.; Morotti, A. Mechanisms of p53 Functional De-Regulation: Role of the IκB-α/p53 Complex. Int. J. Mol. Sci. 2016, 17, 1997. https://doi.org/10.3390/ijms17121997
Carrà G, Crivellaro S, Taulli R, Guerrasio A, Saglio G, Morotti A. Mechanisms of p53 Functional De-Regulation: Role of the IκB-α/p53 Complex. International Journal of Molecular Sciences. 2016; 17(12):1997. https://doi.org/10.3390/ijms17121997
Chicago/Turabian StyleCarrà, Giovanna, Sabrina Crivellaro, Riccardo Taulli, Angelo Guerrasio, Giuseppe Saglio, and Alessandro Morotti. 2016. "Mechanisms of p53 Functional De-Regulation: Role of the IκB-α/p53 Complex" International Journal of Molecular Sciences 17, no. 12: 1997. https://doi.org/10.3390/ijms17121997
APA StyleCarrà, G., Crivellaro, S., Taulli, R., Guerrasio, A., Saglio, G., & Morotti, A. (2016). Mechanisms of p53 Functional De-Regulation: Role of the IκB-α/p53 Complex. International Journal of Molecular Sciences, 17(12), 1997. https://doi.org/10.3390/ijms17121997