
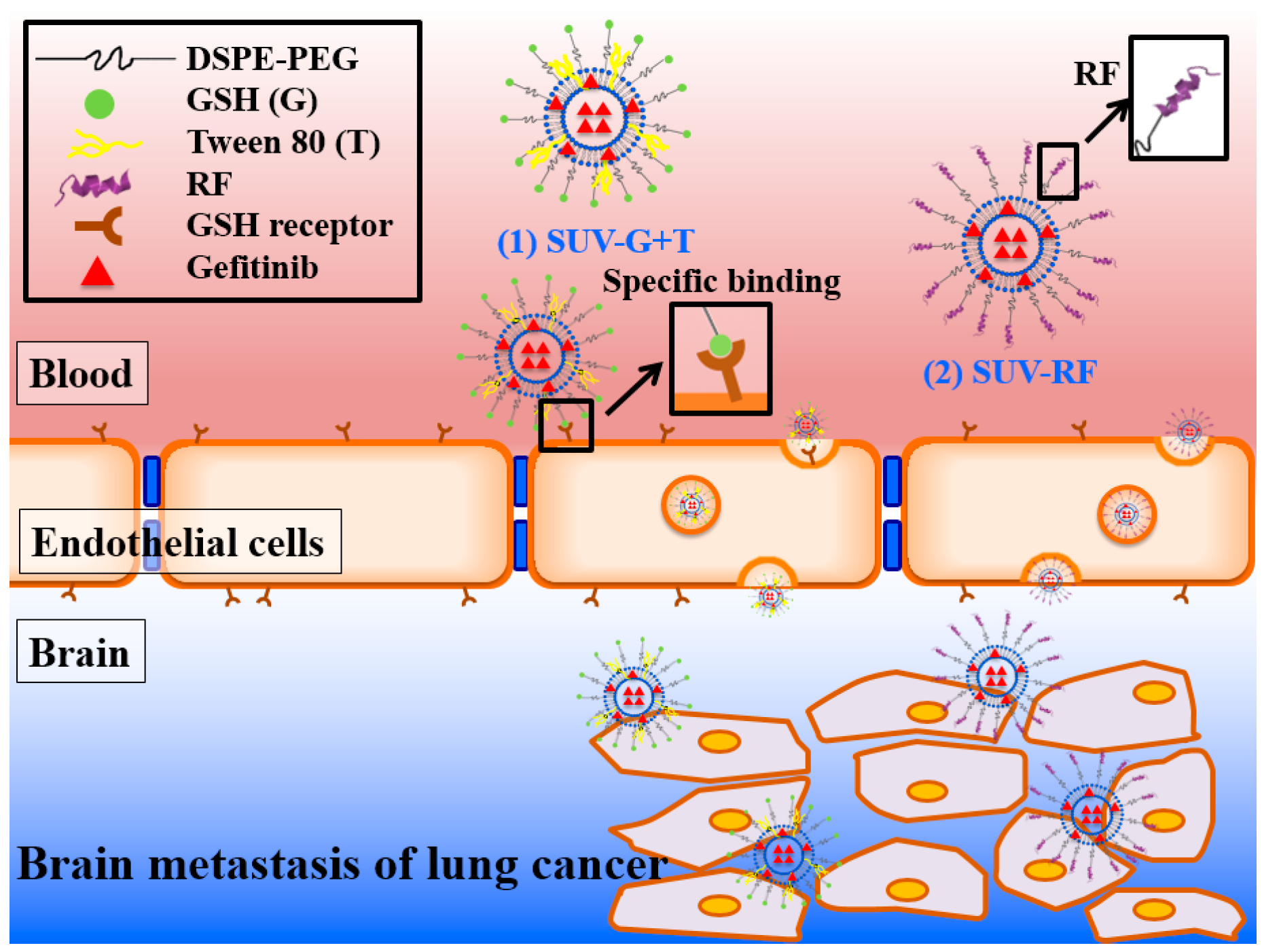
Enhancing Anticancer Effect of Gefitinib across the Blood–Brain Barrier Model Using Liposomes Modified with One α-Helical Cell-Penetrating Peptide or Glutathione and Tween 80

Abstract
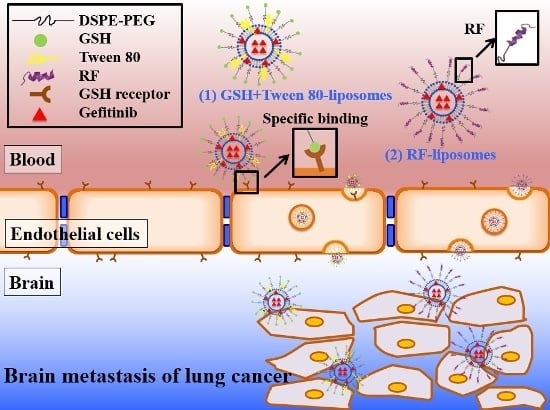
:
1. Introduction
2. Results
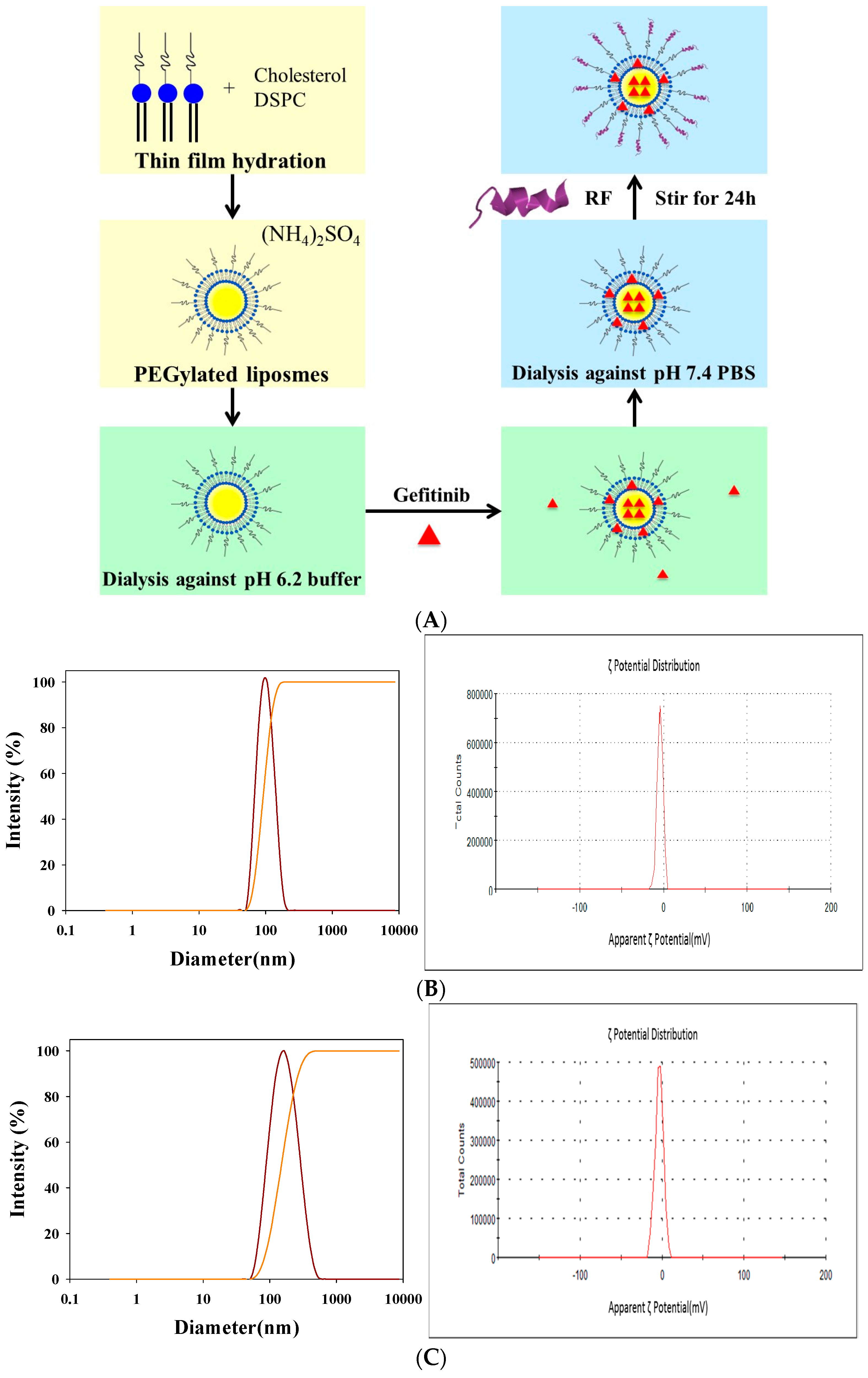
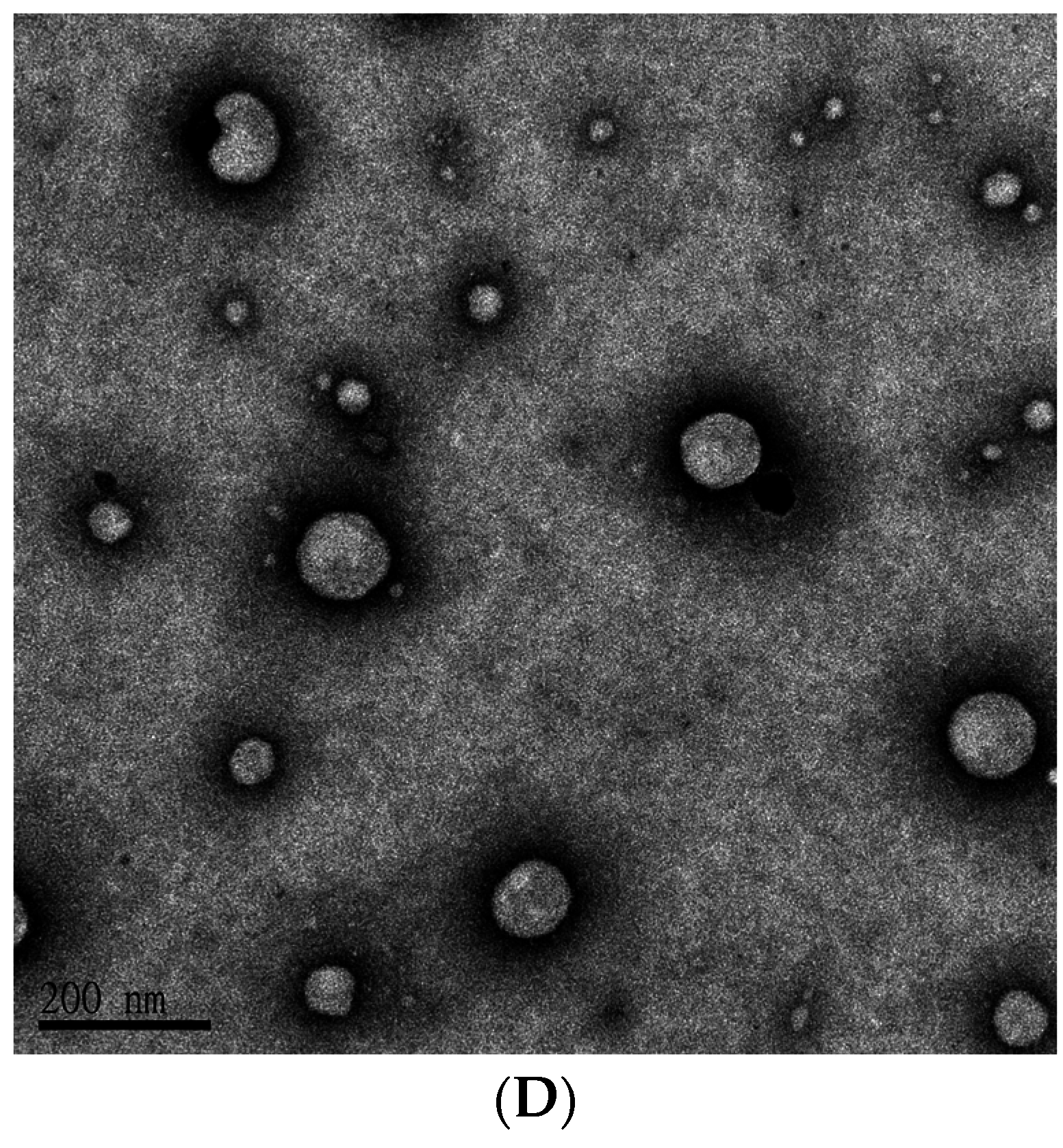
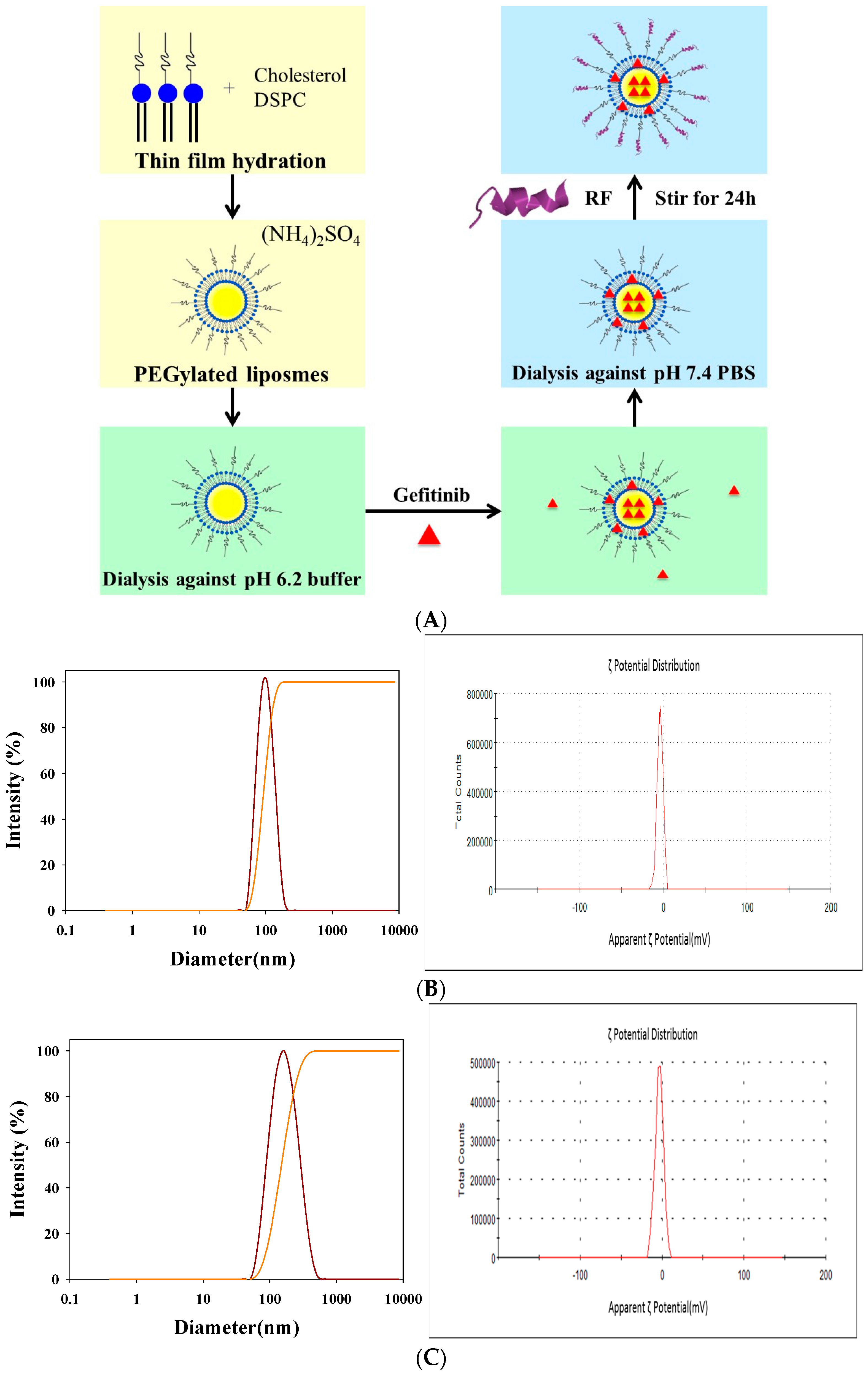
2.1. Determination of Encapsulation Efficiency (EE)%, Particle Size, and ζ Potential of PEGylated Liposomal Gefitinib
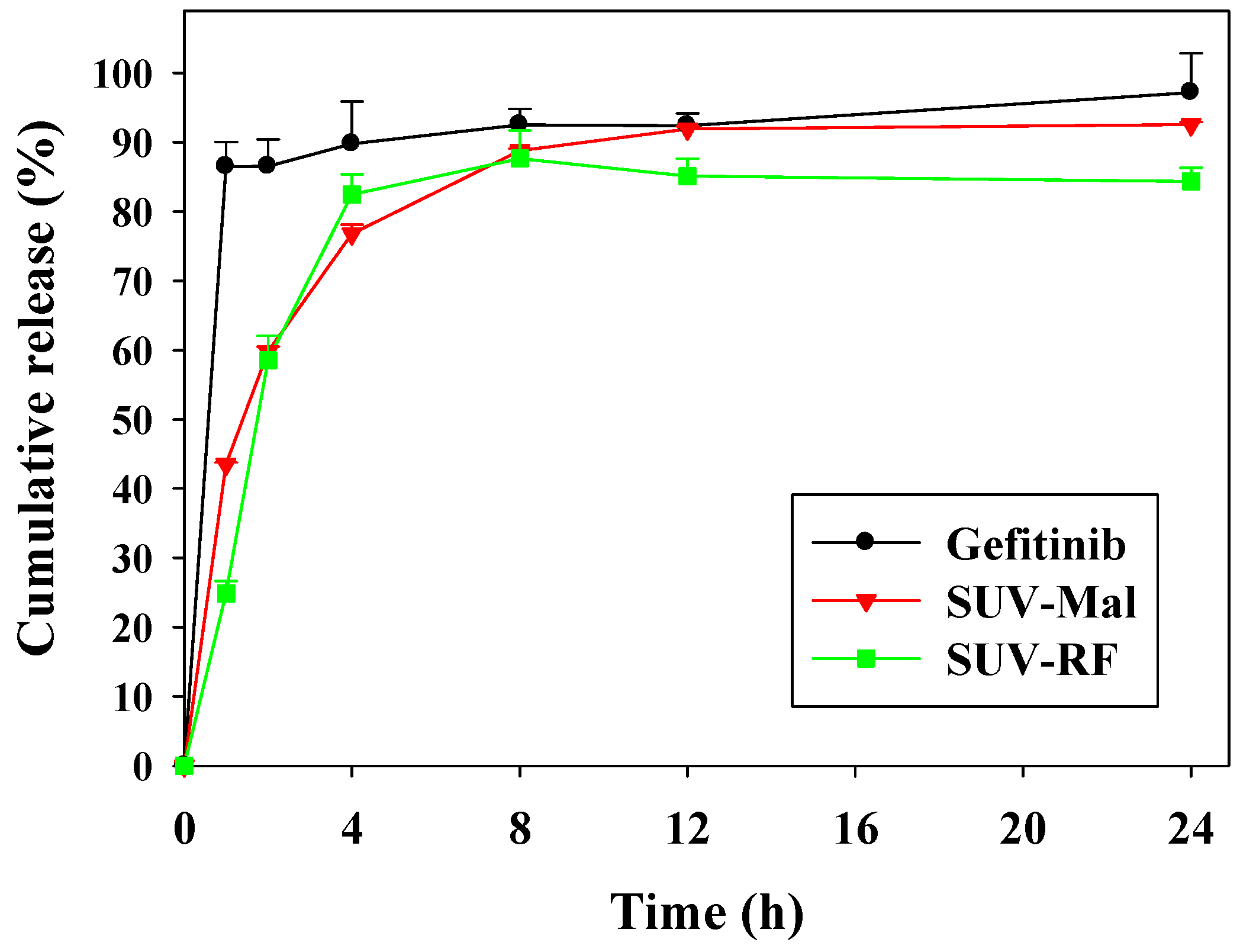
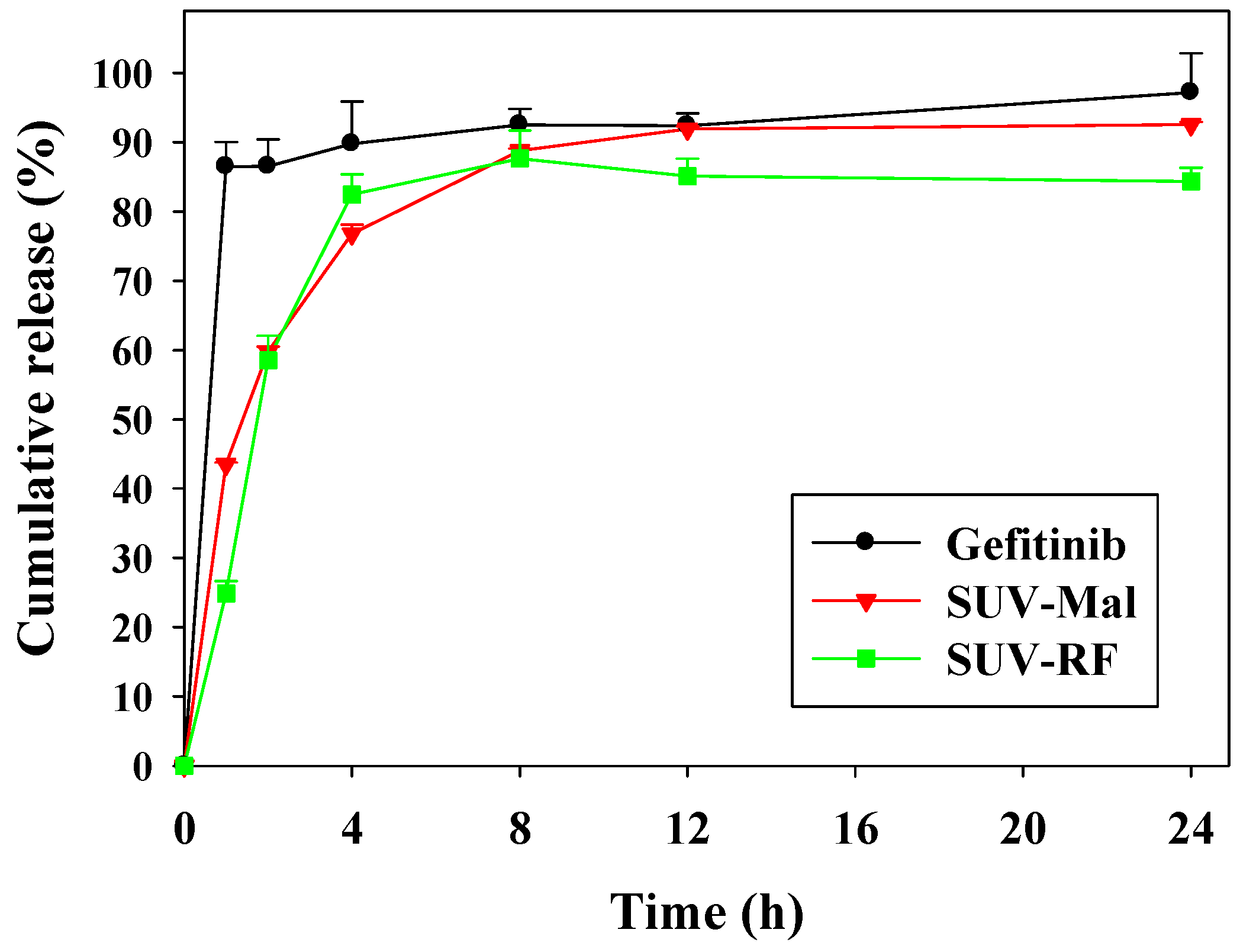
2.2. In Vitro Release of Gefitinib from Small Unilamellar Vesicle (SUV)
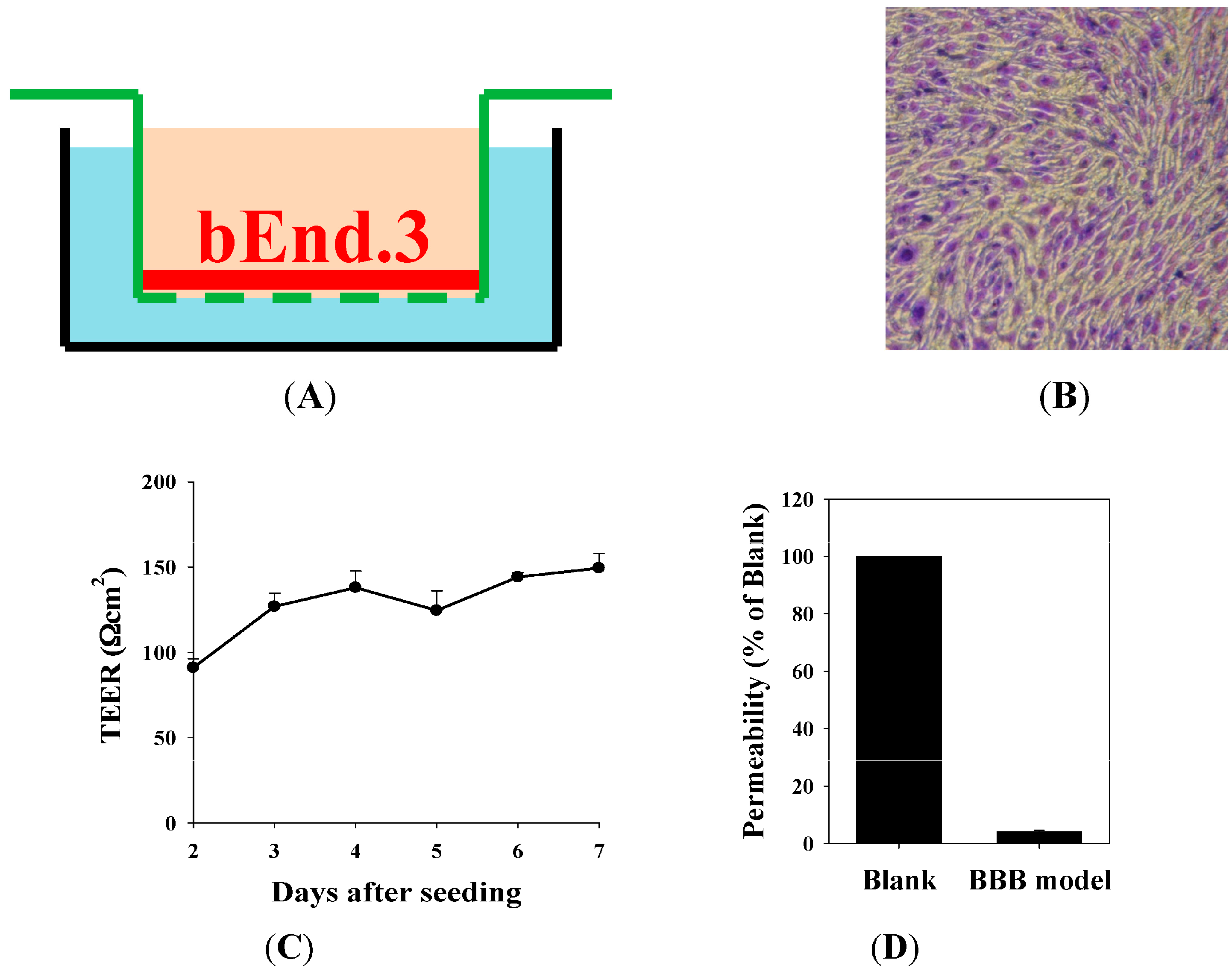
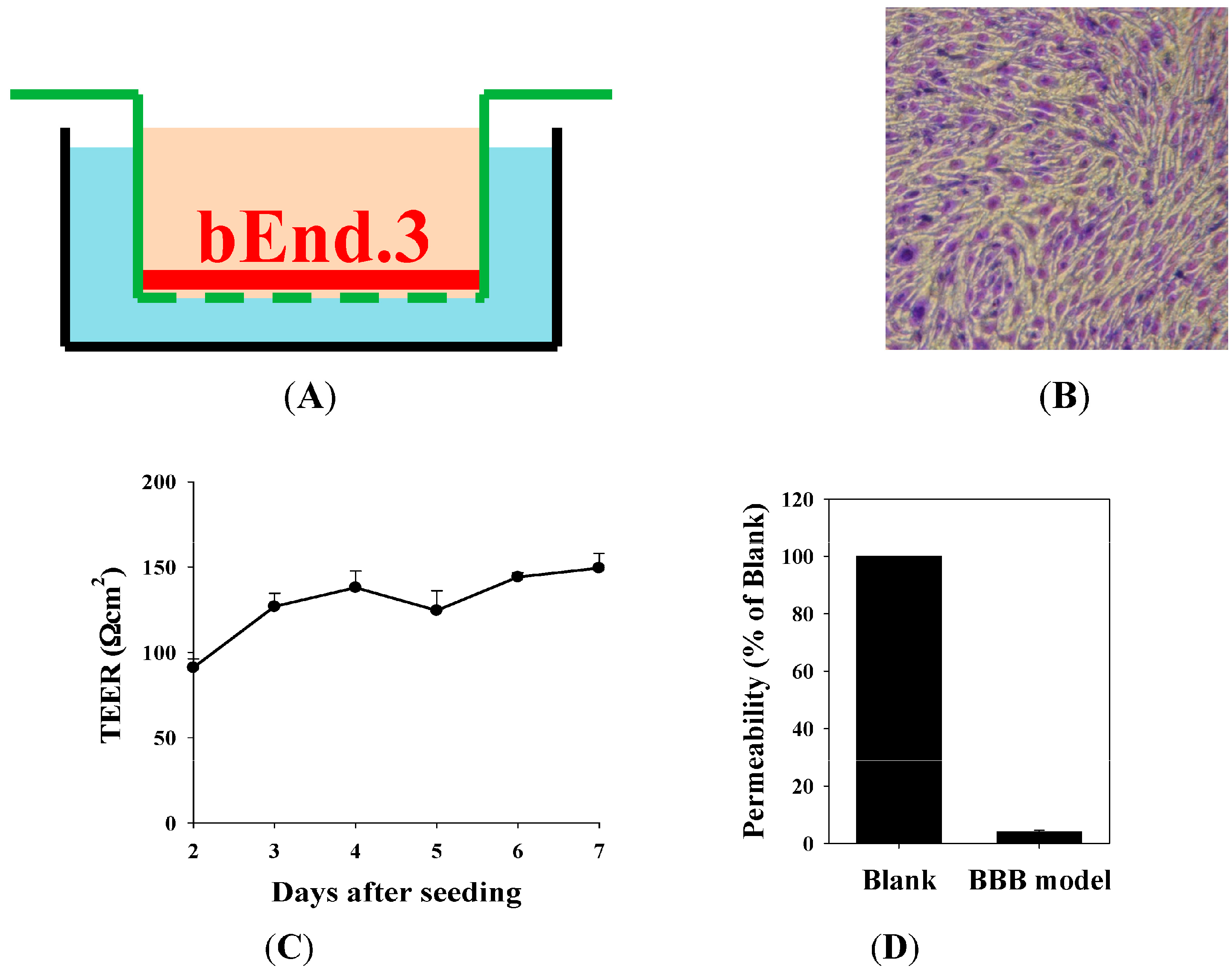
2.3. The Blood–Brain Barrier (BBB) Barrier Integrity
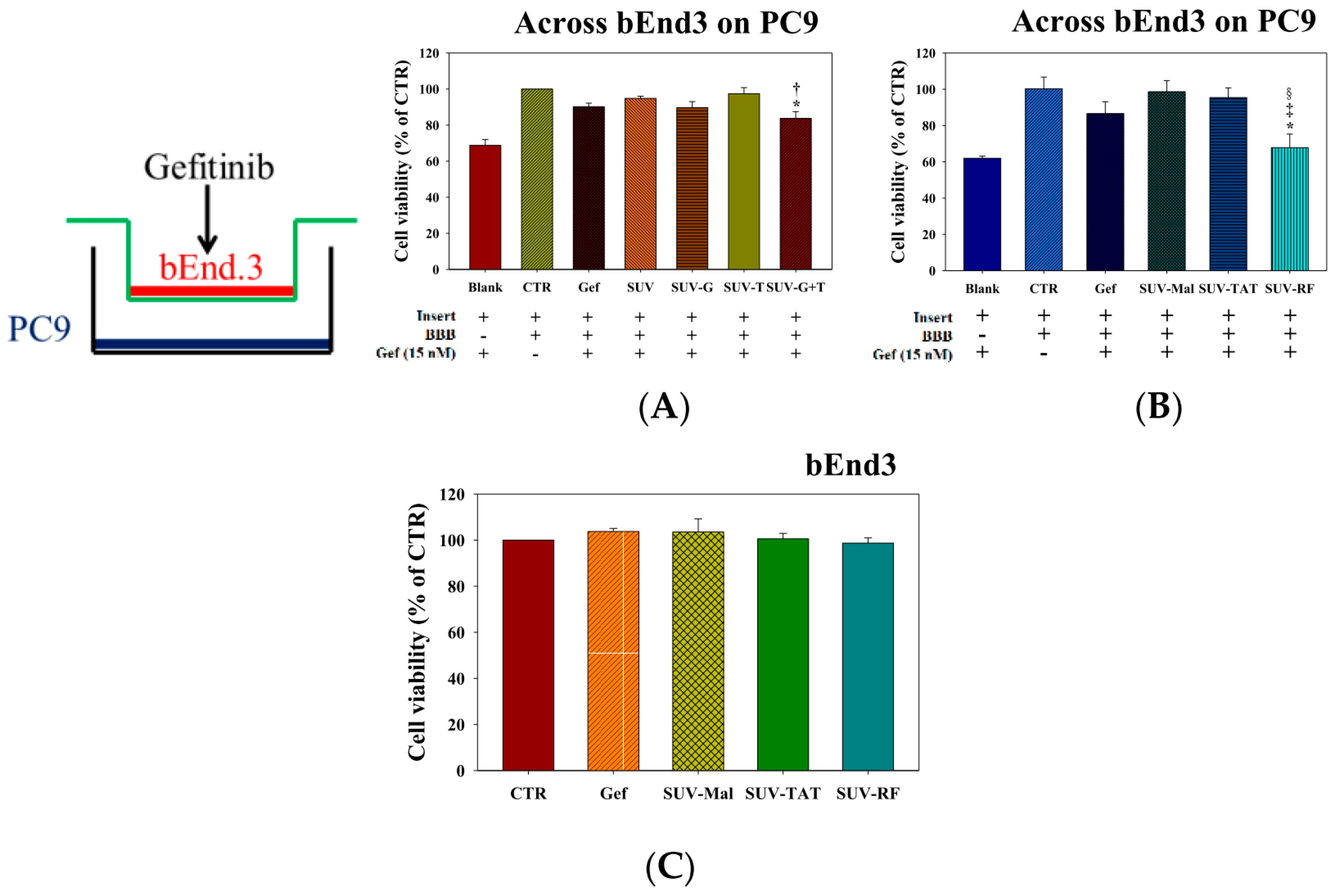
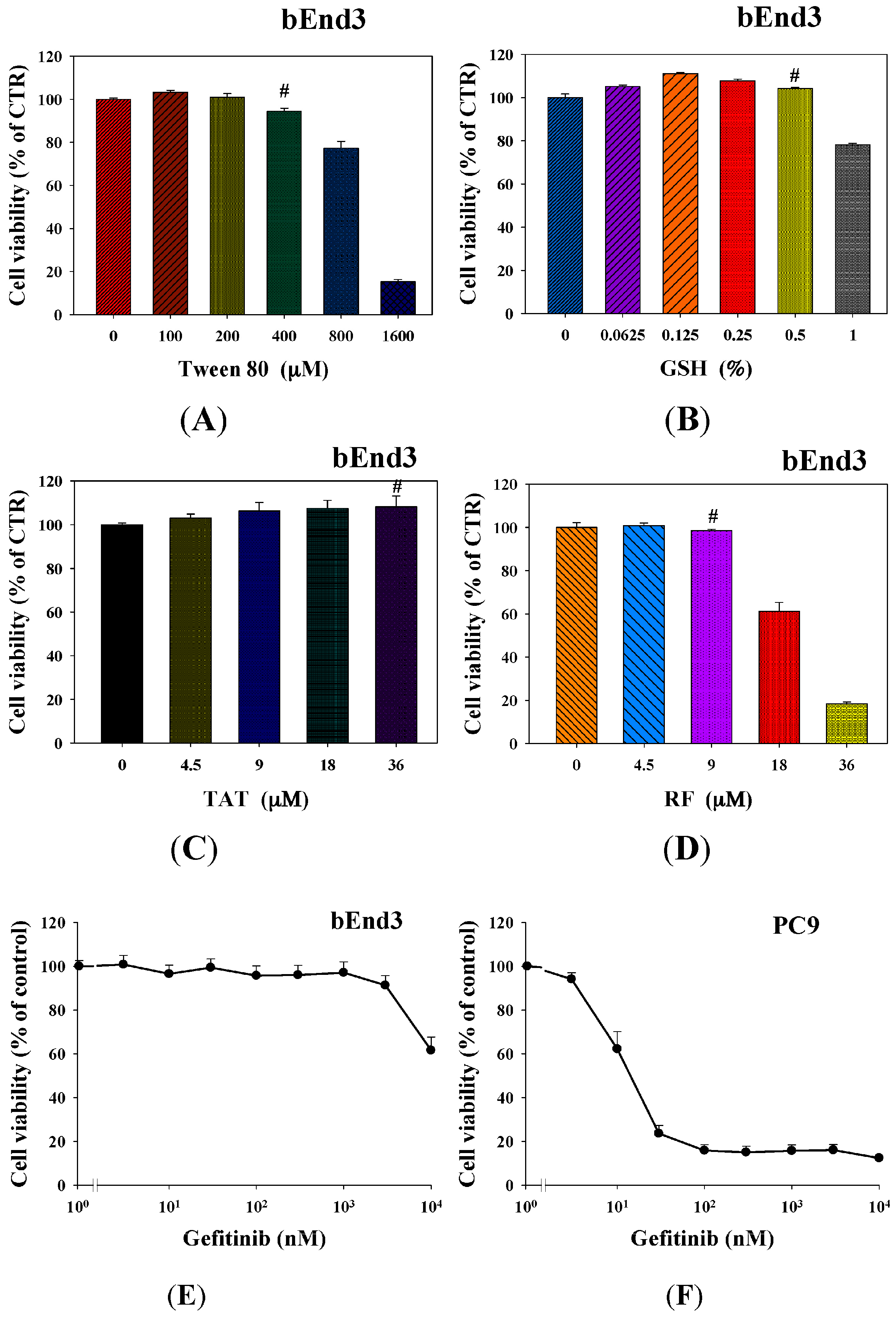
2.4. Cytotoxicity of Tween 80, GSH, RF, TAT, and Gefitinib on bEnd.3 and/or PC9 Cells
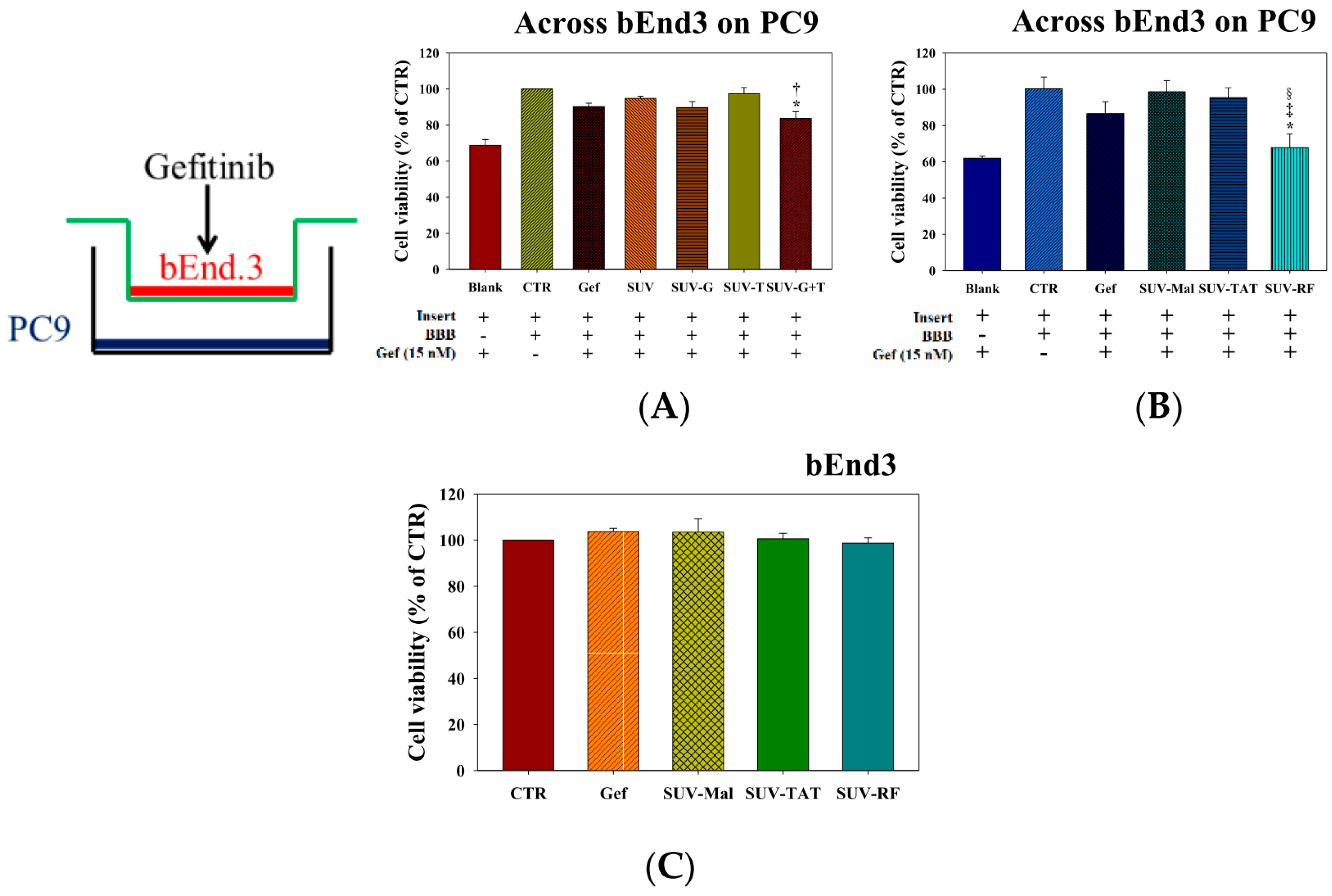
2.5. Cytotoxicity of Gefitinib in SUV-G, SUV-T, SUV-G+T across the BBB on PC9 Cells
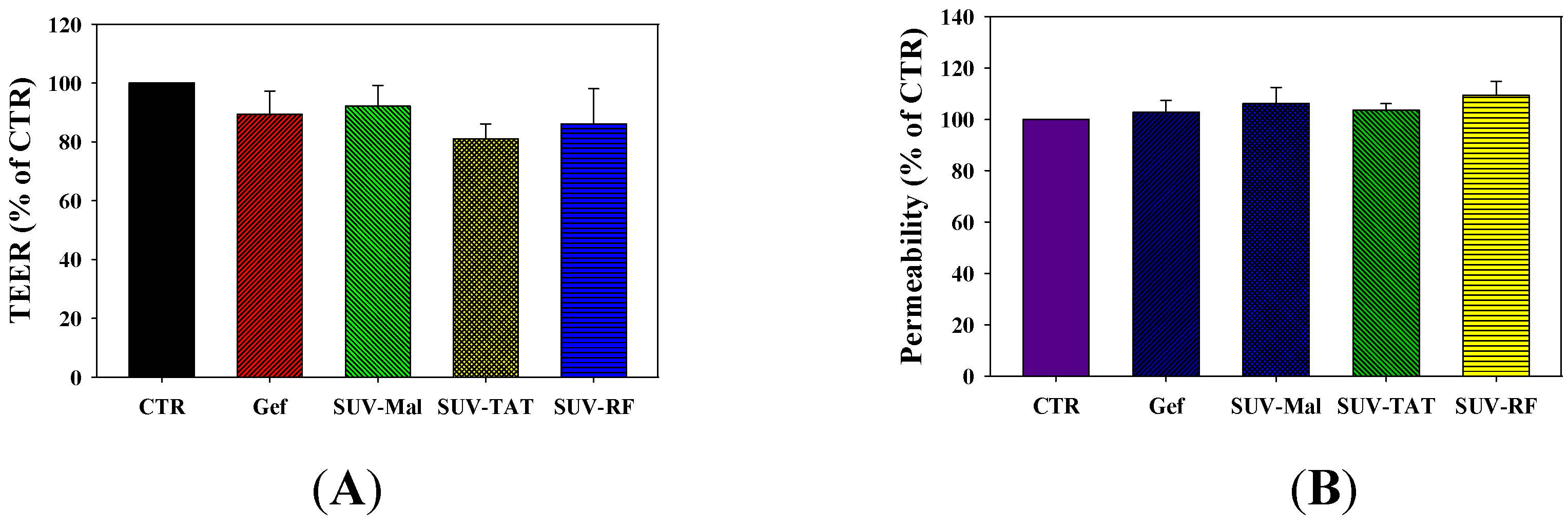
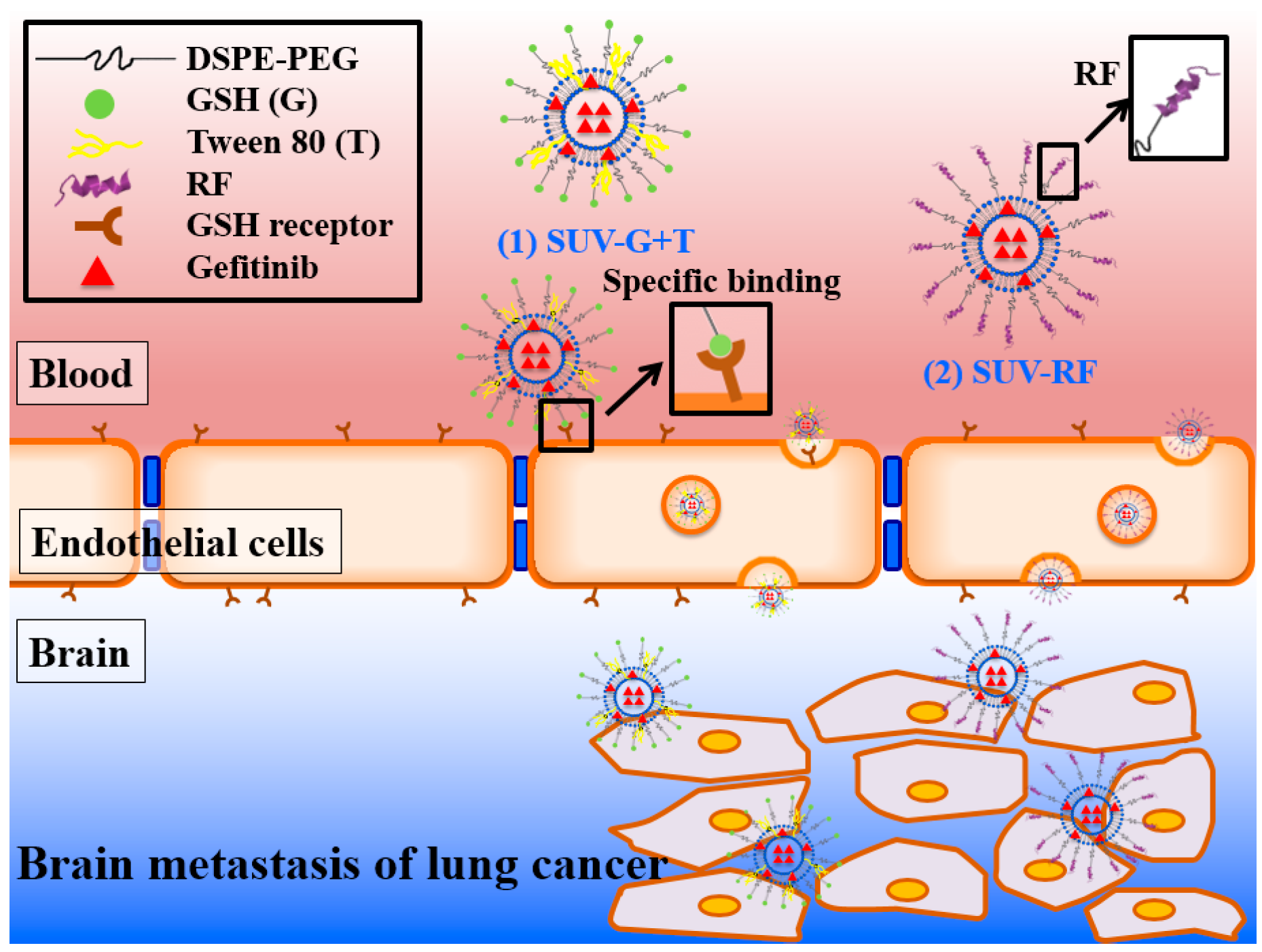
2.6. Cytotoxicity of Gefitinib in SUV-Mal, SUV-TAT, or SUV-RF across the BBB on PC9 Cells
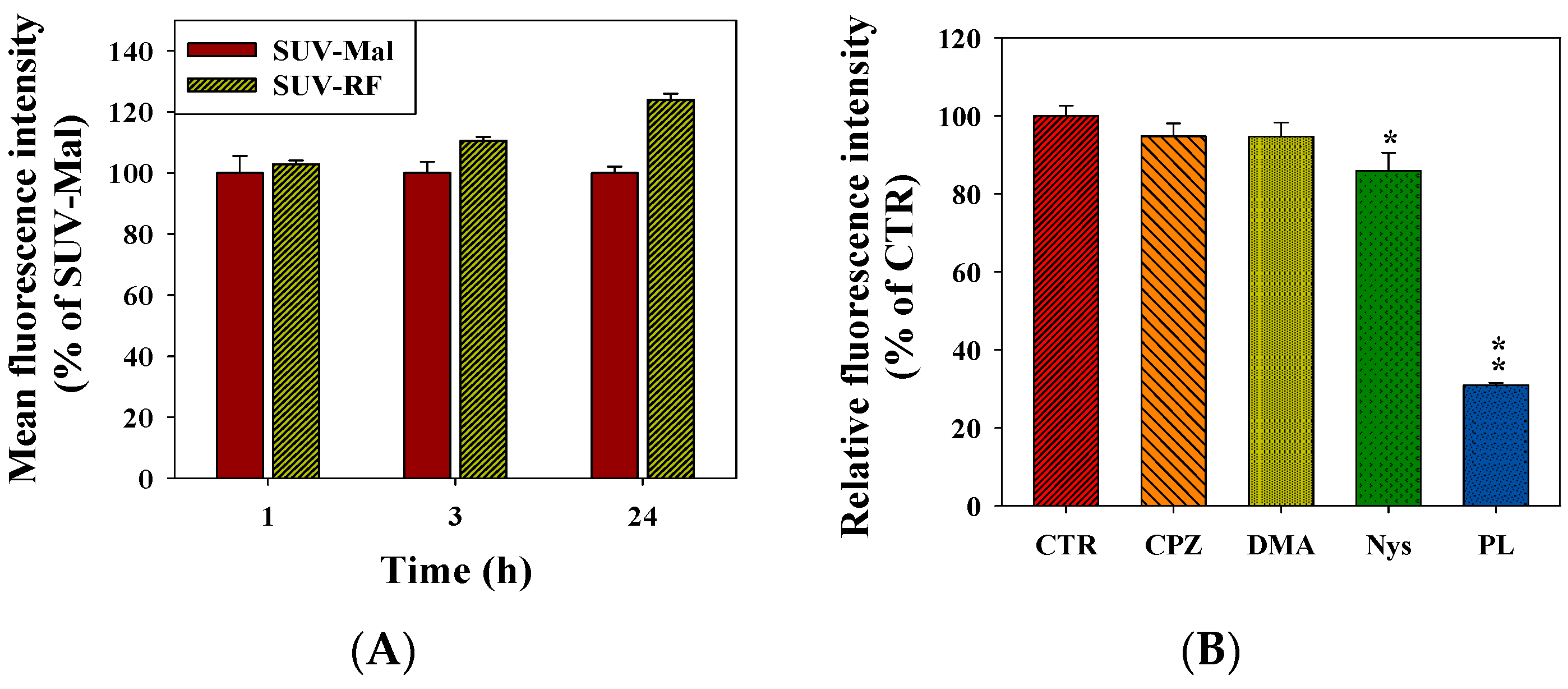
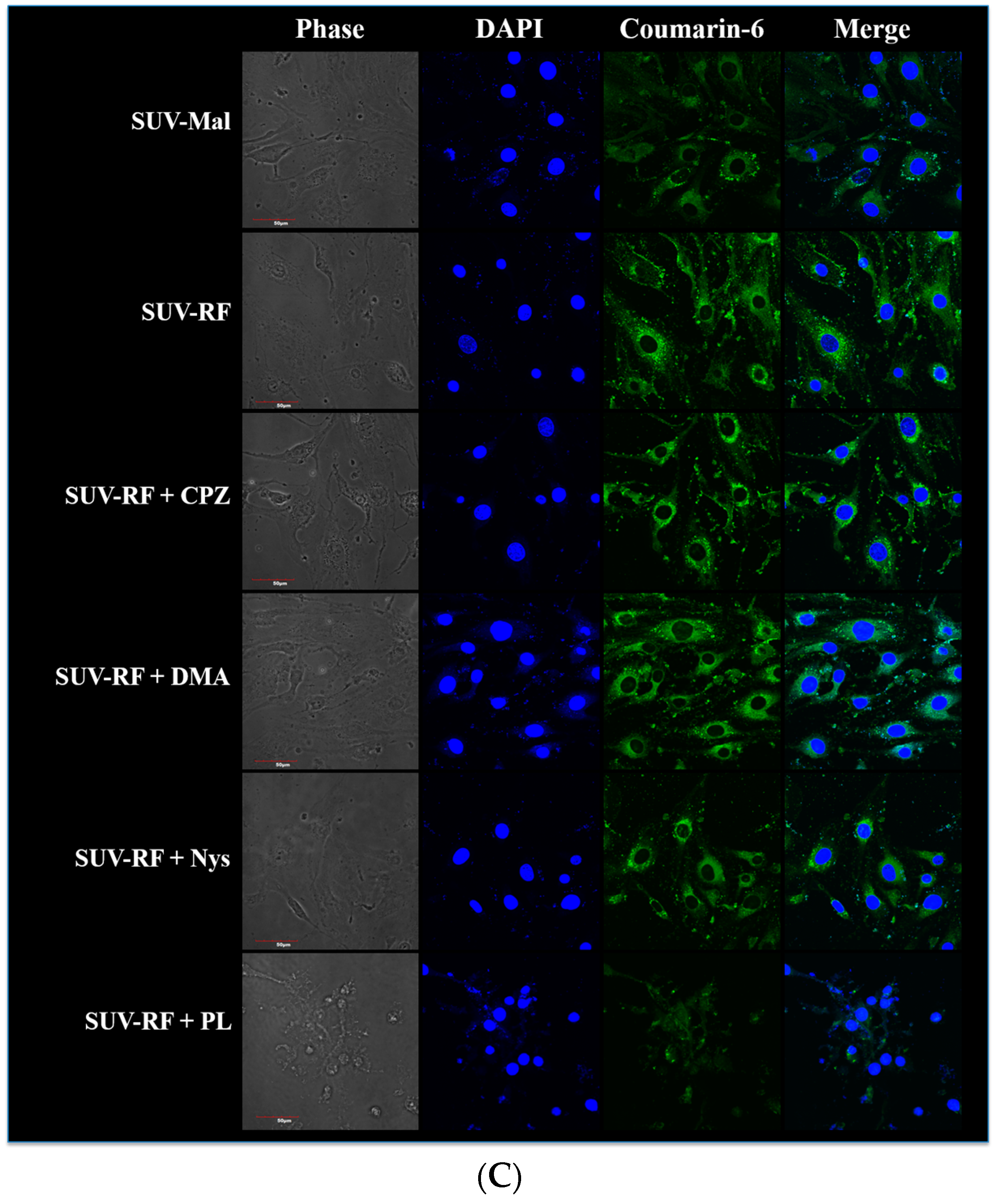
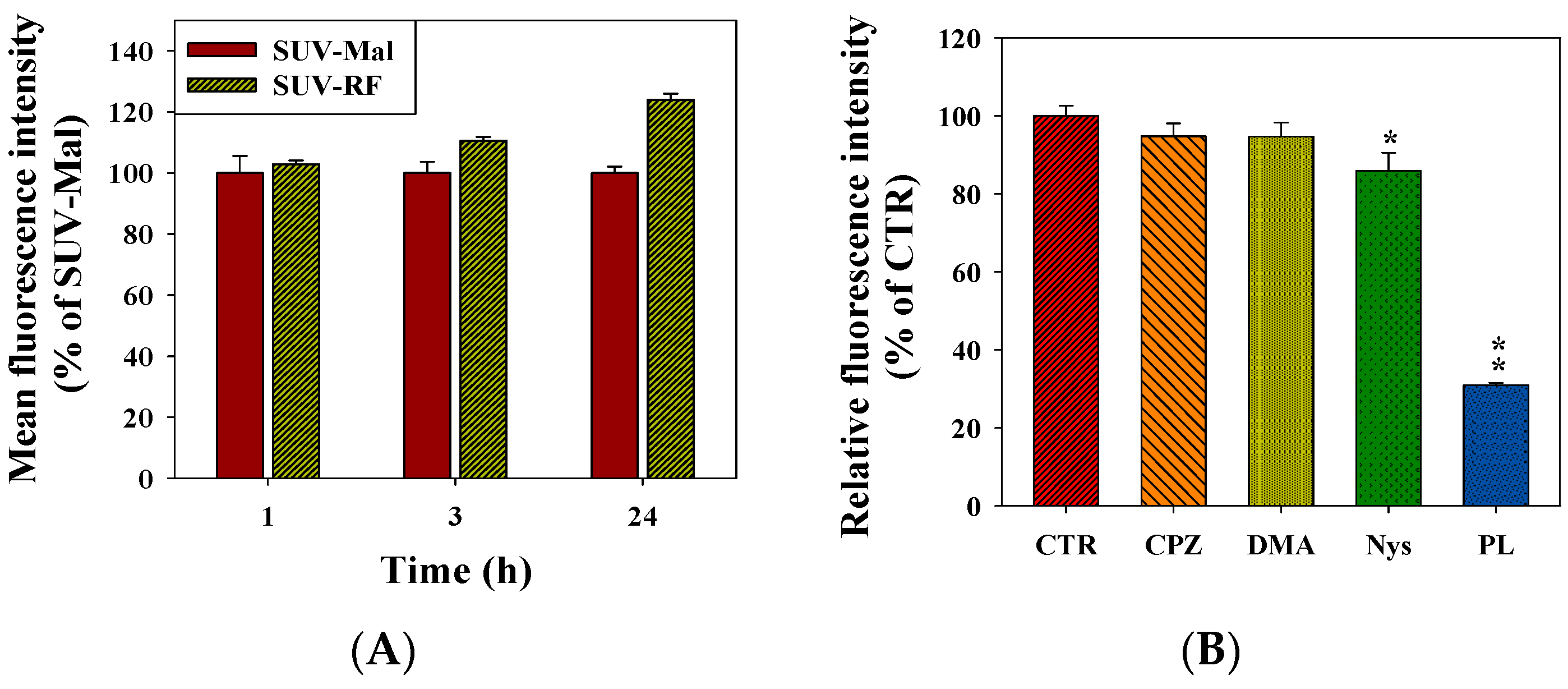
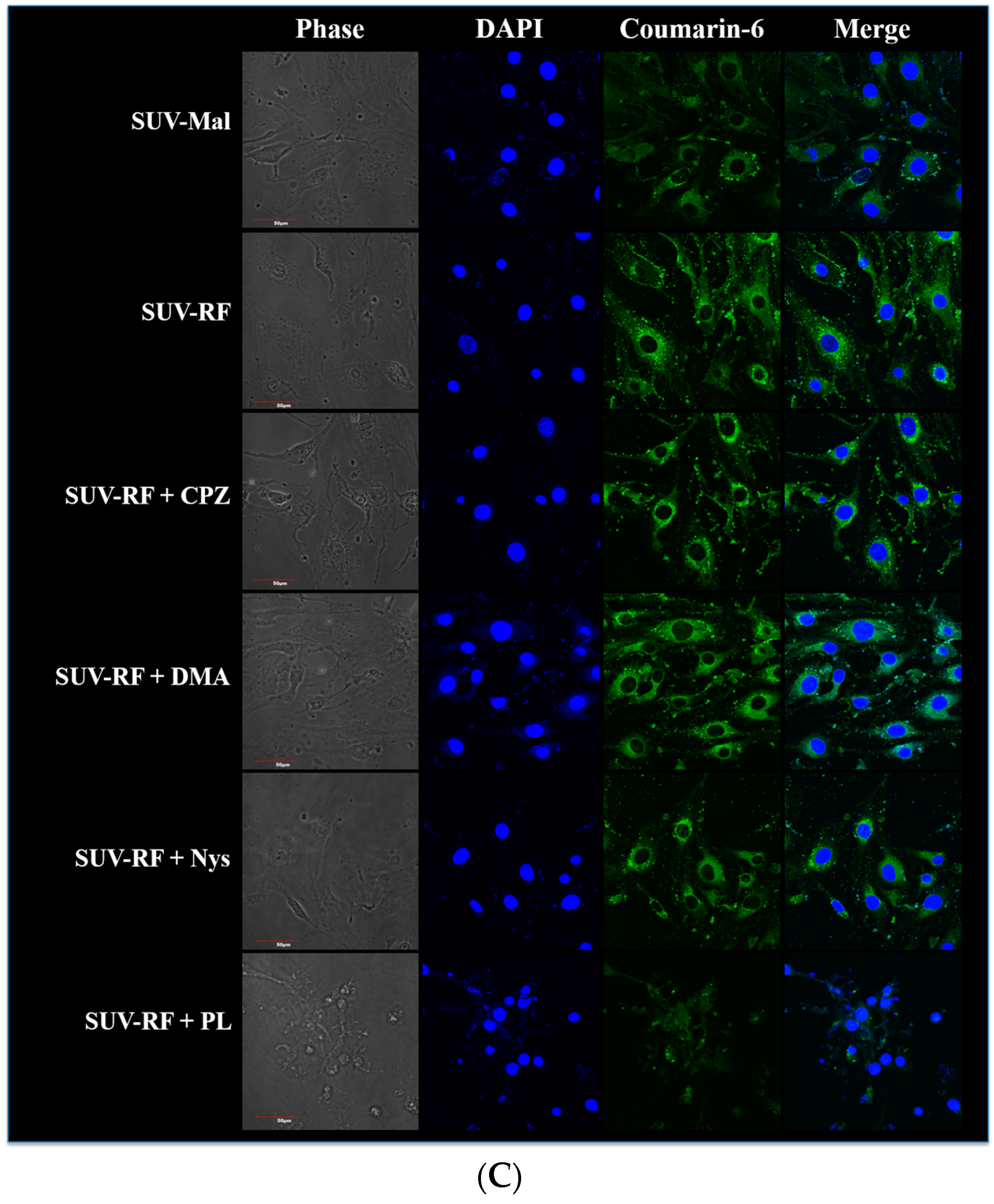
2.7. Quantitative Analysis of Cellular Uptake and Transcytosis Mechanisms of SUV-RF
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of PEGylated Liposomal Gefitinib-Formulations
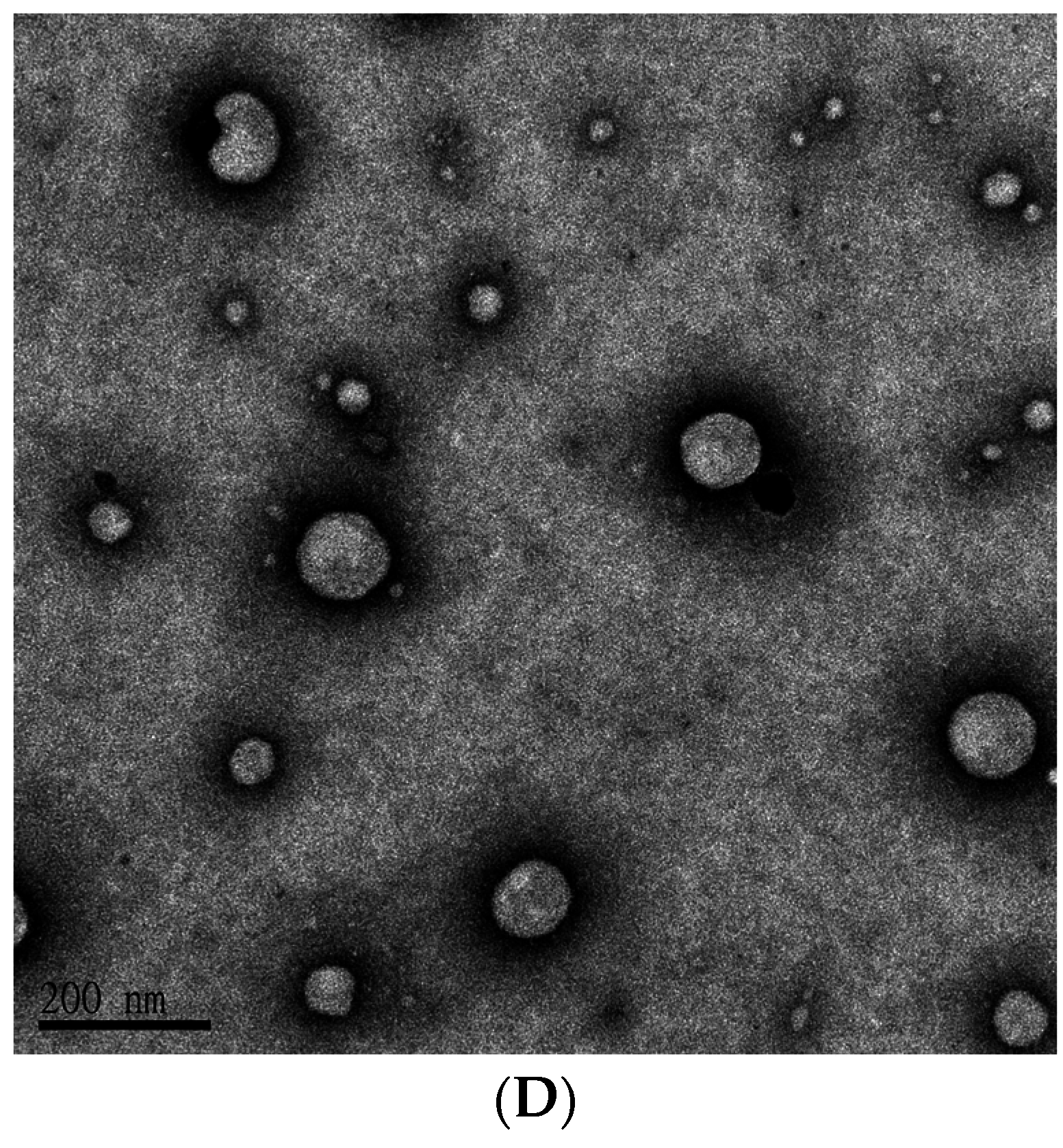
4.3. Characterization of PEGylated Liposomal Gefitinib-Formulations: Encapsulation Efficiency, Size Distribution, Zeta Potential, and Transmission Electron Microscopic Image
4.4. In Vitro Release of Gefitinib from SUV
4.5. Culture of Human Lung Adenocarcinoma PC-9 Cells and Murine Brain Endothelial bEnd.3 Cells
4.6. Establishment of In Vitro BBB Model: Morphology of Barrier Integrity, Transendothelial Electrical Resistance (TEER) Measurement, and Permeability Measurement
4.7. Cell Viability by the SRB Assay
4.8. Endocytic Uptake Mechanisms of SUV-RF
4.9. Intracellular Uptake of SUV-RF by Confocal Laser Scanning Microscope (CLSM)
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tang, M.-C.; Wu, M.-Y.; Hwang, M.-H.; Chang, Y.-T.; Huang, H.-J.; Lin, A.M.-Y.; Yang, J.C.-H. Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells. PLoS ONE 2015, 10, e0119135. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The biology of brain metastasis: Challenges for therapy. Cancer J. 2015, 21, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.D.; Liao, H.; Qin, T.; Zhang, L.; Wei, W.D.; Liang, J.Z.; Xu, F.; Dinglin, X.X.; Ma, S.X.; Chen, L.K. Blood-brain barrier permeability of gefitinib in patients with brain metastases from non-small-cell lung cancer before and during whole brain radiation therapy. Oncotarget 2015, 6, 8366–8376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Miller, D.W. Transporter-based delivery of anticancer drugs to the brain: Improving brain penetration by minimizing drug efflux at the blood-brain barrier. Curr. Pharm. Des. 2014, 20, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.H.; Lee, J.H.; Chang, Y.J.; Tsai, H.H.; Lin, Y.L.; Lin, A.M.; Yang, J.C. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol. Oncol. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, J.; Sun, X.; Meng, X. Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of central nerve system metastases from non-small cell lung cancer. Cancer Lett. 2014, 351, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Spicer, J. Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of epidermal growth factor receptor-mutant non-small cell lung cancer metastatic to the brain. Clin. Cancer Res. 2012, 18, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Haringhuizen, A.; van Tinteren, H.; Vaessen, H.F.; Baas, P.; van Zandwijk, N. Gefitinib as a last treatment option for non-small-cell lung cancer: Durable disease control in a subset of patients. Ann. Oncol. 2004, 15, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T. Pharmacology and toxicology of Cremophor EL diluent. Ann. Pharmacother. 1994, 28, S11–S14. [Google Scholar] [PubMed]
- Masserini, M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013, 2013, 238428. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Hirani, A.; Pathak, Y.; Sutariya, V. Brain-targeted delivery of docetaxel by glutathione-coated nanoparticles for brain cancer. AAPS PharmSciTech 2014, 15, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.; Wehrung, D.; Groshev, A.; Hirani, A.; Sutariya, V. Brain-targeted delivery of doxorubicin using glutathione-coated nanoparticles for brain cancers. Pharm. Dev. Technol. 2015, 20, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; Appeldoorn, C.C.; Dorland, R.; van Kregten, J.; Manca, F.; Vugts, D.J.; Windhorst, B.; van Dongen, G.A.; de Vries, H.E.; Maussang, D.; et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101). PLoS ONE 2014, 9, e82331. [Google Scholar] [CrossRef] [PubMed]
- Birngruber, T.; Raml, R.; Gladdines, W.; Gatschelhofer, C.; Gander, E.; Ghosh, A.; Kroath, T.; Gaillard, P.J.; Pieber, T.R.; Sinner, F. Enhanced doxorubicin delivery to the brain administered through glutathione PEGylated liposomal doxorubicin (2B3-101) as compared with generic Caelyx®/Doxil®—A cerebral open flow microperfusion pilot study. J. Pharm. Sci. 2014, 103, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Henrich-Noack, P.; Kockentiedt, S.; Hintz, W.; Tomas, J.; Sabel, B.A. Surfactants, not size or zeta-potential influence blood-brain barrier passage of polymeric nanoparticles. Eur. J. Pharm. Biopharm. 2014, 87, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P.; Schoubben, A.; Traina, G.; Manfroni, G.; Barberini, L.; Alberti, P.F.; Cirotto, C.; Ricci, M. Lipid nanoparticles for brain targeting III. Long-term stability and in vivo toxicity. Int. J. Pharm. 2013, 454, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kolter, M.; Ott, M.; Hauer, C.; Reimold, I.; Fricker, G. Nanotoxicity of poly(N-butylcyano-acrylate) nanoparticles at the blood-brain barrier, in human whole blood and in vivo. J. Control. Release 2015, 197, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Jose, S.; Sowmya, S.; Cinu, T.A.; Aleykutty, N.A.; Thomas, S.; Souto, E.B. Surface modified PLGA nanoparticles for brain targeting of Bacoside-A. Eur. J. Pharm. Sci. 2014, 63, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Van der Meel, R.; Vehmeijer, L.J.; Kok, R.J.; Storm, G.; van Gaal, E.V. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Zong, T.; Mei, L.; Gao, H.; Cai, W.; Zhu, P.; Shi, K.; Chen, J.; Wang, Y.; Gao, F.; He, Q. Synergistic dual-ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals. Mol. Pharm. 2014, 11, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Wang, K.; Zhao, Z.; Zhang, Y.; Sun, T.; Zhang, F.; Wu, J.; Fu, Y.; Du, Y.; Zhang, L.; et al. Brain-targeted delivery of trans-activating transcriptor-conjugated magnetic PLGA/lipid nanoparticles. PLoS ONE 2014, 9, e106652. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, W.J.; Kim, Y.H.; Choi, J.M. Identification of a novel cell-penetrating peptide from human phosphatidate phosphatase LPIN3. Mol. Cells 2012, 34, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Tsutsumi, H.; Mihara, H. Cell-selective intracellular drug delivery using doxorubicin and α-helical peptides conjugated to gold nanoparticles. Biomaterials 2014, 35, 3480–3487. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Kakiyama, T.; Tomizaki, K.Y.; Mie, M.; Kobatake, E.; Mihara, H. Cell fingerprint patterns using designed α-helical peptides to screen for cell-specific toxicity. Bioorgan. Med. Chem. Lett. 2011, 21, 6281–6284. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Kikuchi, T.; Mie, M.; Kobatake, E.; Mihara, H. Systematic screening of the cellular uptake of designed α-helix peptides. Bioorgan. Med. Chem. 2013, 21, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Tsutsumi, H.; Mihara, H. Cell penetration and cell-selective drug delivery using α-helix peptides conjugated with gold nanoparticles. Biomaterials 2013, 34, 4872–4879. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Morris, A.P.; O′Neil, R.G. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007, 1130, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jin, X.; Liu, K.J.; Liu, W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood–brain barrier damage in early ischemic stroke stage. J. Neurosci. 2012, 32, 3044–3057. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Simon, M.J.; Cancel, L.M.; Shi, Z.D.; Ji, X.; Tarbell, J.M.; Morrison, B., 3rd; Fu, B.M. Permeability of endothelial and astrocyte cocultures: In vitro blood-brain barrier models for drug delivery studies. Ann. Biomed. Eng. 2010, 38, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tsutsumi, H.; Furuta, T.; Sakurai, M.; Mihara, H. Interaction of amphiphilic α-helical cell-penetrating peptides with heparan sulfate. Org. Biomol. Chem. 2014, 12, 4673–4681. [Google Scholar] [CrossRef] [PubMed]
- Trabulo, S.; Mano, M.; Faneca, H.; Cardoso, A.L.; Duarte, S.; Henriques, A.; Paiva, A.; Gomes, P.; Simoes, S.; de Lima, M.C. S413-PV cell penetrating peptide and cationic liposomes act synergistically to mediate intracellular delivery of plasmid DNA. J. Gene Med. 2008, 10, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Trabulo, S.; Cardoso, A.L.; Cardoso, A.M.; Morais, C.M.; Jurado, A.S.; Pedroso de Lima, M.C. Cell-penetrating peptides as nucleic acid delivery systems: From biophysics to biological applications. Curr. Pharm. Des. 2013, 19, 2895–2923. [Google Scholar] [CrossRef] [PubMed]
- Niego, B.; Freeman, R.; Puschmann, T.B.; Turnley, A.M.; Medcalf, R.L. t-PA-specific modulation of a human blood-brain barrier model involves plasmin-mediated activation of the Rho kinase pathway in astrocytes. Blood 2012, 119, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, R.; Suzuka, S.; Oda, K.; Nagai, J.; Takano, M. Endocytic uptake of FITC-albumin by human alveolar epithelial cell line A549. Drug Metab. Pharmacokinet. 2012, 27, 336–343. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Description | Particle Size (nm) | PDI b | ζ Potential (mV) |
|---|---|---|---|---|
| SUV-Mal | Small unilamellar vesicles of DSPC, cholesterol, and DSPE-PEG2000-maleimide | 95.5 ± 2.2 | 0.16 ± 0.01 | −3.38 ± 0.78 |
| SUV-RF | Small unilamellar vesicles of DSPC, cholesterol, DSPE-PEG2000-maleimide, and conjugated with RF | 147.1 ± 3.9 | 0.10 ± 0.02 | −3.42 ± 0.64 |
| SUV | Small unilamellar vesicles of DSPC, cholesterol, and DSPE-PEG-NH2 | 105.5 ± 6.6 | 0.12 ± 0.03 | −3.25 ± 0.27 |
| SUV-G | Small unilamellar vesicles of DSPC, cholesterol, DSPE-PEG-NH2, and coated with GSH | 102.3 ± 6.3 | 0.06 ± 0.01 | −1.70 ± 0.16 |
| SUV-T | Small unilamellar vesicles of DSPC, cholesterol, DSPE-PEG-NH2, and modified with Tween 80 | 85.8 ± 3.7 | 0.13 ± 0.03 | −3.09 ± 0.75 |
| SUV-G+T | Small unilamellar vesicles of DSPC, cholesterol, DSPE-PEG-NH2, and modified with GSH and Tween 80 | 94.2 ± 0.7 | 0.26 ± 0.01 | −3.82 ± 0.85 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, K.-H.; Hong, S.-T.; Wang, H.-T.; Lo, Y.-L.; Lin, A.M.-Y.; Yang, J.C.-H. Enhancing Anticancer Effect of Gefitinib across the Blood–Brain Barrier Model Using Liposomes Modified with One α-Helical Cell-Penetrating Peptide or Glutathione and Tween 80. Int. J. Mol. Sci. 2016, 17, 1998. https://doi.org/10.3390/ijms17121998
Lin K-H, Hong S-T, Wang H-T, Lo Y-L, Lin AM-Y, Yang JC-H. Enhancing Anticancer Effect of Gefitinib across the Blood–Brain Barrier Model Using Liposomes Modified with One α-Helical Cell-Penetrating Peptide or Glutathione and Tween 80. International Journal of Molecular Sciences. 2016; 17(12):1998. https://doi.org/10.3390/ijms17121998
Chicago/Turabian StyleLin, Kuan-Hung, Shu-Ting Hong, Hsiang-Tsui Wang, Yu-Li Lo, Anya Maan-Yuh Lin, and James Chih-Hsin Yang. 2016. "Enhancing Anticancer Effect of Gefitinib across the Blood–Brain Barrier Model Using Liposomes Modified with One α-Helical Cell-Penetrating Peptide or Glutathione and Tween 80" International Journal of Molecular Sciences 17, no. 12: 1998. https://doi.org/10.3390/ijms17121998