
PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
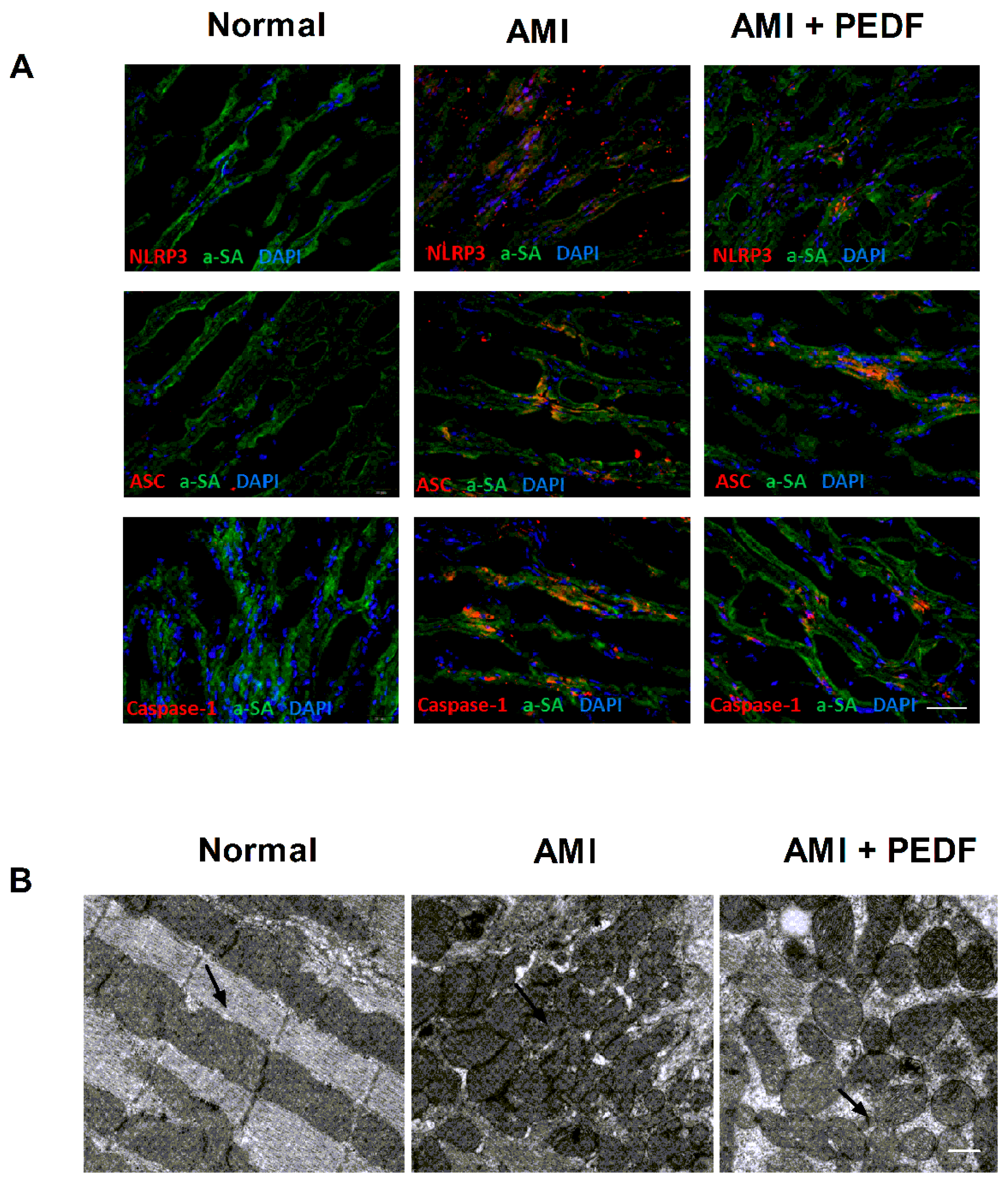
2.1. PEDF Decreased the Expression of the NLRP3 Inflammasome and Mitochondrial Fission in the Rat Heart during AMI
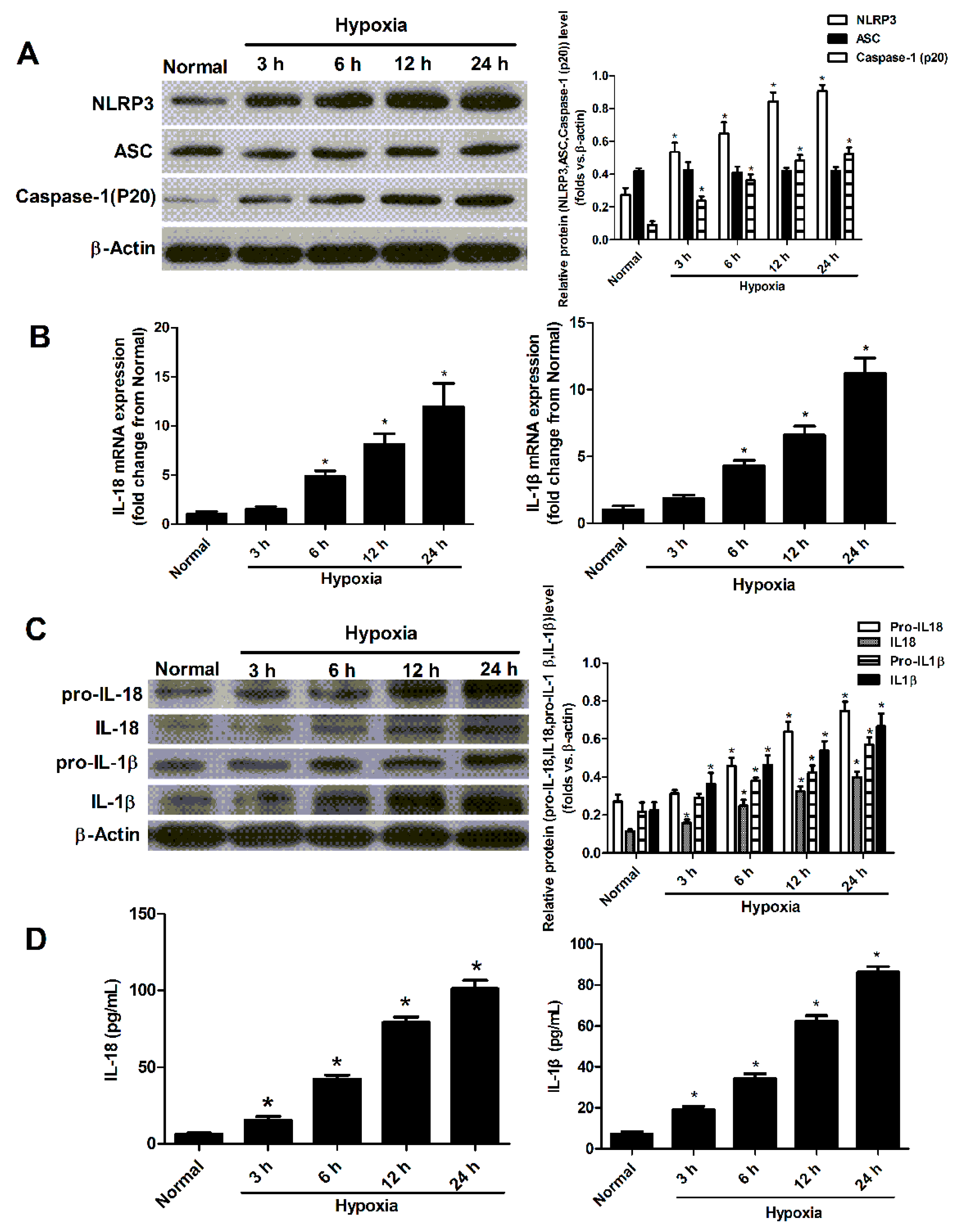
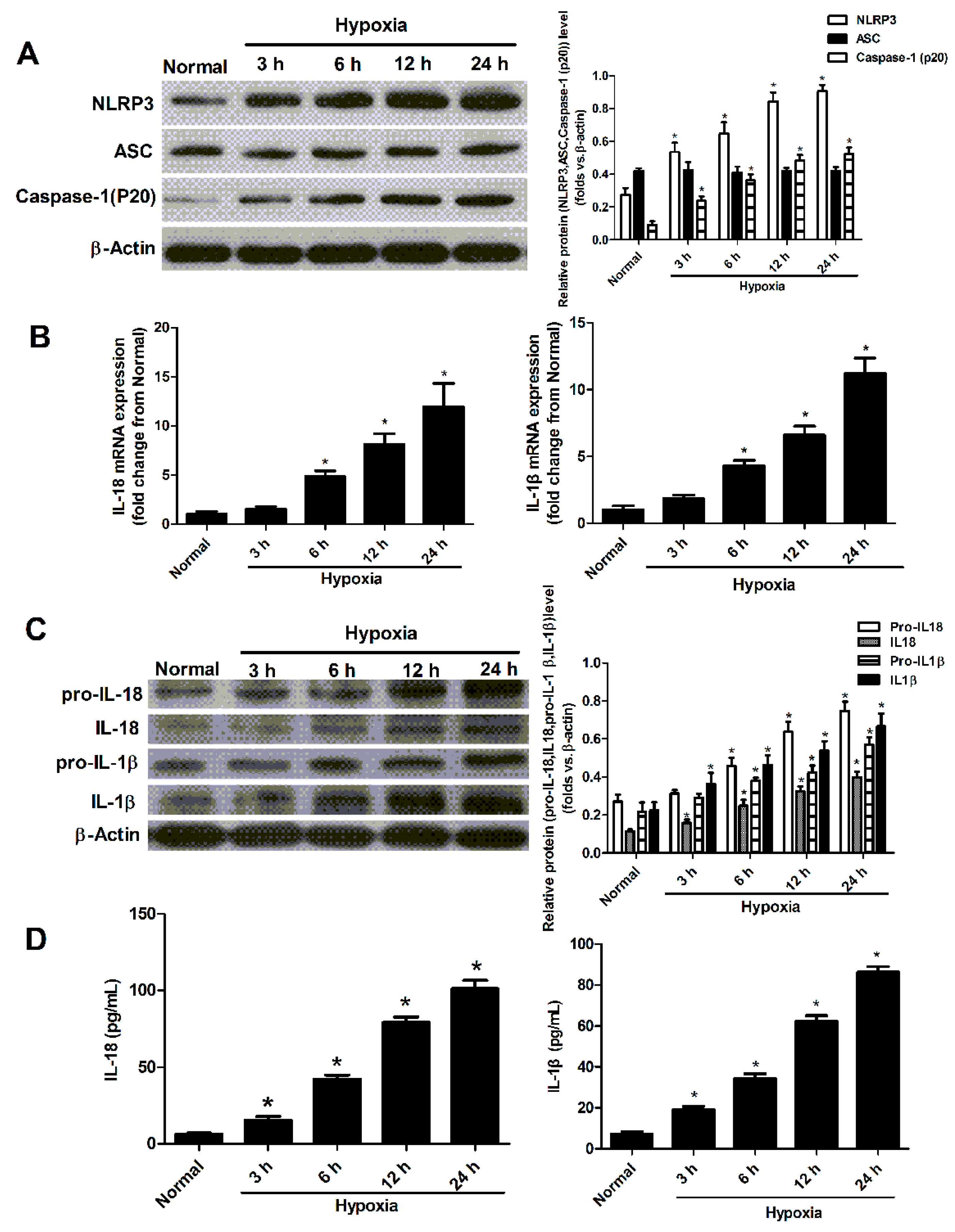
2.2. Hypoxia Induced the Activation of NLRP3 Inflammasome in Cultured Neonatal Cardiomyocytes
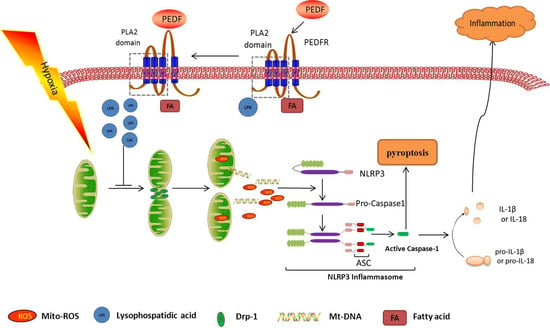
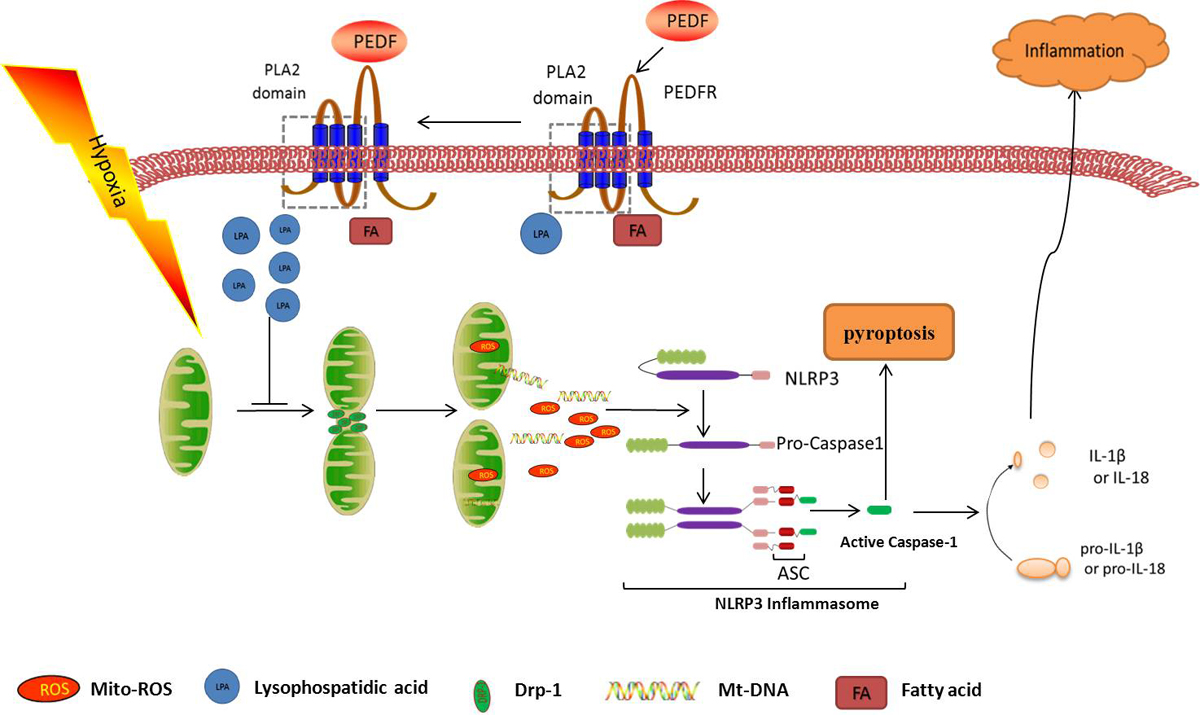
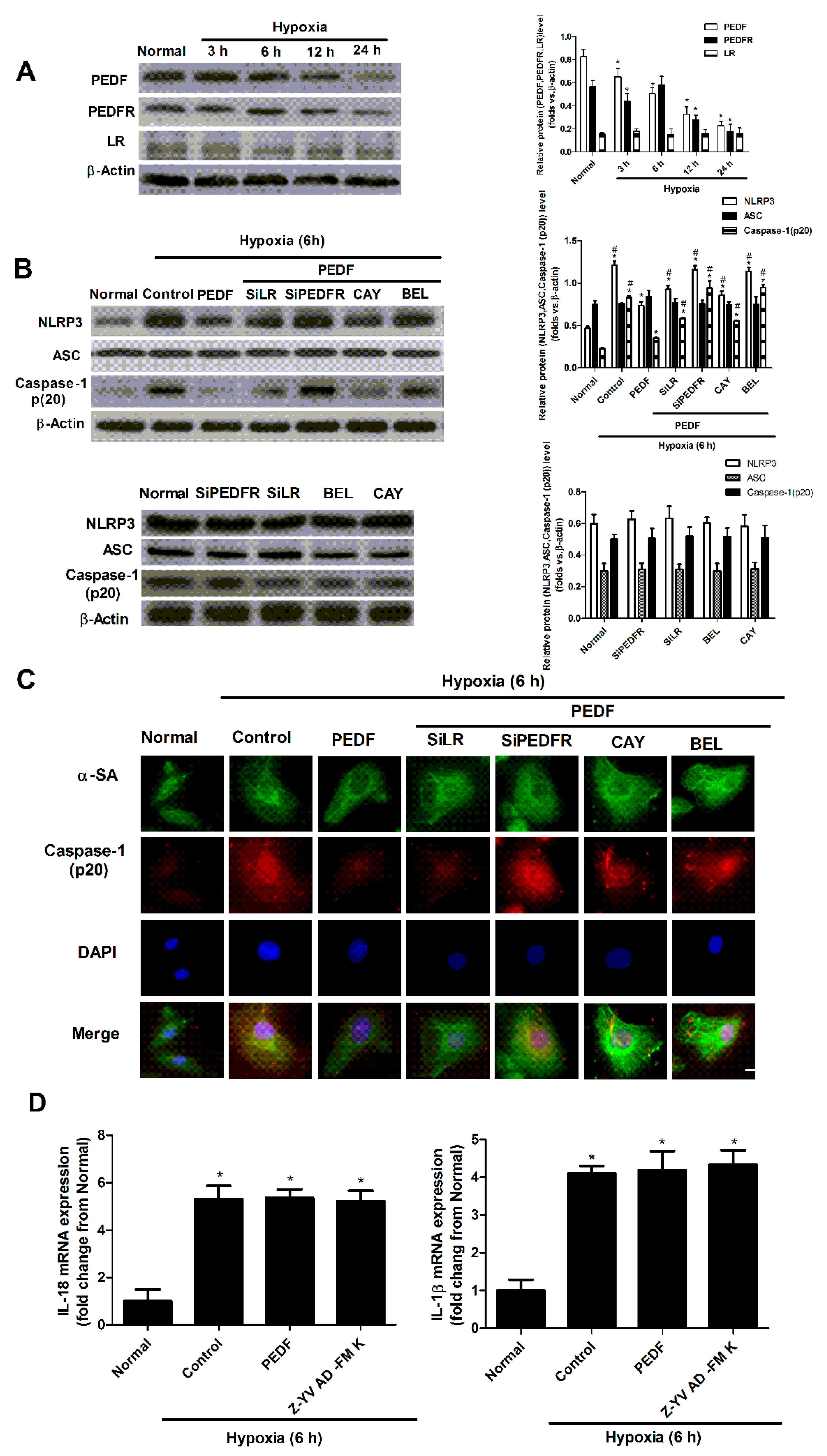
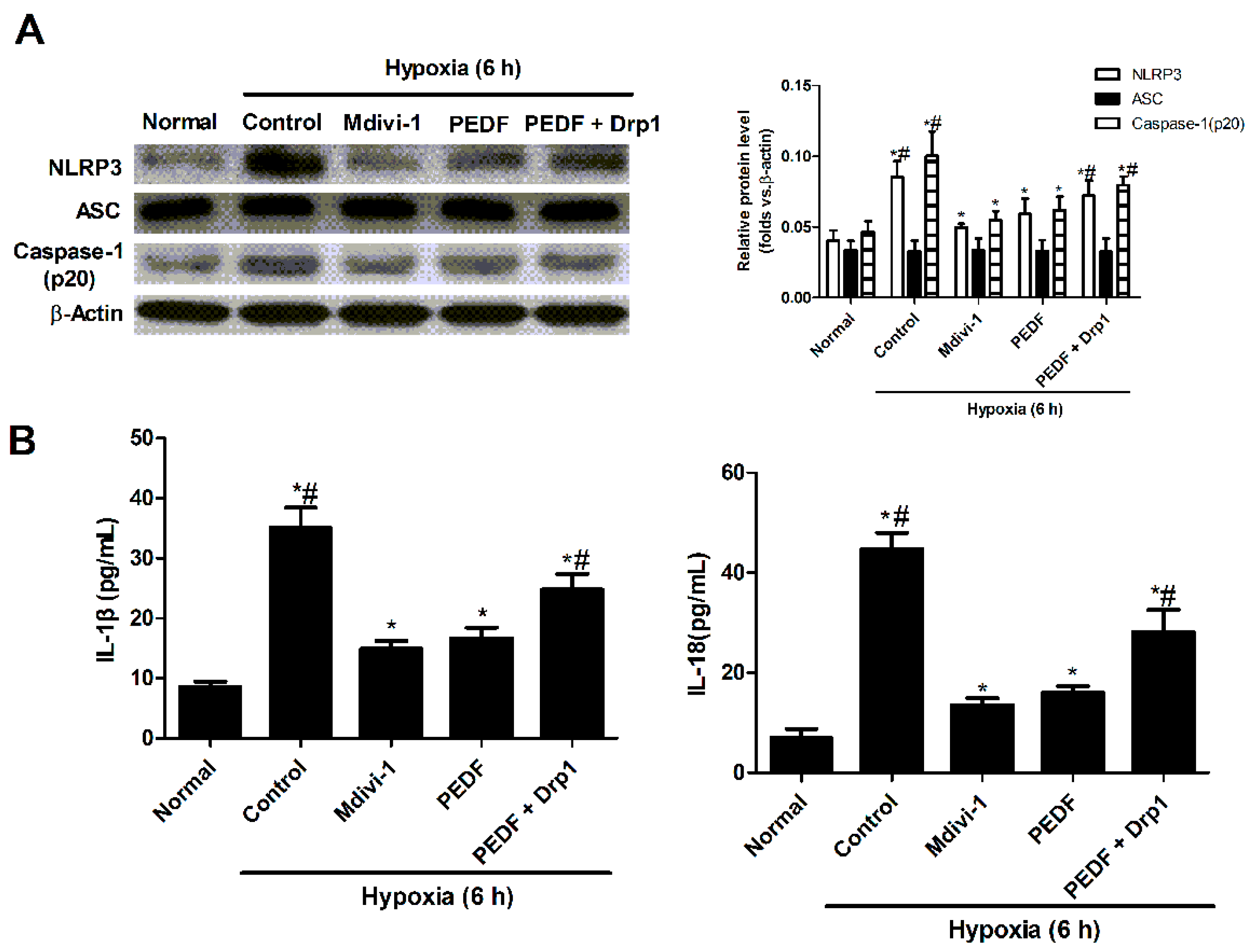
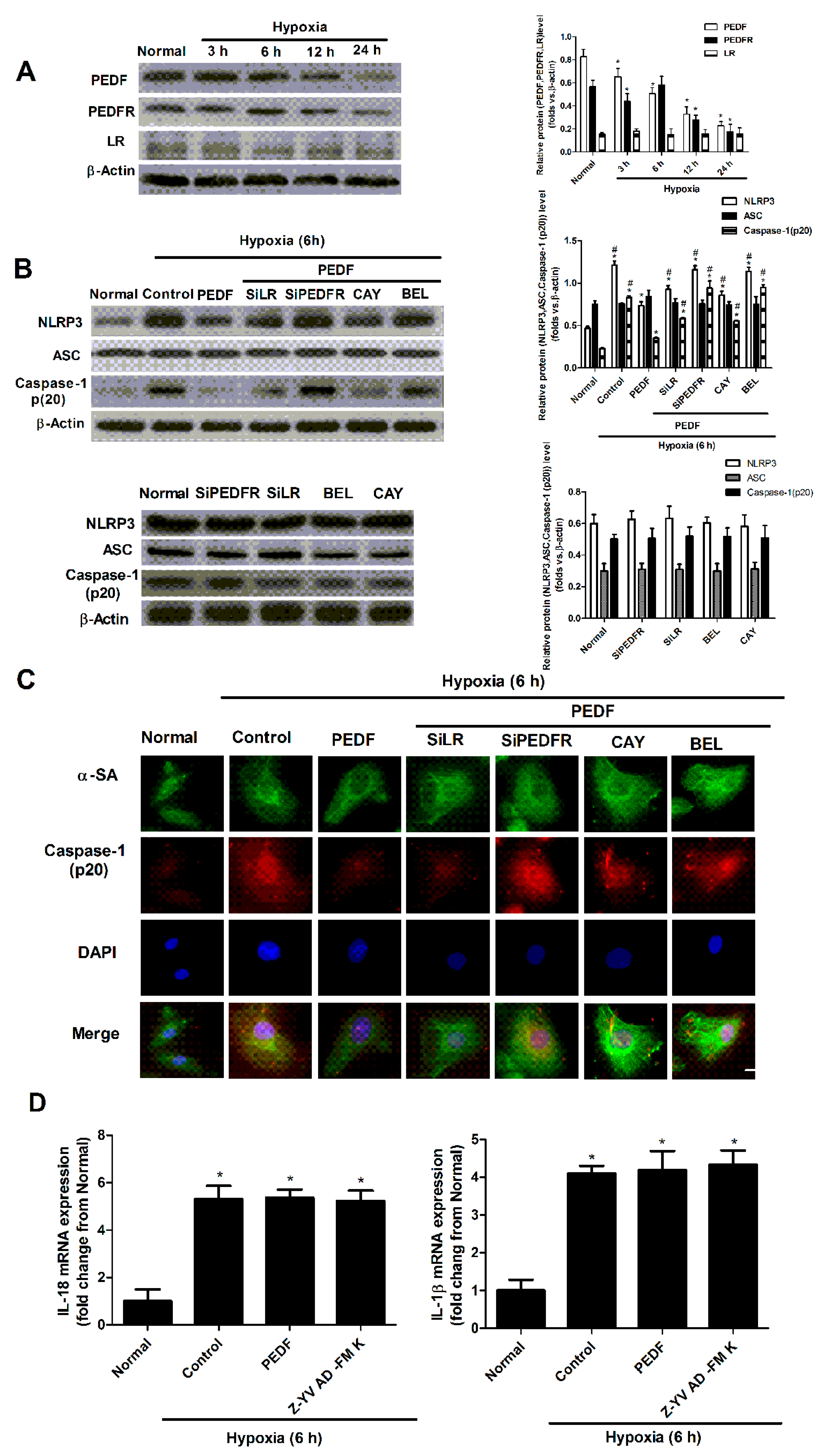
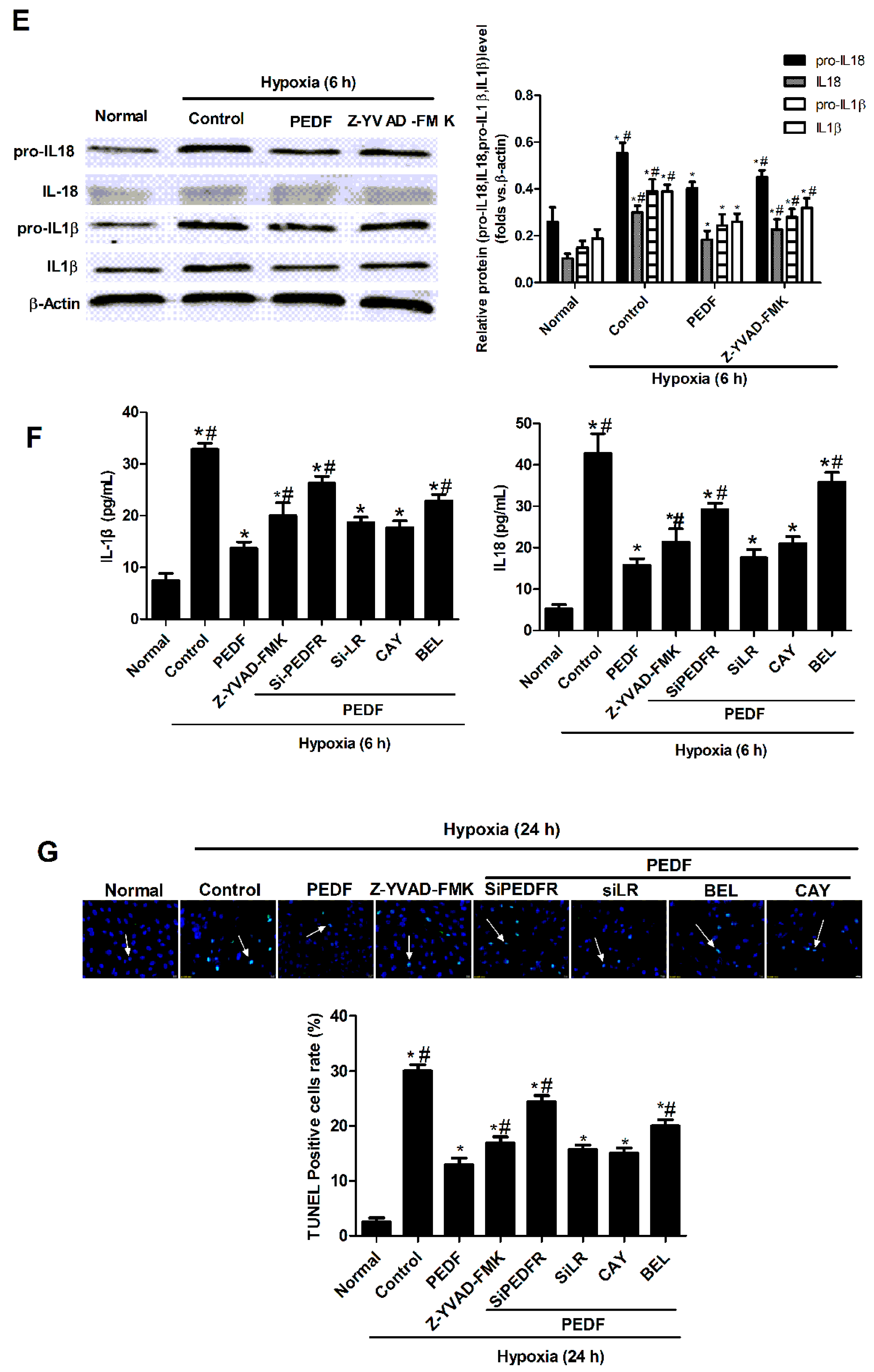
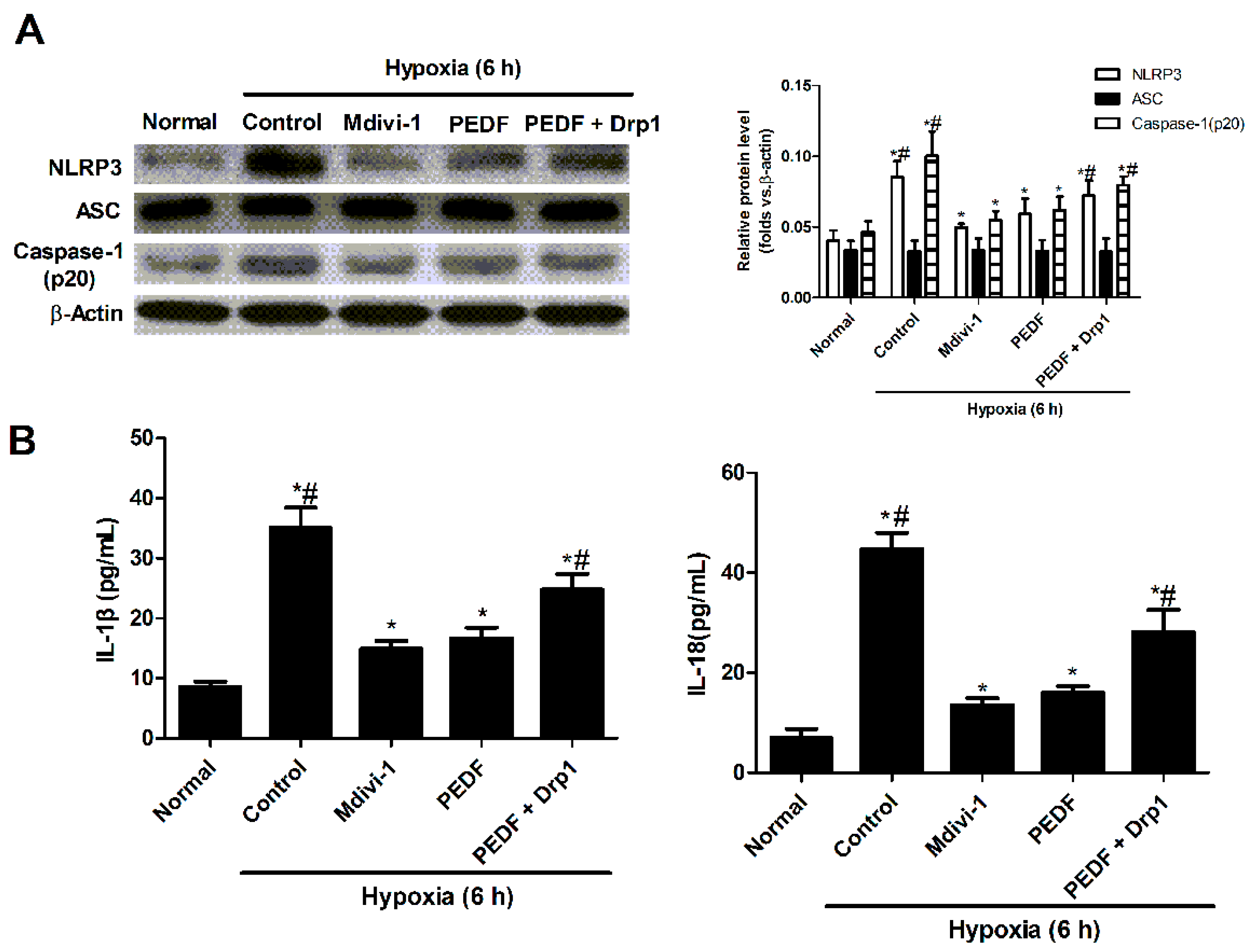
2.3. PEDF Inhibited Hypoxia-Induced NLRP3 Inflammasome Activation via PEDF-R/iPLA2 in the Neonatal Cardiomyocytes
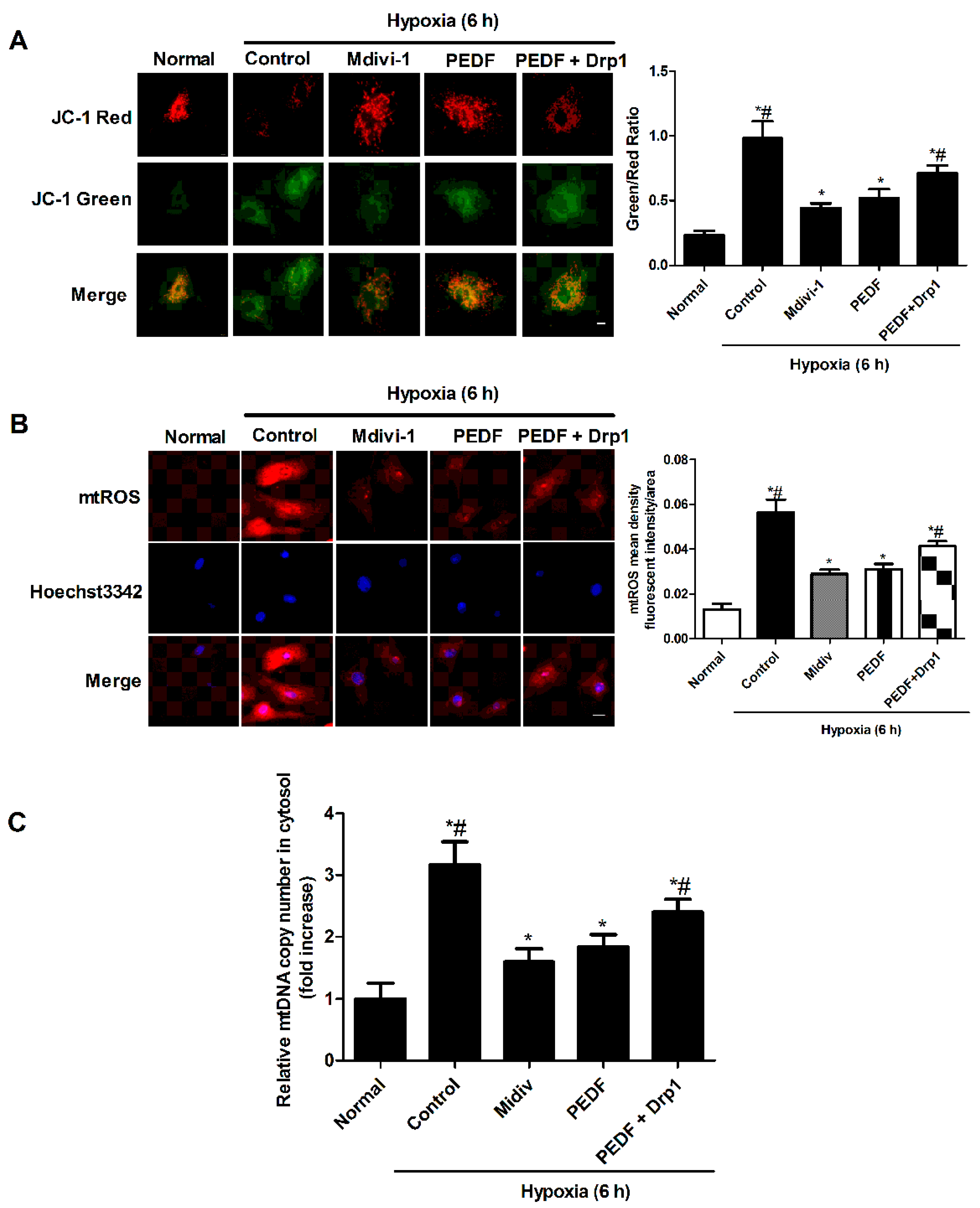
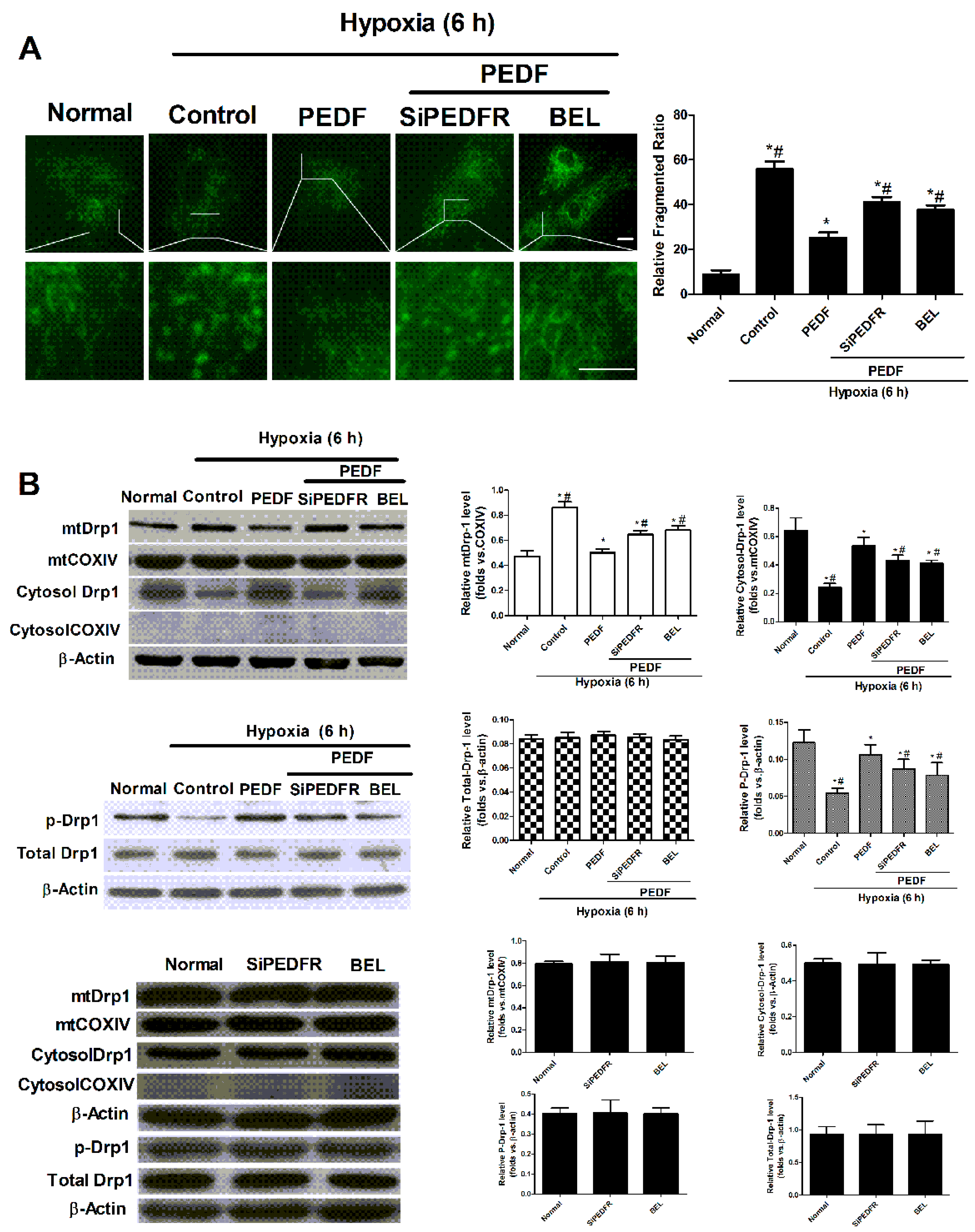
2.4. PEDF Inhibited Hypoxia-Induced Mitochondrial Fission via PEDF-R/iPLA2
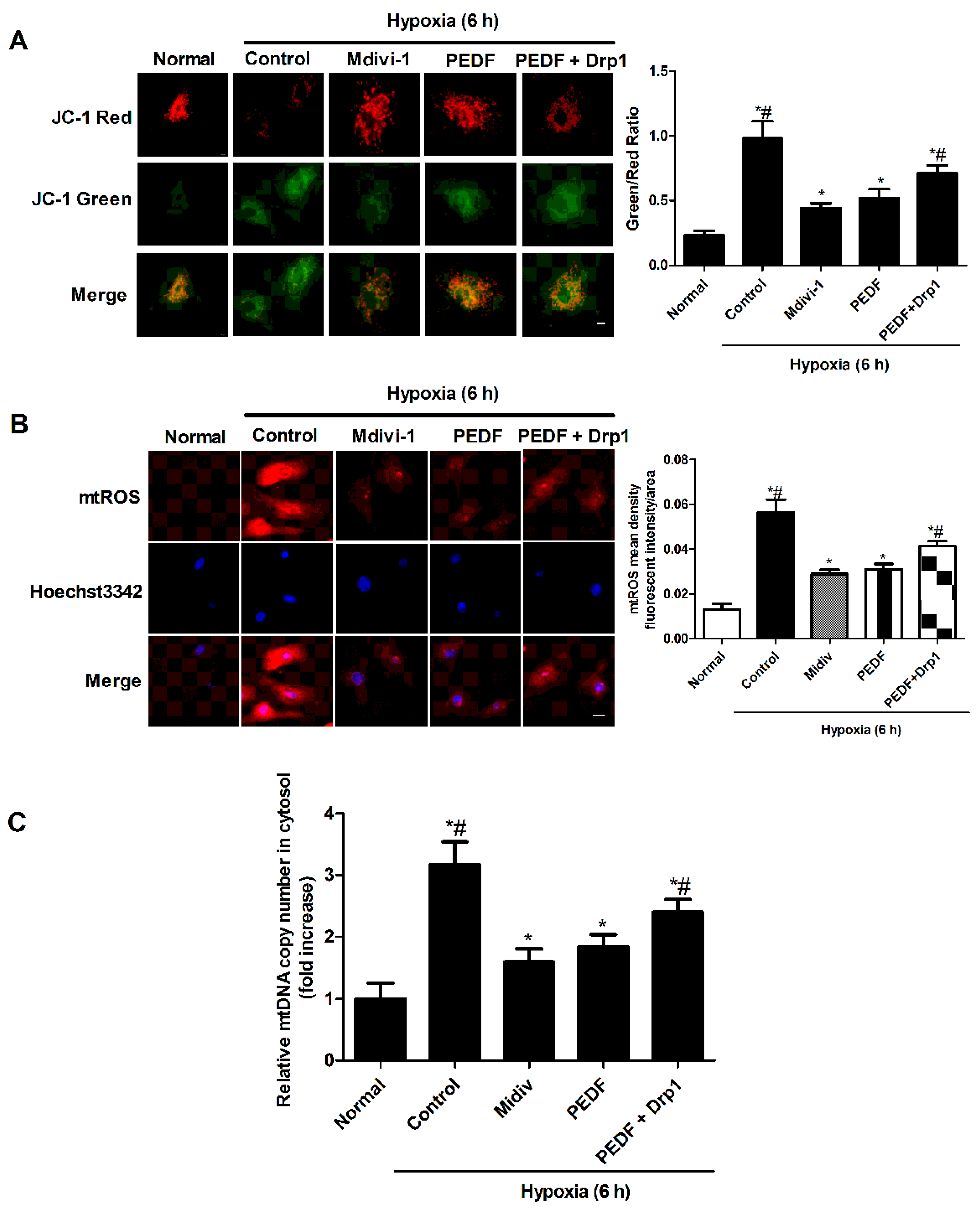
2.5. PEDF Inhibited the Hypoxia-Induced Decrease of Mitochondrial Function
2.6. PEDF Inhibited Mitochondrial Fission-Induced NLRP3 Inflammasome Activation in Neonatal Cardiomyocytes under the Hypoxic Condition
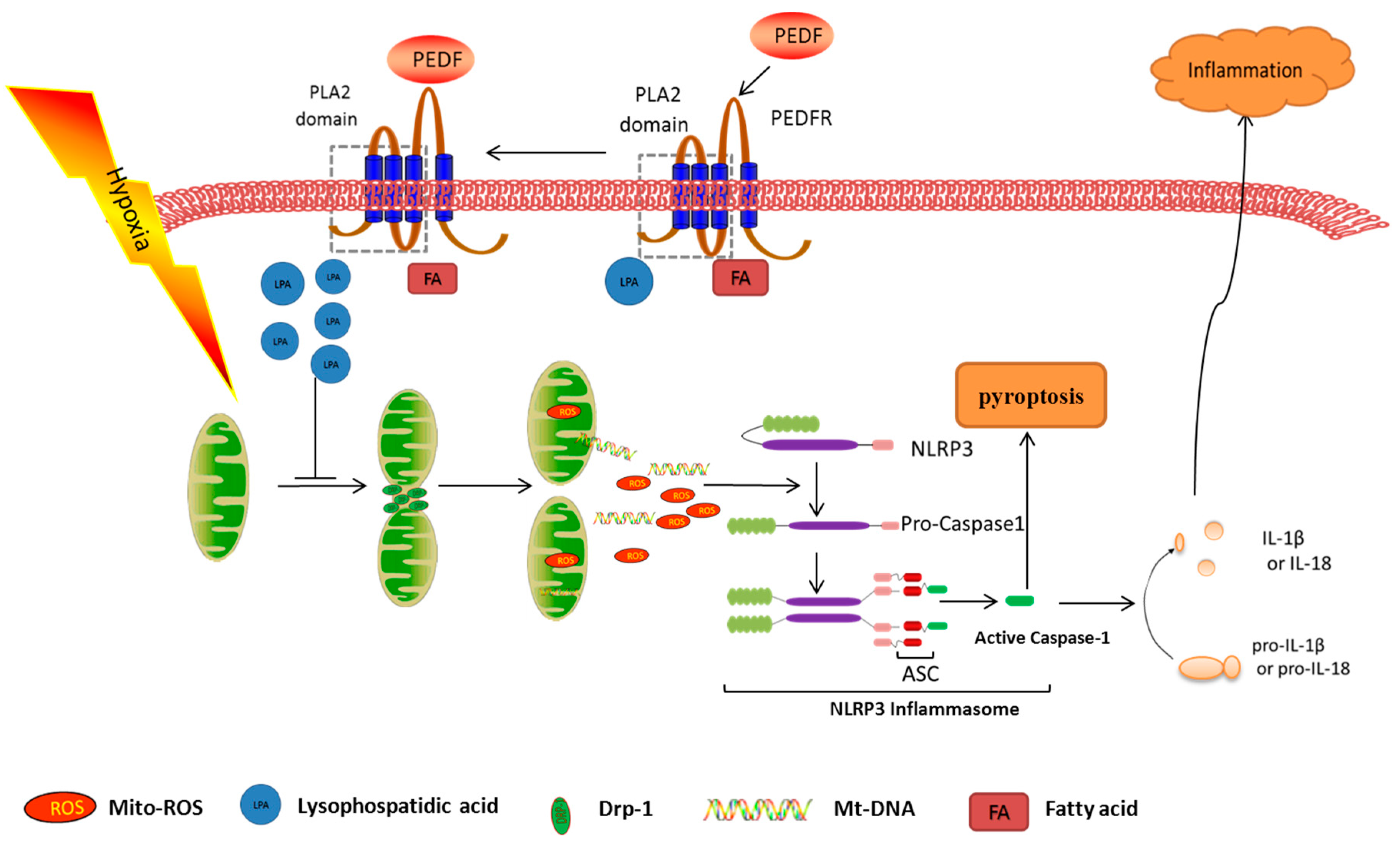
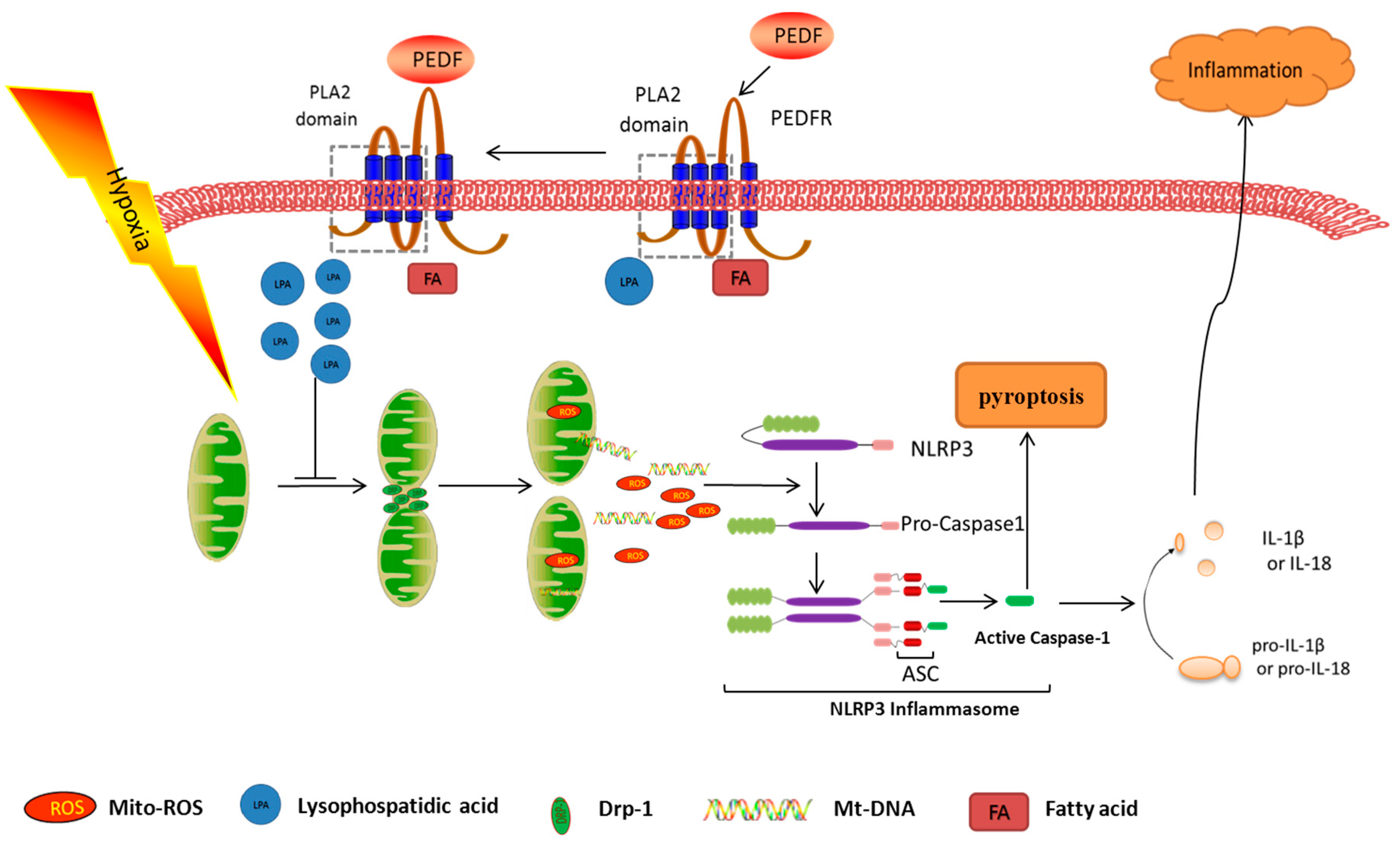
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Recombinant Lentivirus Constructs and Viral Production
4.3. Preparations of PEDF Protein
4.4. Animal Model and Intramyocardial Gene Delivery
4.5. Neonatal Cardiomyocytes Isolation, Culture and Transfection
4.6. Measurement of mtDNA in Cytosol by Quantitative Real-Time PCR
4.7. Quantitative qPCR-Based Gene Expression
4.8. Detection of Mitochondrial ROS Production
4.9. Staining of Cells with a Mitochondrion-Selective Dye, Mito-Tracker® Green
4.10. Measurement of Mitochondrial Membrane Potential
4.11. Western Blotting Analysis
4.12. Immunofluorescence
4.13. Transmission Electron Microscopy
4.14. Enzyme-Linked Immunosorbent Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Im, M.S.; Kim, H.L.; Kim, S.H.; Lim, W.H.; Seo, J.B.; Chung, W.Y.; Zo, J.H.; Kim, M.A.; Park, K.W.; Koo, B.K.; et al. Different prognostic factors according to left ventricular systolic function in patients with acute myocardial infarction. Int. J. Cardiol. 2016, 221, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Boersma, E.; Mercado, N.; Poldermans, D.; Gardien, M.; Vos, J.; Simoons, M.L. Acute myocardial infarction. Lancet 2003, 361, 847–858. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Christia, P.; Frangogiannis, N.G. Targeting inflammatory pathways in myocardial infarction. Eur. J. Clin. Investig. 2013, 43, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. (Warsz) 2009, 57, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Tombran-Tink, J.; Chader, G.G.; Johnson, L.V. PEDF: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res. 1991, 53, 411–414. [Google Scholar] [CrossRef]
- Filleur, S.; Nelius, T.; de Riese, W.; Kennedy, R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Subramanian, P.; Shen, D.; Tuo, J.; Becerra, S.P.; Chan, C.C. Pigment epithelium-derived factor reduces apoptosis and pro-inflammatory cytokine gene expression in a murine model of focal retinal degeneration. ASN Neuro 2013, 5, e00126. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Locatelli-Hoops, S.; Kenealey, J.; DesJardin, J.; Notari, L.; Becerra, S.P. Pigment epithelium-derived factor (PEDF) prevents retinal cell death via PEDF Receptor (PEDF-R): Identification of a functional ligand binding site. J. Biol. Chem. 2013, 288, 23928–23942. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Z.; Feng, S.J.; Xu, L.; Shi, H.X.; Chen, L.L.; Yuan, G.D.; Yan, W.; Zhuang, W.; Zhang, Y.Q.; et al. PEDF improves cardiac function in rats with acute myocardial infarction via inhibiting vascular permeability and cardiomyocyte apoptosis. Int. J. Mol. Sci. 2015, 16, 5618–5634. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Zhuang, W.; Yuan, G.; Sun, T.; Jiang, X.; Zhou, Z.; Yuan, H.; Zhang, Z.; Dong, H. PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Sci. Rep. 2014, 4, 5637. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Yamagishi, S. Laminin receptor mediates anti-inflammatory and anti-thrombogenic effects of pigment epithelium-derived factor in myeloma cells. Biochem. Biophys. Res. Commun. 2014, 443, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Notari, L.; Baladron, V.; Aroca-Aguilar, J.D.; Balko, N.; Heredia, R.; Meyer, C.; Notario, P.M.; Saravanamuthu, S.; Nueda, M.L.; Sanchez-Sanchez, F.; et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem. 2006, 281, 38022–38037. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Lu, P.; Zhang, H.; Li, Y.; Dong, H.; Zhang, Z. PEDF attenuates hypoxia-induced apoptosis and necrosis in H9c2 cells by inhibiting p53 mitochondrial translocation via PEDF-R. Biochem. Biophys. Res. Commun. 2015, 465, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.; Johnson, C.L.; Nelius, T.; Kennedy, R.; Riese, W.; Filleur, S. PEDF inhibits IL8 production in prostate cancer cells through PEDF receptor/phospholipase A2 and regulation of NFκB and PPARγ. Cytokine 2011, 55, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, L.; Pérez, R.; Nieto, M.L.; Balsinde, J.; Balboa, M.A. Bromoenol lactone promotes cell death by a mechanism involving phosphatidate phosphohydrolase-1 rather than calcium-independent phospholipase A2. J. Biol. Chem. 2003, 278, 44683–44690. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.M.; McCollum, G.W.; Penn, J.S. Role of cytosolic phospholipase A(2) in retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. NLRP3 in myocardial ischaemia-reperfusion injury: Inflammasome-dependent or -independent role in different cell types. Cardiovasc. Res. 2013, 99, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Gao-Li, J.; Franco, C.A.; Bouceba, T.; Huet, A.; Li, Z. Laminin receptor involvement in the anti-angiogenic activity of pigment epithelium-derived factor. J. Biol. Chem. 2009, 284, 10480–10490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, T.; Jiang, X.; Yu, H.; Wang, M.; Wei, T.; Cui, H.; Zhuang, W.; Liu, Z.; Zhang, Z.; et al. PEDF and PEDF-derived peptide 44mer stimulate cardiac triglyceride degradation via ATGL. J. Transl. Med. 2015, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Zhang, H.; Pan, J.; Li, Z.; Wei, T.; Cui, H.; Liu, Z.; Guan, Q.; Dong, H.; Zhang, Z. PEDF and PEDF-derived peptide 44mer inhibit oxygen-glucose deprivation-induced oxidative stress through upregulating PPARγ via PEDF-R in H9c2 cells. Biochem. Biophys. Res. Commun. 2016, 472, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.; Witte, K.; Mayers, J.R.; Schuh, A.L.; Audhya, A. Roles of acidic phospholipids and nucleotides in regulating membrane binding and activity of a calcium-independent phospholipase A2 isoform. J. Biol. Chem. 2012, 287, 38824–38834. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Scorrano, L. Targeting cell death. Clin. Pharmacol. Ther. 2007, 82, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimer Dis. 2014, 40, 245–256. [Google Scholar] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Maczewski, M.; Mackiewicz, U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc. Res. 2008, 79, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Ouzounian, M.; de Couto, G.; Chen, M.Y.; Yan, R.; Fukuoka, M.; Li, G.H.; Moon, M.; Liu, Y.; Gramolini, A.; et al. Cathepsin-L Ameliorates Cardiac Hypertrophy Through Activation of the Autophagy-Lysosomal Dependent Protein Processing Pathways. J. Am. Heart Assoc. 2013, 2, e000191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Morita-Fujimura, Y.; Kawase, M.; Copin, J.C.; Calagui, B.; Epstein, C.J.; Chan, P.H. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 3414–3422. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Wang, Z.; Guan, Q.; Qiu, F.; Li, Y.; Liu, Z.; Zhang, H.; Dong, H.; Zhang, Z. PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2. Int. J. Mol. Sci. 2016, 17, 2064. https://doi.org/10.3390/ijms17122064
Zhou Z, Wang Z, Guan Q, Qiu F, Li Y, Liu Z, Zhang H, Dong H, Zhang Z. PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2. International Journal of Molecular Sciences. 2016; 17(12):2064. https://doi.org/10.3390/ijms17122064
Chicago/Turabian StyleZhou, Zhongxin, Zhu Wang, Qiuhua Guan, Fan Qiu, Yufeng Li, Zhiwei Liu, Hao Zhang, Hongyan Dong, and Zhongming Zhang. 2016. "PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2" International Journal of Molecular Sciences 17, no. 12: 2064. https://doi.org/10.3390/ijms17122064