
Genetic Variations Involved in Vitamin E Status



Abstract
:1. Vitamin E: The Major Lipid-Soluble Antioxidant in the Human Body
2. Vitamin E and Human Health
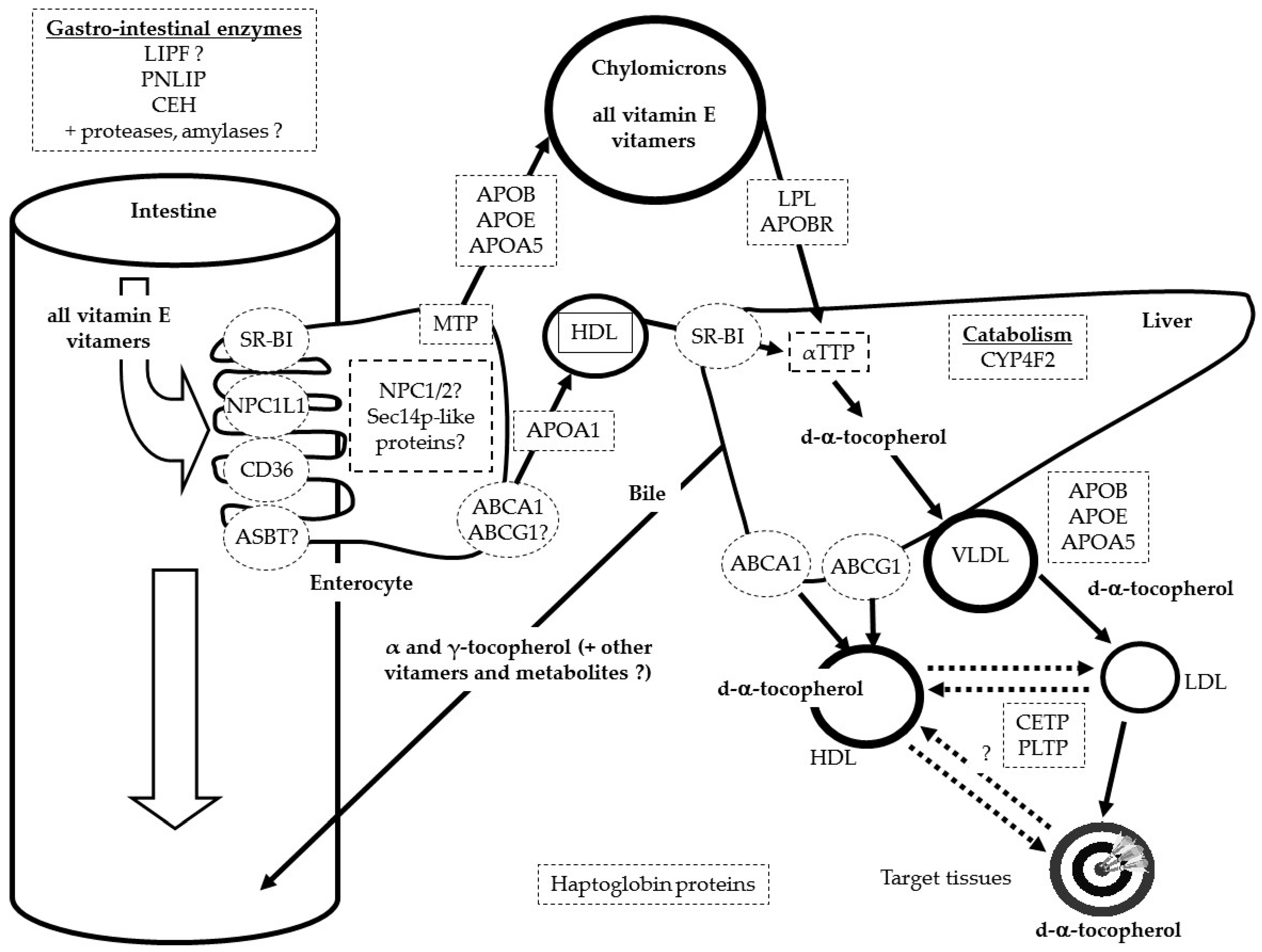
3. Proteins Involved in Vitamin E Status
4. Genetic Variations Associated with the Variability in Vitamin E Status
5. Genetic Variations Associated with the Variability in Vitamin E Bioavailability
6. Other Genetic Variations Potentially Involved in Vitamin E Status
Acknowledgments
Conflicts of Interest
References
- McBurney, M.I. Majority of Americans not consuming vitamin E RDA. J. Nutr. 2011, 141, 1920. [Google Scholar] [CrossRef] [PubMed]
- Polito, A.; Intorre, F.; Andriollo-Sanchez, M.; Azzini, E.; Raguzzini, A.; Meunier, N.; Ducros, V.; O’Connor, J.M.; Coudray, C.; Roussel, A.M.; et al. Estimation of intake and status of vitamin A, vitamin E and folate in older European adults: The ZENITH. Eur. J. Clin. Nutr. 2005, 59, S42–S47. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Friedel, A.; Roos, F.F.; Wyss, A.; Eggersdorfer, M.; Hoffmann, K.; Weber, P. A systematic review of global α-tocopherol status as assessed by nutritional intake levels and blood serum concentrations. Int. J. Vitam. Nutr. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M.; Azzi, A. Non-antioxidant activities of vitamin E. Curr. Med. Chem. 2004, 11, 1113–1133. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Gouranton, E.; Reboul, E.; Cardinault, N.; El Yazidi, C.; Malezet-Desmoulins, C.; Andre, M.; Nowicki, M.; Souidi, M.; Borel, P. Vitamin E decreases endogenous cholesterol synthesis and apo-AI-mediated cholesterol secretion in Caco-2 cells. J. Nutr. Biochem. 2010, 21, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M. Vitamin E: A role in signal transduction. Annu. Rev. Nutr. 2015, 35, 135–173. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wong, J.; Fyrst, H.; Saba, J.D.; Ames, B.N. γ-tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 17825–17830. [Google Scholar] [CrossRef] [PubMed]
- Mabile, L.; Bruckdorfer, K.R.; RiceEvans, C. Moderate supplementation with natural α-tocopherol decreases platelet aggregation and low-density lipoprotein oxidation. Atherosclerosis 1999, 147, 177–185. [Google Scholar] [CrossRef]
- McCary, C.A.; Yoon, Y.; Panagabko, C.; Cho, W.; Atkinson, J.; Cook-Mills, J.M. Vitamin E isoforms directly bind PKCα and differentially regulate activation of PKCα. Biochem. J. 2012, 441, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Pruthi, S.; Allison, T.G.; Hensrud, D.D. Vitamin E supplementation in the prevention of coronary heart disease. Mayo Clin. Proc. 2001, 76, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Negis, Y.; Zingg, J.M.; Libinaki, R.; Meydani, M.; Azzi, A. Vitamin E and cancer. Nutr. Cancer 2009, 61, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Papas, A.; Vos, E. Vitamin E, cancer, and apoptosis. Am. J. Clin. Nutr. 2001, 73, 1113. [Google Scholar] [PubMed]
- Traber, M.G. Does vitamin E decrease heart attack risk? Summary and implications with respect to dietary recommendations. J. Nutr. 2001, 131, 395S–397S. [Google Scholar] [PubMed]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer: The women’s health study: A randomized controlled trial. JAMA 2005, 294, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.; Gebretsadik, T.; Abdala-Valencia, H.; Green, J.; Larkin, E.K.; Dupont, W.D.; Shu, X.O.; Gross, M.; Bai, C.; Gao, Y.T.; et al. Interaction of vitamin E isoforms on asthma and allergic airway disease. Thorax 2016, 71, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Avila, P.C. Vitamin E and D regulation of allergic asthma immunopathogenesis. Int. Immunopharmacol. 2014, 23, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M.; Azzi, A.; Meydani, M. Genetic polymorphisms as determinants for disease-preventive effects of vitamin E. Nutr. Rev. 2008, 66, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.; Vardi, M.; Brown, J.B.; Russell, A.; Milman, U.; Shapira, C.; Levy, N.S.; Miller-Lotan, R.; Asleh, R.; Levy, A.P. Vitamin E reduces cardiovascular disease in individuals with diabetes mellitus and the haptoglobin 2-2 genotype. Pharmacogenomics 2010, 11, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Milman, U.; Blum, S.; Shapira, C.; Aronson, D.; Miller-Lotan, R.; Anbinder, Y.; Alshiek, J.; Bennett, L.; Kostenko, M.; Landau, M.; et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: A prospective double-blinded clinical trial. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Doring, F.; Rimbach, G.; Lodge, J.K. In silico search for single nucleotide polymorphisms in genes important in vitamin E homeostasis. IUBMB Life 2004, 56, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Moussa, M.; Reboul, E.; Lyan, B.; Defoort, C.; Vincent-Baudry, S.; Maillot, M.; Gastaldi, M.; Darmon, M.; Portugal, H.; et al. Human fasting plasma concentrations of vitamin E and carotenoids, and their association with genetic variants in apo C-III, cholesteryl ester transfer protein, hepatic lipase, intestinal fatty acid binding protein and microsomal triacylglycerol transfer protein. Br. J. Nutr. 2009, 101, 680–687. [Google Scholar] [PubMed]
- Borel, P.; Moussa, M.; Reboul, E.; Lyan, B.; Defoort, C.; Vincent-Baudry, S.; Maillot, M.; Gastaldi, M.; Darmon, M.; Portugal, H.; et al. Human plasma levels of vitamin E and carotenoids are associated with genetic polymorphisms in genes involved in lipid metabolism. J. Nutr. 2007, 137, 2653–2659. [Google Scholar] [PubMed]
- Borel, P.; Desmarchelier, C.; Nowicki, M.; Bott, R.; Tourniaire, F. Can genetic variability in α-tocopherol bioavailability explain the heterogeneous response to α-tocopherol supplements? Antioxid. Redox Signal. 2015, 22, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Preveraud, D.; Desmarchelier, C. Bioavailability of vitamin E in humans: An update. Nutr. Rev. 2013, 71, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Schmolz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 7, 14–43. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, C.; Tourniaire, F.; Prévéraud, D.; Samson-Kremser, C.; Crenon, I.; Rosilio, V.; Borel, P. The distribution and relative hydrolysis of tocopheryl acetate in the different matrices co-existing in the lumen of the small intestine during digestion could explain its low bioavaialbility. Mol. Nutr. Food Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Borel, P. Proteins involved in uptake, intracellular transport and basolateral secretion of fat-soluble vitamins and carotenoids by mammalian enterocytes. Prog. Lipid Res. 2011, 50, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Klein, A.; Bietrix, F.; Gleize, B.; Malezet-Desmoulins, C.; Schneider, M.; Margotat, A.; Lagrost, L.; Collet, X.; Borel, P. Scavenger receptor class B type I (SR-BI) is involved in vitamin E transport across the enterocyte. J. Biol. Chem. 2006, 281, 4739–4745. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Soayfane, Z.; Goncalves, A.; Cantiello, M.; Bott, R.; Nauze, M.; Terce, F.; Collet, X.; Comera, C. Respective contributions of intestinal Niemann-Pick C1-like 1 and scavenger receptor class B type I to cholesterol and tocopherol uptake: In vivo v. In vitro studies. Br. J. Nutr. 2012, 107, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Narushima, K.; Takada, T.; Yamanashi, Y.; Suzuki, H. Niemann-Pick C1-like 1 mediates α-tocopherol transport. Mol. Pharmacol. 2008, 74, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Roi, S.; Nowicki, M.; Niot, I.; Reboul, E. Cluster-determinant 36 (CD36) impacts on vitamin E postprandial response. Mol. Nutr. Food Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Anwar, K.; Iqbal, J.; Hussain, M.M. Mechanisms involved in vitamin E transport by primary enterocytes and in vivo absorption. J. Lipid Res. 2007, 48, 2028–2038. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Reboul, E.; Malezet-Desmoulin, C.; Lorec, A.M.; Ghiringhelli, O.; Borel, P. Comparison of different vehicles to study the effect of tocopherols on gene expression in Caco-2 cells. Free Radic. Res. 2008, 42, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Trompier, D.; Moussa, M.; Klein, A.; Landrier, J.F.; Chimini, G.; Borel, P. ATP-binding cassette transporter A1 is significantly involved in the intestinal absorption of α- and γ-tocopherol but not in that of retinyl palmitate in mice. Am. J. Clin. Nutr. 2009, 89, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Sattler, W.; Levakfrank, S.; Radner, H.; Kostner, G.M.; Zechner, R. Muscle-specific overexpression of lipoprotein lipase in transgenic mice results in increased α-tocopherol levels in skeletal muscle. Biochem. J. 1996, 318, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Stocker, A.; Zimmer, S.; Spycher, S.E.; Azzi, A. Identification of a novel cytosolic tocopherol-binding protein: Structure, specificity, and tissue distribution. IUBMB Life 1999, 48, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Panagabko, C.; Morley, S.; Hernandez, M.; Cassolato, P.; Gordon, H.; Parsons, R.; Manor, D.; Atkinson, J. Ligand specificity in the CRAL-TRIO protein family. Biochemistry 2003, 42, 6467–6474. [Google Scholar] [CrossRef] [PubMed]
- Grebenstein, N.; Schumacher, M.; Graeve, L.; Frank, J. α-Tocopherol transfer protein is not required for the discrimination against γ-tocopherol in vivo but protects it from side-chain degradation in vitro. Mol. Nutr. Food Res. 2014, 58, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Sontag, T.J.; Parker, R.S. Cytochrome p450 ω-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J. Biol. Chem. 2002, 277, 25290–25296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Tall, A.R.; Qin, S.; Lin, M.; Schneider, M.; Lalanne, F.; Deckert, V.; Desrumaux, C.; Athias, A.; Witztum, J.L.; et al. Phospholipid transfer protein deficiency protects circulating lipoproteins from oxidation due to the enhanced accumulation of vitamin E. J. Biol. Chem. 2002, 277, 31850–31856. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, J.; Olkkonen, V.M.; Jauhiainen, M.; Ehnholm, C. The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis 2001, 155, 269–281. [Google Scholar] [CrossRef]
- Hacquebard, M.; Vandenbranden, M.; Malaisse, W.J.; Ruysschaert, J.M.; Deckelbaum, R.J.; Carpentier, Y.A. Vitamin E transfer from lipid emulsions to plasma lipoproteins: Mediation by multiple mechanisms. Lipids 2008, 43, 663–671. [Google Scholar] [CrossRef] [PubMed]
- El-Sohemy, A.; Baylin, A.; Ascherio, A.; Kabagambe, E.; Spiegelman, D.; Campos, H. Population-based study of α- and γ-tocopherol in plasma and adipose tissue as biomarkers of intake in Costa Rican adults. Am. J. Clin. Nutr. 2001, 74, 356–363. [Google Scholar] [PubMed]
- Schafer, L.; Overvad, K. Subcutaneous adipose-tissue fatty acids and vitamin E in humans: Relation to diet diet and sampling site. Am. J. Clin. Nutr. 1990, 52, 486–490. [Google Scholar] [PubMed]
- Parker, R.S. Carotenoid and tocopherol composition of human adipose tissue. Am. J. Clin. Nutr. 1988, 47, 33–36. [Google Scholar] [PubMed]
- Christen, S.; Woodall, A.A.; Shigenaga, M.K.; Southwell-Keely, P.T.; Duncan, M.W.; Ames, B.N. γ-Tocopherol traps mutagenic electrophiles such as NOx and complements α-tocopherol: Physiological implications. Proc. Natl. Acad. Sci. USA 1997, 94, 3217–3222. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.; Williams, J.A.; Kucerka, N.; Atkinson, J.; Wassall, S.R.; Katsaras, J.; Harroun, T.A. Tocopherol activity correlates with its location in a membrane: A new perspective on the antioxidant vitamin E. J. Am. Chem. Soc. 2013, 135, 7523–7533. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Deckert, V.; Schneider, M.; Dutrillaux, F.; Hammann, A.; Athias, A.; Le Guern, N.; Pais de Barros, J.P.; Desrumaux, C.; Masson, D.; et al. α-Tocopherol modulates phosphatidylserine externalization in erythrocytes: Relevance in phospholipid transfer protein-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.A.; McQueen, J. Reverse cholesterol transport—A review of the process and its clinical implications. Clin. Biochem. 1997, 30, 517–525. [Google Scholar] [CrossRef]
- Major, J.M.; Yu, K.; Chung, C.C.; Weinstein, S.J.; Yeager, M.; Wheeler, W.; Snyder, K.; Wright, M.E.; Virtamo, J.; Chanock, S.; et al. Genome-wide association study identifies three common variants associated with serologic response to vitamin E supplementation in men. J. Nutr. 2012, 142, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Major, J.M.; Yu, K.; Wheeler, W.; Zhang, H.; Cornelis, M.C.; Wright, M.E.; Yeager, M.; Snyder, K.; Weinstein, S.J.; Mondul, A.; et al. Genome-wide association study identifies common variants associated with circulating vitamin E levels. Hum. Mol. Genet. 2011, 20, 3876–3883. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Perry, J.R.; Matteini, A.; Perola, M.; Tanaka, T.; Silander, K.; Rice, N.; Melzer, D.; Murray, A.; Cluett, C.; et al. Common variation in the β-carotene 15,15′-monooxygenase 1 gene affects circulating levels of carotenoids: A genome-wide association study. Am. J. Hum. Genet. 2009, 84, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Strobel, P.; Miranda, S.; Leighton, F.; Quinones, V.; Amigo, L.; Rozowski, J.; Krieger, M.; Rigotti, A. α-Tocopherol metabolism is abnormal in scavenger receptor class b type I (SR-BI)-deficient mice. J. Nutr. 2002, 132, 443–449. [Google Scholar] [PubMed]
- Zanon-Moreno, V.; Asensio-Marquez, E.M.; Ciancotti-Oliver, L.; Garcia-Medina, J.J.; Sanz, P.; Ortega-Azorin, C.; Pinazo-Duran, M.D.; Ordovas, J.M.; Corella, D. Effects of polymorphisms in vitamin E-, vitamin C-, and glutathione peroxidase-related genes on serum biomarkers and associations with glaucoma. Mol. Vis. 2013, 19, 231–242. [Google Scholar] [PubMed]
- Athinarayanan, S.; Wei, R.; Zhang, M.; Bai, S.; Traber, M.G.; Yates, K.; Cummings, O.W.; Molleston, J.; Liu, W.; Chalasani, N. Genetic polymorphism of cytochrome P450 4F2, vitamin E level and histological response in adults and children with nonalcoholic fatty liver disease who participated in PIVENS and TONIC clinical trials. PLoS ONE 2014, 9, e95366. [Google Scholar] [CrossRef] [PubMed]
- Girona, J.; Guardiola, M.; Cabre, A.; Manzanares, J.M.; Heras, M.; Ribalta, J.; Masana, L. The apolipoprotein A5 gene—1131T→C polymorphism affects vitamin E plasma concentrations in type 2 diabetic patients. Clin. Chem. Lab. Med. 2008, 46, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Lecompte, S.; Szabo de Edelenyi, F.; Goumidi, L.; Maiani, G.; Moschonis, G.; Widhalm, K.; Molnar, D.; Kafatos, A.; Spinneker, A.; Breidenassel, C.; et al. Polymorphisms in the CD36/FAT gene are associated with plasma vitamin E concentrations in humans. Am. J. Clin. Nutr. 2011, 93, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.R.; Perry, J.R.; Tanaka, T.; Hernandez, D.G.; Zheng, H.F.; Melzer, D.; Gibbs, J.R.; Nalls, M.A.; Weedon, M.N.; Spector, T.D.; et al. Imputation of variants from the 1000 Genomes Project modestly improves known associations and can identify low-frequency variant-phenotype associations undetected by HapMap based imputation. PLoS ONE 2013, 8, e64343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmarchelier, C.; Martin, J.C.; Planells, R.; Gastaldi, M.; Nowicki, M.; Goncalves, A.; Valero, R.; Lairon, D.; Borel, P. The postprandial chylomicron triacylglycerol response to dietary fat in healthy male adults is significantly explained by a combination of single nucleotide polymorphisms in genes involved in triacylglycerol metabolism. J. Clin. Endocrinol. Metab. 2014, 99, E484–E488. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Bott, G.R.; Frisdal, E.; Nowick, M.; Plengpanich, W.; Desmarchelier, C.; Roi, S.; Quinn, C.M.; Gelissen, I.; Jessup, W.; et al. ABCG1 is involved in vitamin E efflux. Biochim. Biophys. Acta 2014, 1841, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Thap, S.; Perrot, E.; Amiot, M.J.; Lairon, D.; Borel, P. Effect of the main dietary antioxidants (carotenoids, γ-tocopherol, polyphenols, and vitamin C) on α-tocopherol absorption. Eur. J. Clin. Nutr. 2007, 61, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Mekki, N.; Boirie, Y.; Partier, A.; Grolier, P.; Alexandre-Gouabau, M.C.; Beaufrere, B.; Armand, M.; Lairon, D.; Azais-Braesco, V. Postprandial chylomicron and plasma vitamin E responses in healthy older subjects compared with younger ones. Eur. J. Clin. Investig. 1997, 27, 812–821. [Google Scholar] [CrossRef]


{kind=link}
| SNP | Global MAF 1 | Nearest Gene | Trait | Reference | Study Type |
|---|---|---|---|---|---|
| rs12272004 | 0.085 | APOA5 | FBα | [52] | GWAS |
| rs964184 | 0.222 | APOA5 | FBα(αS) | [50] | GWAS |
| rs2108622 | 0.237 | CYP4F2 | FBα(αS) | [50] | GWAS |
| rs11057830 | 0.139 | SCARB1 | FBα(αS) | [50] | GWAS |
| rs7834588 | 0.433 | NKAIN3 | FBα(αS) | [50] | GWAS |
| rs10401969 | 0.118 | SUGP1 | FBα | [58] | GWAS |
| rs58542926 | 0.067 | TM6SF2 | FBα | [58] | GWAS |
| Rs675 | 0.099 | APOA4 | FBαγ | [22] | CGAS |
| E2, E3, E4 | – | APOE | FBα | [22] | CGAS |
| rs4238001 | 0.064 | SCARB1 | FBγ | [22] | CGAS |
| rs5888 | 0.323 | SCARB1 | FBα | [22] | CGAS |
| rs662799 | 0.163 | APOA5 | FBα | [56] | CGAS |
| rs5128 | 0.234 | APOC3 | FBα | [21] | CGAS |
| rs708272 | 0.378 | CETP | FBα | [21] | CGAS |
| rs1800588 | 0.387 | LIPC | FBγ | [21] | CGAS |
| rs1527479 | 0.349 | CD36 | FBα | [57] | CGAS |
| rs6994076 | 0.349 | TTPA | FBα | [54] | CGAS |
| rs2108622 | 0.237 | CYP4F2 | FBα(αS) | [55] | CGAS |
| rs3093105 | 0.157 | CYP4F2 | FBα(αS) | [55] | CGAS |
| rs468320 | 0.234 | ABCG1 | α-B 2 | [23] | CGAS |
| rs2915775 | 0.257 | PNLIP | α-B | [23] | CGAS |
| rs3010494 | 0.294 | PNLIP | α-B | [23] | CGAS |
| rs1571513 | 0.240 | SLC10A2 | α-B | [23] | CGAS |
| rs9558203 | 0.198 | SLC10A2 | α-B | [23] | CGAS |
| rs16961116 | 0.162 | SLC10A2 | α-B | [23] | CGAS |
| rs12874168 | 0.210 | SLC10A2 | α-B | [23] | CGAS |
| rs2065550 | 0.160 | SLC10A2 | α-B | [23] | CGAS |
| rs2839715 | 0.168 | SREBF2 | α-B | [23] | CGAS |
| rs4822062 | 0.153 | SREBF2 | α-B | [23] | CGAS |
| rs4149314 * | 0.069 | ABCA1 | α-B | [23] | CGAS |
| rs11789603 * | 0.117 | ABCA1 | α-B | [23] | CGAS |
| rs2274873 * | 0.082 | ABCA1 | α-B | [23] | CGAS |
| rs4149297 * | 0.084 | ABCA1 | α-B | [23] | CGAS |
| rs4643493 * | 0.082 | APOB | α-B | [23] | CGAS |
| rs1042031 * | 0.128 | APOB | α-B | [23] | CGAS |
| rs1713222 * | 0.155 | APOB | α-B | [23] | CGAS |
| rs10464587 * | 0.297 | BET1 | α-B | [23] | CGAS |
| rs1316328 * | 0.134 | IRS1 | α-B | [23] | CGAS |
| rs4238329 * | 0.148 | LIPC | α-B | [23] | CGAS |
| rs8041525 * | 0.086 | LIPC | α-B | [23] | CGAS |
| rs7164909 * | 0.153 | LIPC | α-B | [23] | CGAS |
| rs8035357 * | 0.150 | LIPC | α-B | [23] | CGAS |
| rs12591216 * | 0.084 | LIPC | α-B | [23] | CGAS |
| rs12593880 * | 0.068 | LIPC | α-B | [23] | CGAS |
| rs4921920 * | 0.101 | NAT2 | α-B | [23] | CGAS |
| rs7296124 * | 0.107 | ZNF664 | α-B | [23] | CGAS |
| rs1048497 * | 0.061 | ZNF664 | α-B | [23] | CGAS |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borel, P.; Desmarchelier, C. Genetic Variations Involved in Vitamin E Status. Int. J. Mol. Sci. 2016, 17, 2094. https://doi.org/10.3390/ijms17122094
Borel P, Desmarchelier C. Genetic Variations Involved in Vitamin E Status. International Journal of Molecular Sciences. 2016; 17(12):2094. https://doi.org/10.3390/ijms17122094
Chicago/Turabian StyleBorel, Patrick, and Charles Desmarchelier. 2016. "Genetic Variations Involved in Vitamin E Status" International Journal of Molecular Sciences 17, no. 12: 2094. https://doi.org/10.3390/ijms17122094
APA StyleBorel, P., & Desmarchelier, C. (2016). Genetic Variations Involved in Vitamin E Status. International Journal of Molecular Sciences, 17(12), 2094. https://doi.org/10.3390/ijms17122094