
Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine

Abstract
:1. Introduction
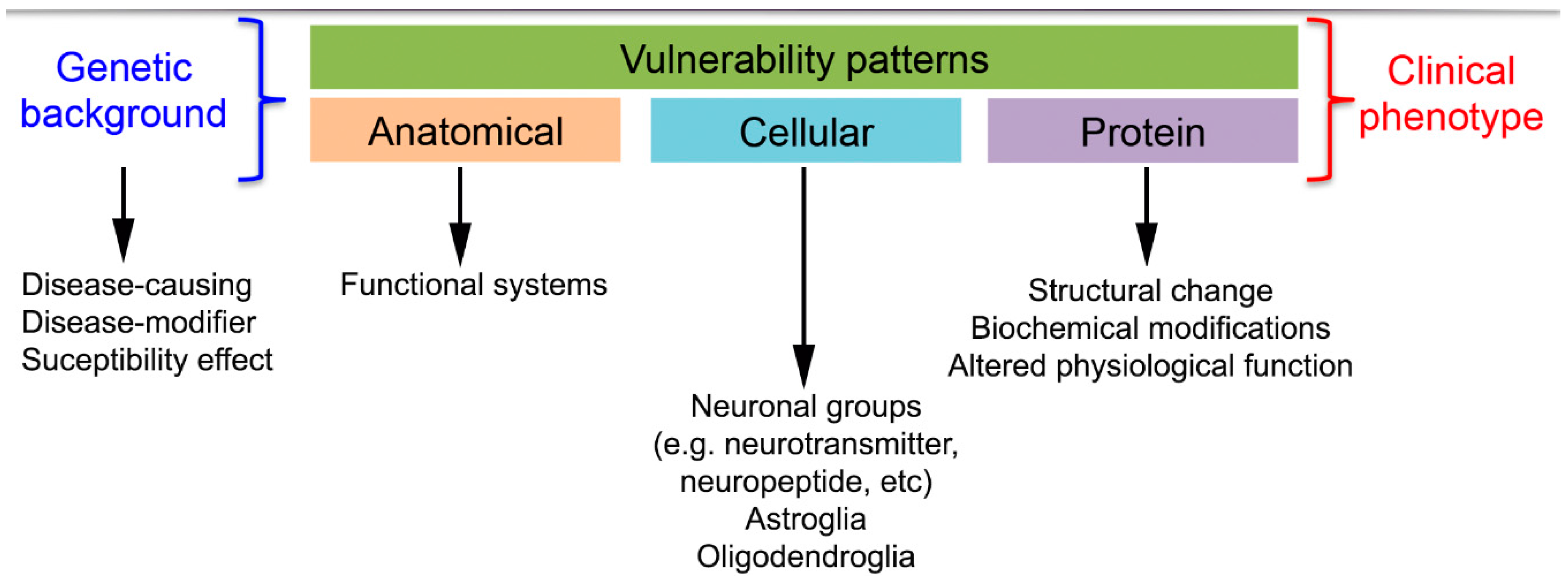
2. Concepts of Disease Classification

2.1. Anatomical Involvement of Neuronal Loss Underlying Clinical Symptomatology
2.2. Neuropathological-Biochemical Classification
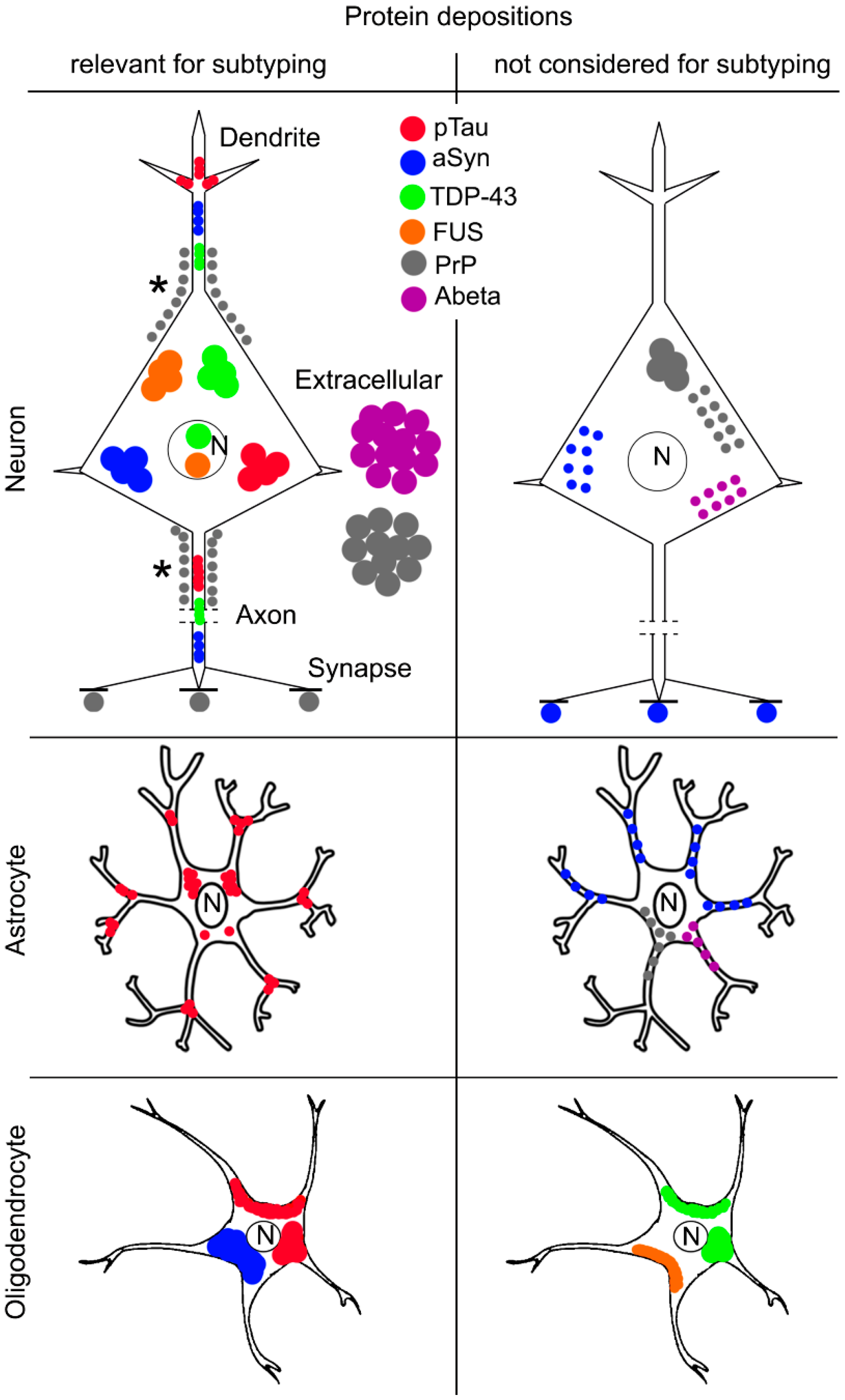
3. Altered Proteins in Neurodegenerative Diseases
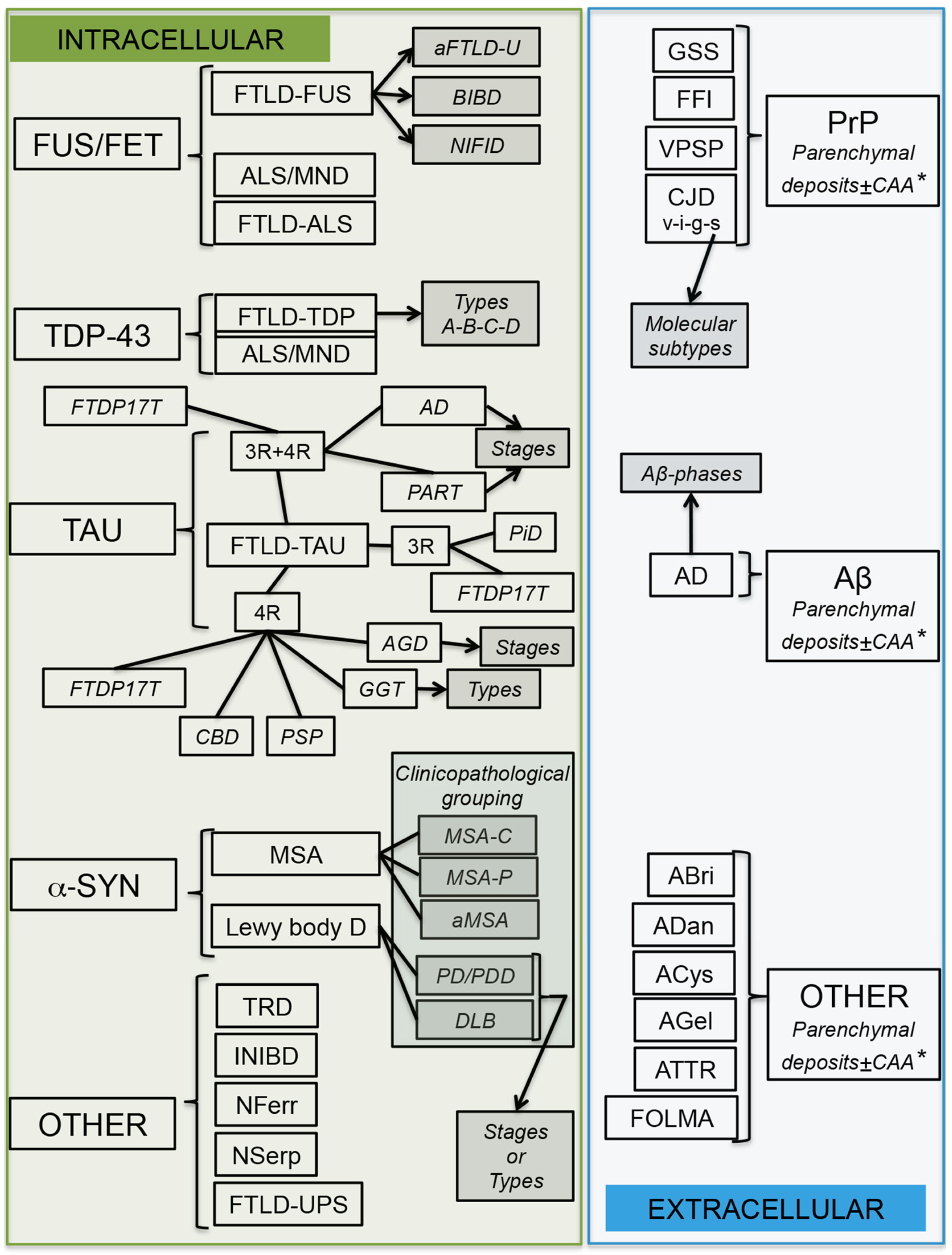
4. Molecular Pathological Subtyping

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Group | Protein | Disease Type | Form | Phenotype |
|---|---|---|---|---|
| AD | Tau, Aβ | AD | SP/GEN | DEM |
| Tauopathy (FTLD-Tau *) | Tau | PiD | SP | FTD |
| GGT | SP | FTD | ||
| CBD | SP | MD/FTD | ||
| PSP | SP | MD/FTD | ||
| AGD | SP | DEM | ||
| NFT-dementia/PART | SP | DEM | ||
| FTDP-17T | GEN | FTD/MD | ||
| TDP-43 proteinopathy | TDP-43 | FTLD-TDP (type A–D) | SP/GEN | FTD |
| MND-TDP | SP/GEN | MD | ||
| FTLD-MND-TDP | SP/GEN | FTD-MD | ||
| FUS (FET)-proteinopathy FTLD/MND-FUS (FET) | FUS/FET | aFTLD-U, NIFID, BIBD | SP | FTD/MD |
| MND-FUS | GEN | MD | ||
| α-Synucleinopathy | α-Synuclein | PD | SP/GEN | MD |
| DLB | SP/GEN | DEM/MD | ||
| MSA | SP | MD | ||
| Prion disease | PrP | sCJD, VPSPr, sFI | SP | DEM/MD |
| iCJD | ACQ | DEM/MD | ||
| vCJD | ACQ | DEM/MD | ||
| Kuru | ACQ | DEM/MD | ||
| gCJD, GSS, FFI, PrP-CAA | GEN | DEM/MD | ||
| TRD ** | Huntingtin | HD | GEN | MD |
| Ataxin 1, 2, 3, 7, CACNA1A, TBP | SCA 1, 2, 3, 6, 7, 17 | GEN | MD | |
| FMRP | FXTAS | GEN | MD | |
| ARP | SBMA | GEN | MD | |
| Atrophin-1 | DRPLA | GEN | MD | |
| Other forms | Ferritin | Hereditary ferritinopathy | GEN | DEM/MD |
| Neuroserpin | Neuroserpinopathy | GEN | DEM | |
| ABri, ADan, gelsolin, cystatin, transthyretin, Aβ | Hereditary amyloidoses/CAA | GEN | DEM | |
| Only UPS | FTLD-UPS | GEN | FTD | |
| Not determined | FTLD-ni | SP | FTD | |
| Tau, α-Synuclein | NBIA | GEN | DEM/MD | |
| Tau, a-Synuclein, TDP-43 | Various genetic and sporadic diseases (“secondary” proteinopathy forms) | SP/GEN | DEM/MD |
| Protein | Remarks on Modifications with Potential Relevance for Classification |
|---|---|
| Aβ | Aβ peptides are produced by the sequential cleavage by different proteases (e.g., β- followed by γ-secretases) |
| Aβ 1-40/1-42 peptides are the most abundant components of Aβ deposits | |
| N-terminal truncation of soluble and insoluble Aβ peptide species as well as C-terminally truncated Aβ species (1-37/38/39) have been also described | |
| Aβ deposits may have distinct PK resistance | |
| Further aspects: pyroglutamate modifications at residues 3 or 11 (AβN3pE and AβN11pE); isomerization/racemization (D-Asp or L-isoAsp at N1, N7); glycosylation; phosphorylation at Serine residue 8 and 26 (pSer8Aβ and pSer26Aβ) | |
| PrP | The physiological cellular form of PrP (PrPC) is a detergent soluble, PK sensitive protein that has endogenously truncated fragments, while the disease-associated PrPSc is detergent-insoluble and resistant to PK treatment (termed PrPres) |
| Based on differences in electrophoretic mobility and N-terminal sequence of the core fragments, different forms of PrPres were distinguished. The most common PrPres species is PrP27-30. Further fragments are for example PrP 11, PrP7-8, PrP14, PrP-CTF12/13, PrP16-17, and PrP17.5-18 | |
| Oligomer form of PrP has been also described | |
| Tau | Alternative splicing generates six isoforms, which are present in the adult human brain; based on the absence or presence of exon 10 tau isoforms, either three or four repeat (3R, 4R) domains are distinguished |
| Hyperphosphorylation is a common modification | |
| Tauopathies are distinguished based on the ratio of 3R- and 4R-tau and two or three major phospho-tau bands (60, 64, and 68 kDa) in Western blot of sarkosyl-insoluble fractions | |
| Further aspects: N- and C-terminal truncation, glycosylation, glycation, nitration of tyrosine residues, transglutamination, deamidation; acetylation; oligomer; the banding patterns of C-terminal fragments of tau and the trypsin-resistant band patterns are distinct among tauopathies | |
| a-Syn | Phosphorylation at serine 87 and 129 (most relevant) and at tyrosine 125 residue |
| Further aspects: Nitration, glycosylation, C-terminally truncated species; oligomer forms (also physiological native oligomers called multimers: α-synuclein exists in various conformations and oligomeric states in a dynamic equilibrium); PK-resistant form is also described | |
| TDP-43 | Phosphorylation on serine 379 (S379), S403, S404, S409, S410 residues |
| Further aspects: ubiquitinylation and abnormal cleavage; oligomer; C-terminal fragments detected in disease | |
| FUS | FUS was detected in the SDS soluble fractions of a subset of FTLD cases |
4.1. Alzheimer Disease
4.1.1. Overview of Neuropathological Features
4.1.2. Aspects of Classification
4.2. Prion Diseases
4.2.1. Overview of Neuropathological Features
4.2.2. Aspects of Classification
4.3. Tauopathies
4.3.1. Overview of Neuropathological Features
4.3.2. Aspects of Classification
4.4. α-Synucleinopathies
4.4.1. Overview of Neuropathological Features
4.4.2. Aspects of Classification
4.5. TDP-43 Proteinopathies
4.5.1. Overview of Neuropathological Features
4.5.2. Aspects of Classification
4.6. FUS-Proteinopathies
4.7. Rare Forms of Hereditary Neurodegenerative Conditions with Protein Deposition
5. Synthesis of Biochemistry, Genetics, and Morphology
| Disease | Molecular Pathological Features | |
|---|---|---|
| Disease Group | Currently Used for Subtyping | Disease-Specific but Not (yet) Crucial for Subtyping |
| AD-related pathology | Anatomical distribution of neuronal tau pathology | Truncated Aβ species |
| Anatomical distribution of extracellular Aβ deposits | Pyroglutamate modifications | |
| Presence and ditribution of CAA | Phosphorylation patterns of Aβ and tau | |
| / | Subtyping based on predominance of NFT | |
| Prion disease | Morphology of PrP deposition | Oligomer forms |
| Glycosylation pattern and electrophoretic mobility of PK-resistant PrP (only WB) | / | |
| Codon 129 polymorphism | / | |
| Aetiology if known | / | |
| Tauopathies | Morphology of neuronal or glial protein deposits | Detecting phosphorylation epitopes |
| Distinguishing 3R and 4R isoforms | Acetylation | |
| Anatomical distribution of protein deposits | Truncated forms (i.e., C-terminal) | |
| / | Trypsin-resistant band patterns | |
| / | Oligomer forms | |
| α-Synucleinopathies | Morphology of neuronal or glial protein deposits | Phosphorylation |
| Anatomical distribution of protein deposits | Nitration | |
| / | Oligomer forms | |
| / | Predominance of soluble/insoluble form | |
| / | Truncated forms | |
| / | Detection of PK-resistant form * | |
| TDP-43 proteinopathies | Morphology and subcellular distribution of protein deposits in neurons | Phosphorylation |
| Anatomical distribution of protein deposits | C-terminal fragments | |
| / | Glial inclusions | |
| FUS proteinopathies | Morphology, subcellular and anatomical distribution of protein deposits | Different immunoreactivity for FET proteins |
| / | Glial inclusions | |



6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| AβPP | Aβ-precursor protein |
| AD | Alzheimer disease |
| aFTLD-U | atypical FTLD-U |
| AGD | argyrophilic grain disease |
| ALS | amyotrophic lateral sclerosis |
| ARTAG | aging-related tau astrogliopathy |
| BIBD | basophilic inclusion body disease |
| BSE | bovine spongiform encephalopathy |
| CBD | corticobasal degeneration |
| CJD | Creutzfeldt-Jakob disease |
| CNS | central nervous system |
| DCTN1 | dynactin |
| DLB | dementia with Lewy bodies |
| DN | dystrophic neurities |
| EWS | Ewing’s sarcoma RNA-binding protein 1 |
| ELS | endosomal-lysosomal system |
| FFI | Fatal familial insomnia |
| FTD | frontotemporal dementia |
| FTLD | frontotemporal lobar degeneration |
| FUS | fused in sarcoma |
| GFA | granular/fuzzy astrocytes |
| GGT | globular glial tauopathies |
| GAI | Globular astroglial inclusions |
| GOI | globular oligodendroglial inclusions |
| GSS | Gerstmann-Sträussler-Scheinker disease |
| GRN | granulin |
| HD | Huntington disease |
| INIBD | intranuclear inclusion body disease |
| MAPT | microtubule associated protein tau |
| NCI | neuronal cytoplasmic inclusion |
| NDDs | Neurodegenerative diseases |
| NFT | neurofibrillary tangle |
| NIFID | neuronal intermediate filament inclusion disease |
| NII | neuronal intranuclear inclusion |
| OPTN | optineurin |
| PART | primary age-related tauopathy |
| PET-blot | paraffin-embedded-tissue blot |
| PD | Parkinson disease |
| PiD | Pick disease |
| PK | proteinase K |
| PrP | prion protein |
| PRNP | gene of PrP |
| PSP | progressive supranuclear palsy |
| R | repeat |
| SCA | spinocerebellar ataxia |
| SOD1 | superoxide dismutase |
| SNCA | α-Synuclein gene |
| SQSTM1 | sequestome-1 |
| TAF15 | TATA-binding protein-associated factor 15 |
| TARDBP | gene of TDP-43 |
| TDP-43 | Transactive response (TAR) DNA-binding protein |
| TSA | thorn-shaped astrocytes |
| VCP | valosin-containing protein |
| VPSPr | variably proteinase sensitive prionopathy |
References
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Nijholt, D.A.; de Kimpe, L.; Elfrink, H.L.; Hoozemans, J.J.; Scheper, W. Removing protein aggregates: The role of proteolysis in neurodegeneration. Curr. Med. Chem. 2011, 18, 2459–2476. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenin, J. Alzheimer’s disease: Redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox. Signal. 2012, 16, 974–1031. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 2009, 41, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Spencer, B.; Masliah, E. Immunotherapeutic Approaches Targeting Amyloid-β, α-Synuclein, and Tau for the Treatment of Neurodegenerative Disorders. Neurotherapeutics 2016, 13, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Molina-Porcel, L.; Falcone, D.; McCluskey, L.; Lee, V.M.; van Deerlin, V.M.; Trojanowski, J.Q. TDP-43 pathology in a case of hereditary spastic paraplegia with a NIPA1/SPG6 mutation. Acta Neuropathol. 2012, 124, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Zuchner, S.; Gierer, S.; Schulte, C.; Schols, L.; Schule, R.; Synofzik, M. Abnormal Paraplegin Expression in Swollen Neurites, τ- and α-Synuclein Pathology in a Case of Hereditary Spastic Paraplegia SPG7 with an Ala510Val Mutation. Int. J. Mol. Sci. 2015, 16, 25050–25066. [Google Scholar] [CrossRef]
- Woehrer, A.; Laszlo, L.; Finsterer, J.; Stollberger, C.; Furtner, J.; Rinner, W.; Molnar, K.; Budka, H.; Kovacs, G.G. Novel crystalloid oligodendrogliopathy in hereditary spastic paraplegia. Acta Neuropathol. 2012, 124, 583–591. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; den Dunnen, W.; Korf, H.W.; Rub, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Current concepts of neurodegenerative diseases. EMJ Neurol. 2014, 1, 78–86. [Google Scholar]
- Kovacs, G.G. Introduction: Classification of neurodegenerative diseases. In Neuropathology of Neurodegenerative Diseases: A Practical Guide; Kovacs, G.G., Ed.; Cambridge University Press: Cambridge, UK, 2015; pp. 1–8. [Google Scholar]
- Kovacs, G.G.; Milenkovic, I.; Wohrer, A.; Hoftberger, R.; Gelpi, E.; Haberler, C.; Honigschnabl, S.; Reiner-Concin, A.; Heinzl, H.; Jungwirth, S.; et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: A community-based autopsy series. Acta Neuropathol. 2013, 126, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Botond, G.; Budka, H. Protein coding of neurodegenerative dementias: The neuropathological basis of biomarker diagnostics. Acta Neuropathol. 2010, 119, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson's disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Budka, H. Molecular pathology of human prion diseases. Int. J. Mol. Sci. 2009, 10, 976–999. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Zou, W.Q.; Gambetti, P. Prion: The chameleon protein. Cell. Mol. Life Sci. 2007, 64, 3266–3270. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fandrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson's disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Leugers, C.J. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012, 107, 263–293. [Google Scholar] [PubMed]
- Sephton, C.F.; Yu, G. The function of RNA-binding proteins at the synapse: Implications for neurodegeneration. Cell. Mol. Life Sci. 2015, 72, 3621–3635. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Selkoe, D.; Bartels, T. New insights into cellular α-synuclein homeostasis in health and disease. Curr. Opin. Neurobiol. 2015, 36, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.; Trojanowski, J.Q.; et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Sergeant, N.; Delacourte, A.; Buee, L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 2005, 1739, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Perry, G.; Luna-Munoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser396−404 is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Watanabe, S.; Arai, T.; Kametani, F.; Nonaka, T.; Masuda-Suzukake, M.; Tarutani, A.; Murayama, S.; Saito, Y.; Arima, K.; Yoshida, M.; et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M.; et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Budka, H. Current concepts of neuropathological diagnostics in practice: Neurodegenerative diseases. Clin. Neuropathol. 2010, 29, 271–288. [Google Scholar] [CrossRef]
- Liberski, P.P.; Hainfellner, J.A.; Sikorska, B.; Budka, H. Prion protein (PrP) deposits in the tectum of experimental Gerstmann-Straussler-Scheinker disease following intraocular inoculation. Folia Neuropathol. 2012, 50, 85–88. [Google Scholar] [PubMed]
- Liberski, P.P.; Yanagihara, R.; Gibbs, C.J., Jr.; Gajdusek, D.C. Spread of Creutzfeldt-Jakob disease virus along visual pathways after intraocular inoculation. Arch. Virol. 1990, 111, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Diamond, M.I.; Duff, K.E.; Hyman, B.T. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013, 70, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: Prion-like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Holtzman, D.M. Biomarker modeling of Alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Rocca, W.A.; Knopman, D.S.; Mielke, M.M.; Lowe, V.J.; Senjem, M.L.; Gunter, J.L.; Preboske, G.M.; et al. Age-specific population frequencies of cerebral β-amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: A cross-sectional study. Lancet Neurol. 2014, 13, 997–1005. [Google Scholar] [CrossRef]
- Baker-Nigh, A.; Vahedi, S.; Davis, E.G.; Weintraub, S.; Bigio, E.H.; Klein, W.L.; Geula, C. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 2015, 138, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal Aβ accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Gouras, G.K.; Willen, K.; Tampellini, D. Critical role of intraneuronal Aβ in Alzheimer’s disease: Technical challenges in studying intracellular Aβ. Life Sci. 2012, 91, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Pensalfini, A.; Albay, R., III; Rasool, S.; Wu, J.W.; Hatami, A.; Arai, H.; Margol, L.; Milton, S.; Poon, W.W.; Corrada, M.M.; et al. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 2014, 71, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Molnar, K.; Keller, E.; Botond, G.; Budka, H.; Laszlo, L. Intraneuronal immunoreactivity for the prion protein distinguishes a subset of E200K genetic from sporadic Creutzfeldt-Jakob Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Arzberger, T.; Al-Sarraj, S.; Bodi, I.; Bogdanovic, N.; Braak, H.; Bugiani, O.; del-Tredici, K.; Ferrer, I.; Gelpi, E.; et al. Staging of neurofibrillary pathology in Alzheimer’s disease: A study of the BrainNet Europe Consortium. Brain Pathol. 2008, 18, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Thomas, A.; Jellinger, K. Correlations between cortical and subcortical tau pathology. Neuropathol. Appl. Neurobiol. 2012, 38, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.T.; Rub, U.; Ferretti, R.E.; Nitrini, R.; Farfel, J.M.; Polichiso, L.; Gierga, K.; Jacob-Filho, W.; Heinsen, H. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol. Appl. Neurobiol. 2009, 35, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, K.; Heinsen, H.; Korf, H.W.; Del Turco, D.; Ghebremedhin, E.; Seidel, K.; Bouzrou, M.; Grinberg, L.T.; Bohl, J.; Wharton, S.B.; et al. Precortical Phase of Alzheimer’s Disease (AD)-Related Tau Cytoskeletal Pathology. Brain Pathol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Ghebremedhin, E.; Rub, U.; Yamaguchi, H.; del Tredici, K.; Braak, H. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Griffin, W.S.; de Vos, R.A.; Ghebremedhin, E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008, 115, 599–609. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef]
- Nelson, P.T.; Kukull, W.A.; Frosch, M.P. Thinking outside the box: Alzheimer-type neuropathology that does not map directly onto current consensus recommendations. J. Neuropathol. Exp. Neurol. 2010, 69, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Horvath, M.C.; Majtenyi, K.; Lutz, M.I.; Hurd, Y.L.; Keller, E. Heroin abuse exaggerates age-related deposition of hyperphosphorylated tau and p62-positive inclusions. Neurobiol. Aging 2015, 36, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Pletnikova, O.; Rudow, G.L.; Hyde, T.M.; Kleinman, J.E.; Ali, S.Z.; Bharadwaj, R.; Gangadeen, S.; Crain, B.J.; Fowler, D.R.; Rubio, A.I.; et al. Alzheimer Lesions in the Autopsied Brains of People 30 to 50 Years of Age. Cogn. Behav. Neurol. 2015, 28, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Head, M.W.; Hegyi, I.; Bunn, T.J.; Flicker, H.; Hainfellner, J.A.; McCardle, L.; Laszlo, L.; Jarius, C.; Ironside, J.W.; et al. Immunohistochemistry for the prion protein: Comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol. 2002, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; de Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.J.; Hoftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef]
- Peden, A.H.; Ironside, J.W. Review: Pathology of variant Creutzfeldt-Jakob disease. Folia Neuropathol. 2004, 42 (Suppl. A), S85–S91. [Google Scholar]
- Kretzschmar, H.A.; Sethi, S.; Foldvari, Z.; Windl, O.; Querner, V.; Zerr, I.; Poser, S. Latrogenic Creutzfeldt-Jakob disease with florid plaques. Brain Pathol. 2003, 13, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, M.; Abela, E.; Maissen, M.; Aguzzi, A. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N. Engl. J. Med. 2003, 349, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Head, M.W.; Northcott, V.; Rennison, K.; Ritchie, D.; McCardle, L.; Bunn, T.J.; McLennan, N.F.; Ironside, J.W.; Tullo, A.B.; Bonshek, R.E. Prion protein accumulation in eyes of patients with sporadic and variant Creutzfeldt-Jakob disease. Investig. Ophthalmol. Vis. Sci. 2003, 44, 342–346. [Google Scholar] [CrossRef]
- Head, M.W.; Ritchie, D.; Smith, N.; McLoughlin, V.; Nailon, W.; Samad, S.; Masson, S.; Bishop, M.; McCardle, L.; Ironside, J.W. Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt-Jakob disease: An immunohistochemical, quantitative, and biochemical study. Am. J. Pathol. 2004, 164, 143–153. [Google Scholar] [CrossRef]
- Notari, S.; Moleres, F.J.; Hunter, S.B.; Belay, E.D.; Schonberger, L.B.; Cali, I.; Parchi, P.; Shieh, W.J.; Brown, P.; Zaki, S.; et al. Multiorgan detection and characterization of protease-resistant prion protein in a case of variant CJD examined in the United States. PLoS ONE 2010, 5, e8765. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.H.; Ritchie, D.L.; Head, M.W.; Ironside, J.W. Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am. J. Pathol. 2006, 168, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.H.; Ritchie, D.L.; Uddin, H.P.; Dean, A.F.; Schiller, K.A.; Head, M.W.; Ironside, J.W. Abnormal prion protein in the pituitary in sporadic and variant Creutzfeldt-Jakob disease. J. Gen. Virol. 2007, 88, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies; WHO Press: Geneva, Switzerland, 2006. [Google Scholar]
- Hilton, D.A.; Sutak, J.; Smith, M.E.; Penney, M.; Conyers, L.; Edwards, P.; McCardle, L.; Ritchie, D.; Head, M.W.; Wiley, C.A.; et al. Specificity of lymphoreticular accumulation of prion protein for variant Creutzfeldt-Jakob disease. J. Clin. Pathol. 2004, 57, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Koperek, O.; Kovacs, G.G.; Ritchie, D.; Ironside, J.W.; Budka, H.; Wick, G. Disease-associated prion protein in vessel walls. Am. J. Pathol. 2002, 161, 1979–1984. [Google Scholar] [CrossRef]
- Ghetti, B.; Tagliavini, F.; Takao, M.; Bugiani, O.; Piccardo, P. Hereditary prion protein amyloidoses. Clin. Lab. Med. 2003, 23, 65–85, viii. [Google Scholar] [CrossRef]
- Gambetti, P.; Dong, Z.; Yuan, J.; Xiao, X.; Zheng, M.; Alshekhlee, A.; Castellani, R.; Cohen, M.; Barria, M.A.; Gonzalez-Romero, D.; et al. A novel human disease with abnormal prion protein sensitive to protease. Ann. Neurol. 2008, 63, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Almer, G.; Hainfellner, J.A.; Brucke, T.; Jellinger, K.; Kleinert, R.; Bayer, G.; Windl, O.; Kretzschmar, H.A.; Hill, A.; Sidle, K.; et al. Fatal familial insomnia: A new Austrian family. Brain 1999, 122, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lugaresi, E.; Medori, R.; Montagna, P.; Baruzzi, A.; Cortelli, P.; Lugaresi, A.; Tinuper, P.; Zucconi, M.; Gambetti, P. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N. Engl. J. Med. 1986, 315, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Medori, R.; Tritschler, H.J.; LeBlanc, A.; Villare, F.; Manetto, V.; Chen, H.Y.; Xue, R.; Leal, S.; Montagna, P.; Cortelli, P.; et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N. Engl. J. Med. 1992, 326, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Peden, A.; Weis, S.; Hoftberger, R.; Berghoff, A.S.; Yull, H.; Strobel, T.; Koppi, S.; Katzenschlager, R.; Langenscheidt, D.; et al. Rapidly progressive dementia with thalamic degeneration and peculiar cortical prion protein immunoreactivity, but absence of proteinase K resistant PrP: A new disease entity? Acta Neuropathol. Commun. 2013, 1, 72. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Gelpi, E.; Ströbel, T.; Ricken, G.; Nyengaard, J.R.; Bernheimer, H.; Budka, H. Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 2007, 66, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.E.; Tipler, C.; Laszlo, L.; Hope, J.; Landon, M.; Mayer, R.J. The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J. Pathol. 1995, 176, 403–411. [Google Scholar] [CrossRef]
- Baron, G.S.; Wehrly, K.; Dorward, D.W.; Chesebro, B.; Caughey, B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrPSc) into contiguous membranes. EMBO J. 2002, 21, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar] [PubMed]
- Caughey, B.; Raymond, G.J.; Ernst, D.; Race, R.E. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): Implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 1991, 65, 6597–6603. [Google Scholar] [PubMed]
- Laszlo, L.; Lowe, J.; Self, T.; Kenward, N.; Landon, M.; McBride, T.; Farquhar, C.; McConnell, I.; Brown, J.; Hope, J.; et al. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J. Pathol. 1992, 166, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Raeber, A.J.; Borchelt, D.R.; Serban, D.; Prusiner, S.B. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 1992, 3, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J.; Tschoke, S.; Kranefuss, N.; Drose, W.; Hause-Reitner, D.; Giese, A.; Groschup, M.H.; Kretzschmar, H.A. The paraffin-embedded tissue blot detects PrPSc early in the incubation time in prion diseases. Am. J. Pathol. 2000, 156, 51–56. [Google Scholar] [CrossRef]
- Quadrio, I.; Perret-Liaudet, A.; Kovacs, G.G. Molecular diagnosis of human prion disease. Expert Opin. Med. Diagn. 2011, 5, 291–306. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Bigio, E.H.; Mackenzie, I.R.; Neumann, M.; Lee, V.M.; Hatanpaa, K.J.; White, C.L., III; Schneider, J.A.; Grinberg, L.T.; Halliday, G.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropath. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef]
- Ahmed, Z.; Bigio, E.H.; Budka, H.; Dickson, D.W.; Ferrer, I.; Ghetti, B.; Giaccone, G.; Hatanpaa, K.J.; Holton, J.L.; Josephs, K.A.; et al. Globular glial tauopathies (GGT): Consensus recommendations. Acta Neuropathol. 2013, 126, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Doherty, K.M.; Silveira-Moriyama, L.; Bandopadhyay, R.; Lashley, T.; Mamais, A.; Hondhamuni, G.; Wray, S.; Newcombe, J.; O'Sullivan, S.S.; et al. Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: An emerging group of 4-repeat tauopathies. Acta Neuropathol. 2011, 122, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999, 9, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Ferrer, I.; Grinberg, L.T.; Alafuzoff, I.; Attems, J.; Budka, H.; Cairns, N.J.; Crary, J.F.; Duyckaerts, C.; Ghetti, B.; et al. Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol. 2016, 131, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Lopez-Gonzalez, I.; Carmona, M.; Arregui, L.; Dalfo, E.; Torrejon-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Rozemuller, A.J.; van Swieten, J.C.; Gelpi, E.; Majtenyi, K.; Al-Sarraj, S.; Troakes, C.; Bodi, I.; King, A.; Hortobagyi, T.; et al. Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: A study of the BrainNet Europe Consortium. Neuropathol. Appl. Neurobiol. 2013, 39, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Tacik, P.; Sanchez-Contreras, M.; Rademakers, R.; Dickson, D.W.; Wszolek, Z.K. Genetic Disorders with Tau Pathology: A Review of the Literature and Report of Two Patients with Tauopathy and Positive Family Histories. Neurodegener. Dis. 2016, 16, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernandez, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.T.; Wang, X.; Wang, C.; Sohn, P.D.; Theofilas, P.; Sidhu, M.; Arevalo, J.B.; Heinsen, H.; Huang, E.J.; Rosen, H.; et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013, 125, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ruberu, N.N.; Sawabe, M.; Arai, T.; Tanaka, N.; Kakuta, Y.; Yamanouchi, H.; Murayama, S. Staging of argyrophilic grains: An age-associated tauopathy. J. Neuropathol. Exp. Neurol. 2004, 63, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Santpere, G.; van Leeuwen, F.W. Argyrophilic grain disease. Brain 2008, 131, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Holton, J.L.; Strand, C.; Pittman, A.; de Silva, R.; Lees, A.J.; Revesz, T. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 2007, 130, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Brettschneider, J.; McMillan, C.T.; Cooper, F.; Olm, C.; Arnold, S.E.; van Deerlin, V.M.; Seeley, W.W.; Miller, B.L.; Lee, E.B.; et al. Deep Clinical and Neuropathological Phenotyping of Pick’s Disease. Ann. Neurol. 2015. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Majtenyi, K.; Spina, S.; Murrell, J.R.; Gelpi, E.; Hoftberger, R.; Fraser, G.; Crowther, R.A.; Goedert, M.; Budka, H.; et al. White matter tauopathy with globular glial inclusions: A distinct sporadic frontotemporal lobar degeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Budka, H. Protein-based neuropathology and molecular classification of human neurodegenerative diseases. In Protein Folding and Misfolding: Neurodegenerative Diseases; Ovadi, J., Orosz, F., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 251–272. [Google Scholar]
- Campbell, B.C.; McLean, C.A.; Culvenor, J.G.; Gai, W.P.; Blumbergs, P.C.; Jakala, P.; Beyreuther, K.; Masters, C.L.; Li, Q.X. The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 2001, 76, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Brudek, T.; Winge, K.; Bredo Rasmussen, N.; Bahl Czarna, J.M.; Tanassi, J.; Klitmoller Agander, T.; Hyde, T.M.; Pakkenberg, B. Altered Α-Synuclein, Parkin, and Synphilin Isoform Levels in Multiple System Atrophy Brains. J. Neurochem. 2016, 136, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Song, Y.J.; Harding, A.J. Striatal β-amyloid in dementia with Lewy bodies but not Parkinson’s disease. J. Neural Transm. 2011, 118, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006, 112, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Parkkinen, L.; Al-Sarraj, S.; Arzberger, T.; Bell, J.; Bodi, I.; Bogdanovic, N.; Budka, H.; Ferrer, I.; Gelpi, E.; et al. Assessment of α-synuclein pathology: A study of the BrainNet Europe Consortium. J. Neuropathol. Exp. Neurol. 2008, 67, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; White, C.L.; Hamilton, R.L.; Duda, J.E.; Iwatsubo, T.; Dickson, D.W.; Leverenz, J.B.; Roncaroli, F.; Buttini, M.; Hladik, C.L.; et al. Evaluation of α-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008, 116, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Wagner, U.; Dumont, B.; Pikkarainen, M.; Osman, A.A.; Streichenberger, N.; Leisser, I.; Verchere, J.; Baron, T.; Alafuzoff, I.; et al. An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012, 124, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Vaikath, N.N.; Majbour, N.K.; Paleologou, K.E.; Ardah, M.T.; van Dam, E.; van de Berg, W.D.; Forrest, S.L.; Parkkinen, L.; Gai, W.P.; Hattori, N.; et al. Generation and characterization of novel conformation-specific monoclonal antibodies for α-synuclein pathology. Neurobiol. Dis. 2015, 79, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Miake, H.; Mizusawa, H.; Iwatsubo, T.; Hasegawa, M. Biochemical characterization of the core structure of α-synuclein filaments. J. Biol. Chem. 2002, 277, 19213–19219. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson's disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Mimura, J.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Proteinase K-resistant α-synuclein is deposited in presynapses in human Lewy body disease and A53T α-synuclein transgenic mice. Acta Neuropathol. 2010, 120, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of α-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138, 1642–1657. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Demonstration of α-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp. Neurol. 2002, 176, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lorincz, P.; Perju-Dumbrava, L.; Giera, R.; Pirker, W.; Lutz, M.; et al. Intracellular processing of disease-associated α-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated α-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of α-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Perrett, R.M.; Alexopoulou, Z.; Tofaris, G.K. The endosomal pathway in Parkinson’s disease. Mol. Cell. Neurosci. 2015, 66, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Walker, D.G.; Adler, C.H.; Shill, H.; Tran, H.; Akiyama, H.; Sue, L.I.; Caviness, J.; Sabbagh, M.N.; Beach, T.G. Biochemical increase in phosphorylated α-synuclein precedes histopathology of Lewy-type synucleinopathies. Brain Pathol. 2012, 22, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; et al. Changes in properties of serine 129 phosphorylated α-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Muntane, G.; Ferrer, I.; Martinez-Vicente, M. α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 2012, 200, 106–119. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Bruck, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and α-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.H.; Dugger, B.N.; Hinni, M.L.; Lott, D.G.; Driver-Dunckley, E.; Hidalgo, J.; Henry-Watson, J.; Serrano, G.; Sue, L.I.; Nagel, T.; et al. Submandibular gland needle biopsy for the diagnosis of Parkinson disease. Neurology 2014, 82, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Dugger, B.N.; Serrano, G.; Hidalgo, J.; Henry-Watson, J.; Shill, H.A.; Sue, L.I.; Sabbagh, M.N.; Akiyama, H.; et al. Submandibular gland biopsy for the diagnosis of Parkinson disease. J. Neuropathol. Exp. Neurol. 2013, 72, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Carew, J.; Serrano, G.; Adler, C.H.; Shill, H.A.; Sue, L.I.; Sabbagh, M.N.; Akiyama, H.; Cuenca, N.; Arizona Parkinson’s Disease Consortium. Phosphorylated α-synuclein-immunoreactive retinal neuronal elements in Parkinson’s disease subjects. Neurosci. Lett. 2014, 571, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Hawkes, C.H.; Ghebremedhin, E.; Braak, H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010, 119, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Marti, M.J.; Hernandez, I.; Valldeoriola, F.; Rene, R.; et al. Multiple organ involvement by α-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Sobotka, S.; Chen, J.; Su, H.; Sanders, I.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Samanta, J.E.; Beach, T.G.; et al. Α-synuclein pathology and axonal degeneration of the peripheral motor nerves innervating pharyngeal muscles in Parkinson disease. J. Neuropathol. Exp. Neurol. 2013, 72, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, J.; Milenkovic, I.; Kovacs, G.G. Patterns of Tau and α-Synuclein Pathology in the Visual System. J. Parkinsons Dis. 2015, 5, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Mahlke, J.; Siswanto, S.; Kruger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.W.; et al. The brainstem pathologies of Parkinson's disease and dementia with Lewy bodies. Brain Pathol. 2015, 25, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Doppler, K.; Weis, J.; Karl, K.; Ebert, S.; Ebentheuer, J.; Trenkwalder, C.; Klebe, S.; Volkmann, J.; Sommer, C. Distinctive distribution of phospho-α-synuclein in dermal nerves in multiple system atrophy. Mov. Disord. 2015, 30, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, M.; Saito, Y.; Sengoku, R.; Sakiyama, Y.; Hatsuta, H.; Kanemaru, K.; Sawabe, M.; Arai, T.; Ito, G.; Iwatsubo, T.; et al. Lewy body pathology involves cutaneous nerves. J. Neuropathol. Exp. Neurol. 2008, 67, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Boettner, M.; Alexoudi, A.; Zorenkov, D.; Deuschl, G.; Wedel, T. Can we use peripheral tissue biopsies to diagnose Parkinson’s disease? A review of the literature. Eur. J. Neurol. 2016, 23, 247–261. [Google Scholar] [CrossRef]
- Zange, L.; Noack, C.; Hahn, K.; Stenzel, W.; Lipp, A. Phosphorylated α-synuclein in skin nerve fibres differentiates Parkinson’s disease from multiple system atrophy. Brain 2015, 138, 2310–2321. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Swallow, D.; Grosset, K.A.; Anichtchik, O.; Spillantini, M.; Grosset, D.G. Α-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2014, 130, 59–72. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of α-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.D.; Coon, E.A.; Powell, S.Z.; Jenkins, S.M.; Benarroch, E.E.; Low, P.A.; Schmeichel, A.M.; Parisi, J.E. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015, 138, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Accumulation of phosphorylated α-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 2015. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Filamentous aggregations of phosphorylated α-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol. Commun. 2015, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Van Rooden, S.M.; Heiser, W.J.; Kok, J.N.; Verbaan, D.; van Hilten, J.J.; Marinus, J. The identification of Parkinson’s disease subtypes using cluster analysis: A systematic review. Mov. Disord. 2010, 25, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kosaka, K.; Tsuchiya, K.; Yoshimura, M. Lewy body disease with and without dementia: A clinicopathological study of 35 cases. Clin. Neuropathol. 1988, 7, 299–305. [Google Scholar] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O'Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.; Lippa, C.F.; Lee, V.M.; Trojanowski, J.Q. Lewy bodies in the amygdala: Increase of α-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch. Neurol. 2004, 61, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Uchikado, H.; Lin, W.L.; DeLucia, M.W.; Dickson, D.W. Alzheimer disease with amygdala Lewy bodies: A distinct form of α-synucleinopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 2008, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Formation and development of Lewy pathology: A critical update. J. Neurol. 2009, 256 (Suppl. 3), S270–S279. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord. 2012, 27, 8–30. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathology of multiple system atrophy: New thoughts about pathogenesis. Mov. Disord. 2014, 29, 1720–1741. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Revesz, T.; the Neuropathology Working Group on MSA. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol. Appl. Neurobiol. 2007, 33, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Boyer, P.J.; Lund, C.; Lin, W.L.; Koga, S.; Ross, O.A.; Weiner, M.; Lipton, A.; Powers, J.M.; White, C.L., III; et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: Frontotemporal lobar degeneration associated with α-synuclein. Acta Neuropathol. 2015, 130, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Rohan, Z.; Rahimi, J.; Weis, S.; Kapas, I.; Auff, E.; Mitrovic, N.; Liberski, P.P.; Sikorska, B.; Matej, R.; Kovacs, G.G. Screening for α-synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD-synuclein). Acta Neuropathol. 2015, 130, 299–301. [Google Scholar] [CrossRef]
- Benussi, A.; Padovani, A.; Borroni, B. Phenotypic Heterogeneity of Monogenic Frontotemporal Dementia. Front Aging Neurosci. 2015, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dachsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009, 41, 163–165. [Google Scholar] [CrossRef]
- Kovacs, G.G.; van der Zee, J.; Hort, J.; Kristoferitsch, W.; Leitha, T.; Hoftberger, R.; Strobel, T.; van Broeckhoven, C.; Matej, R. Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology 2016, 36, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Frick, P.; Neumann, M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014, 127, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, J.; Gijselinck, I.; Dillen, L.; van Langenhove, T.; Theuns, J.; Engelborghs, S.; Philtjens, S.; Vandenbulcke, M.; Sleegers, K.; Sieben, A.; et al. A pan-European study of the C9orf72 repeat associated with FTLD: Geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 2013, 34, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, J.; Van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matej, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Langenhove, T.; van der Zee, J.; van Broeckhoven, C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann. Med. 2012, 44, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Dickson, D.W.; Stoessl, A.J.; Tsuboi, Y.; Chapon, F.; Gutmann, L.; Lechevalier, B.; Calne, D.B.; Personett, D.A.; Hulihan, M.; et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat. Disord. 2009, 15, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kamada, M.; Izumi, Y.; Ayaki, T.; Nakamura, M.; Kagawa, S.; Kudo, E.; Sako, W.; Maruyama, H.; Nishida, Y.; Kawakami, H.; et al. Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology 2014, 34, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Rohan, Z.; Matej, R.; Rusina, R.; Kovacs, G.G. Oligodendroglial response in the spinal cord in TDP-43 proteinopathy with motor neuron involvement. Neurodegener. Dis. 2014, 14, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Espay, A.J.; Spina, S.; Houghton, D.J.; Murrell, J.R.; de Courten-Myers, G.M.; Ghetti, B.; Litvan, I. Rapidly progressive atypical parkinsonism associated with frontotemporal lobar degeneration and motor neuron disease. J. Neurol. Neurosurg. Psychiatry 2011, 82, 751–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Kwong, L.K.; Truax, A.C.; Vanmassenhove, B.; Kretzschmar, H.A.; van Deerlin, V.M.; Clark, C.M.; Grossman, M.; Miller, B.L.; Trojanowski, J.Q.; et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 2007, 66, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Bigio, E.H.; Cairns, N.J.; Womack, K.B.; Weintraub, S.; Morris, J.C.; Foong, C.; Xiao, G.; Hladik, C.; Mantanona, T.Y.; et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: A midwest-southwest consortium for FTLD study. J. Neuropathol. Exp. Neurol. 2008, 67, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Arai, T. Significance and limitation of the pathological classification of TDP-43 proteinopathy. Neuropathology 2014, 34, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ludolph, A.; Thal, D.R.; del Tredici, K. Amyotrophic lateral sclerosis: Dash-like accumulation of phosphorylated TDP-43 in somatodendritic and axonal compartments of somatomotor neurons of the lower brainstem and spinal cord. Acta Neuropathol. 2010, 120, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White, C.L., III; Schneider, J.A.; Kretzschmar, H.A.; et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007, 171, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Cairns, N.J.; Grossman, M.; McMillan, C.T.; Lee, E.B.; van Deerlin, V.M.; Lee, V.M.; Trojanowski, J.Q. Frontotemporal lobar degeneration: Defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015, 129, 469–491. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Rohrer, J.D.; Mead, S.; Revesz, T. Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2015, 41, 858–881. [Google Scholar] [CrossRef]
- Matej, R.; Botond, G.; Laszlo, L.; Kopitar-Jerala, N.; Rusina, R.; Budka, H.; Kovacs, G.G. Increased neuronal Rab5 immunoreactive endosomes do not colocalize with TDP-43 in motor neuron disease. Exp. Neurol. 2010, 225, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.F.; Chen, Y.R.; Liu, X.B.; DeCarli, C.; Seeley, W.W.; Jin, L.W. Detection of TDP-43 oligomers in frontotemporal lobar degeneration-TDP. Ann. Neurol. 2015, 78, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Schmitt, F.A.; Lin, Y.; Abner, E.L.; Jicha, G.A.; Patel, E.; Thomason, P.C.; Neltner, J.H.; Smith, C.D.; Santacruz, K.S.; et al. Hippocampal sclerosis in advanced age: Clinical and pathological features. Brain 2011, 134, 1506–1518. [Google Scholar] [CrossRef]
- Dickson, D.W.; Rademakers, R.; Nicholson, A.M.; Schneider, J.A.; Yu, L.; Bennett, D.A. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 2015, 85, 1354–1355. [Google Scholar] [CrossRef]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; De Jager, P.L.; Yang, J.; Trojanowski, J.Q.; Bennett, D.A.; Schneider, J.A. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 2015, 84, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Martinez-Lage, M.; Kwong, L.K.; Lee, V.M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: The TDP-43 diseases. J. Neurol. 2009, 256, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Mackenzie, I.R.; Hasegawa, M.; Nonoka, T.; Niizato, K.; Tsuchiya, K.; Iritani, S.; Onaya, M.; Akiyama, H. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 125–136. [Google Scholar] [CrossRef]
- Fujishiro, H.; Uchikado, H.; Arai, T.; Hasegawa, M.; Akiyama, H.; Yokota, O.; Tsuchiya, K.; Togo, T.; Iseki, E.; Hirayasu, Y. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol. 2009, 117, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Parisi, J.E.; Petrucelli, L.; Jack, C.R.; Petersen, R.C.; Dickson, D.W. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014, 127, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Murray, M.E.; Ogaki, K.; Fujioka, S.; Rutherford, N.J.; Rademakers, R.; Ross, O.A.; Dickson, D.W. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol. 2015, 129, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Baborie, A.; Pickering-Brown, S.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Neary, D.; Snowden, J.S.; Mann, D.M. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. 2006, 112, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Sampathu, D.M.; Neumann, M.; Kwong, L.K.; Chou, T.T.; Micsenyi, M.; Truax, A.; Bruce, J.; Grossman, M.; Trojanowski, J.Q.; Lee, V.M. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 2006, 169, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; del Tredici, K.; Irwin, D.J.; Grossman, M.; Robinson, J.L.; Toledo, J.B.; Lee, E.B.; Fang, L.; van Deerlin, V.M.; Ludolph, A.C.; et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014, 127, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.G.; Neumann, M.; Kusaka, H.; Yokota, O.; Ishihara, K.; Terada, S.; Kuroda, S.; Mackenzie, I.R. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009, 118, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Roeber, S.; Kretzschmar, H.A.; Rademakers, R.; Baker, M.; Mackenzie, I.R. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009, 118, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Higashi, S.; Kawakami, I.; Kobayashi, Z.; Hosokawa, M.; Katsuse, O.; Togo, T.; Hirayasu, Y.; Akiyama, H. Localization of fused in sarcoma (FUS) protein to the post-synaptic density in the brain. Acta Neuropathol. 2012, 124, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, W.F. Neuropathological diagnostic considerations in hyperkinetic movement disorders. Front. Neurol. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Rub, U.; Hentschel, M.; Stratmann, K.; Brunt, E.; Heinsen, H.; Seidel, K.; Bouzrou, M.; Auburger, G.; Paulson, H.; Vonsattel, J.P.; et al. Huntington’s disease (HD): Degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol. 2014, 24, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Sima, A.A.; Junck, L.; Kluin, K.J.; Koeppe, R.A.; Lohman, M.E.; Little, R. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann. Neurol. 1996, 39, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sato, T.; Tsuji, S.; Takahashi, H. CAG repeat disorder models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hagerman, P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013, 12, 786–798. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Holohan, P.D.; Shrimpton, A.E.; Tatum, A.H.; Daucher, J.; Collins, G.H.; Todd, R.; Bradshaw, C.; Kent, P.; Feiglin, D.; et al. Familial encephalopathy with neuroserpin inclusion bodies. Am. J. Pathol. 1999, 155, 1901–1913. [Google Scholar] [CrossRef]
- Davis, R.L.; Shrimpton, A.E.; Holohan, P.D.; Bradshaw, C.; Feiglin, D.; Collins, G.H.; Sonderegger, P.; Kinter, J.; Becker, L.M.; Lacbawan, F.; et al. Familial dementia caused by polymerization of mutant neuroserpin. Nature 1999, 401, 376–379. [Google Scholar] [CrossRef]
- Gourfinkel-An, I.; Duyckaerts, C.; Camuzat, A.; Meyrignac, C.; Sonderegger, P.; Baulac, M.; Brice, A. Clinical and neuropathologic study of a French family with a mutation in the neuroserpin gene. Neurology 2007, 69, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.C.; Murrell, J.R.; Delisle, M.B.; Andermann, E.; Andermann, F.; Guiot, M.C.; Ghetti, B. Encephalopathy with neuroserpin inclusion bodies presenting as progressive myoclonus epilepsy and associated with a novel mutation in the Proteinase Inhibitor 12 gene. Brain Pathol. 2011, 21, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Takao, M.; Benson, M.D.; Murrell, J.R.; Yazaki, M.; Piccardo, P.; Unverzagt, F.W.; Davis, R.L.; Holohan, P.D.; Lawrence, D.A.; Richardson, R.; et al. Neuroserpin mutation S52R causes neuroserpin accumulation in neurons and is associated with progressive myoclonus epilepsy. J. Neuropathol. Exp. Neurol. 2000, 59, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Ghetti, B.; Takao, M.; Brefel-Courbon, C.; Uro-Coste, E.; Glazier, B.S.; Siani, V.; Benson, M.D.; Calvas, P.; Miravalle, L.; et al. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J. Neuropathol. Exp. Neurol. 2004, 63, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef]
- Takahashi-Fujigasaki, J. Neuronal intranuclear hyaline inclusion disease. Neuropathology 2003, 23, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Tanji, K.; Odagiri, S.; Hattori, M.; Hoshikawa, Y.; Kono, C.; Yasui, K.; Yokoi, S.; Hasegawa, Y.; Kamitani, T.; et al. Ubiquitin-related proteins in neuronal and glial intranuclear inclusions in intranuclear inclusion body disease. Pathol. Int. 2012, 62, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mimuro, M.; Yoshida, M.; Hashizume, Y.; Niwa, H.; Miyao, S.; Ujihira, N.; Akatsu, H. Inclusion-positive cell types in adult-onset intranuclear inclusion body disease: Implications for clinical diagnosis. Acta Neuropathol. 2008, 116, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Murray, M.E.; Lin, W.L.; Kusaka, H.; Dickson, D.W. Optineurin immunoreactivity in neuronal and glial intranuclear inclusions in adult-onset neuronal intranuclear inclusion disease. Am. J. Neurodegener. Dis. 2014, 3, 93–102. [Google Scholar] [PubMed]
- Woulfe, J.; Gray, D.A.; Mackenzie, I.R. FUS-Immunoreactive Intranuclear Inclusions in Neurodegenerative Disease. Brain Pathol. 2010, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Trummert, A.; Unterberger, U.; Strobel, T.; Hortobagyi, T.; Kovacs, G.G. Atypical sporadic CJD-MM phenotype with white matter kuru plaques associated with intranuclear inclusion body and argyrophilic grain disease. Neuropathology 2015, 35, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Alafuzoff, I.; Al-Sarraj, S.; Arzberger, T.; Bogdanovic, N.; Capellari, S.; Ferrer, I.; Gelpi, E.; Kovari, V.; Kretzschmar, H.; et al. Mixed brain pathologies in dementia: The BrainNet Europe consortium experience. Dement. Geriatr. Cogn. Disord. 2008, 26, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G.; Seguin, J.; Quadrio, I.; Hoftberger, R.; Kapas, I.; Streichenberger, N.; Biacabe, A.G.; Meyronet, D.; Sciot, R.; Vandenberghe, R.; et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: Characterization of a complex proteinopathy. Acta Neuropathol. 2011, 121, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Sieczkowski, E.; Milenkovic, I.; Venkataramani, V.; Giera, R.; Strobel, T.; Hoftberger, R.; Liberski, P.P.; Auff, E.; Wirths, O.; Bayer, T.A.; et al. I716F AβPP mutation associates with the deposition of oligomeric pyroglutamate amyloid-β and α-synucleinopathy with Lewy bodies. J. Alzheimer’s Dis. 2015, 44, 103–114. [Google Scholar]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. https://doi.org/10.3390/ijms17020189
Kovacs GG. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. International Journal of Molecular Sciences. 2016; 17(2):189. https://doi.org/10.3390/ijms17020189
Chicago/Turabian StyleKovacs, Gabor G. 2016. "Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine" International Journal of Molecular Sciences 17, no. 2: 189. https://doi.org/10.3390/ijms17020189
APA StyleKovacs, G. G. (2016). Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. International Journal of Molecular Sciences, 17(2), 189. https://doi.org/10.3390/ijms17020189