
Apobec-1 Complementation Factor (A1CF) Inhibits Epithelial-Mesenchymal Transition and Migration of Normal Rat Kidney Proximal Tubular Epithelial Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Apobec-1 Complementation Factor (A1CF) Is Highly Conserved among Species

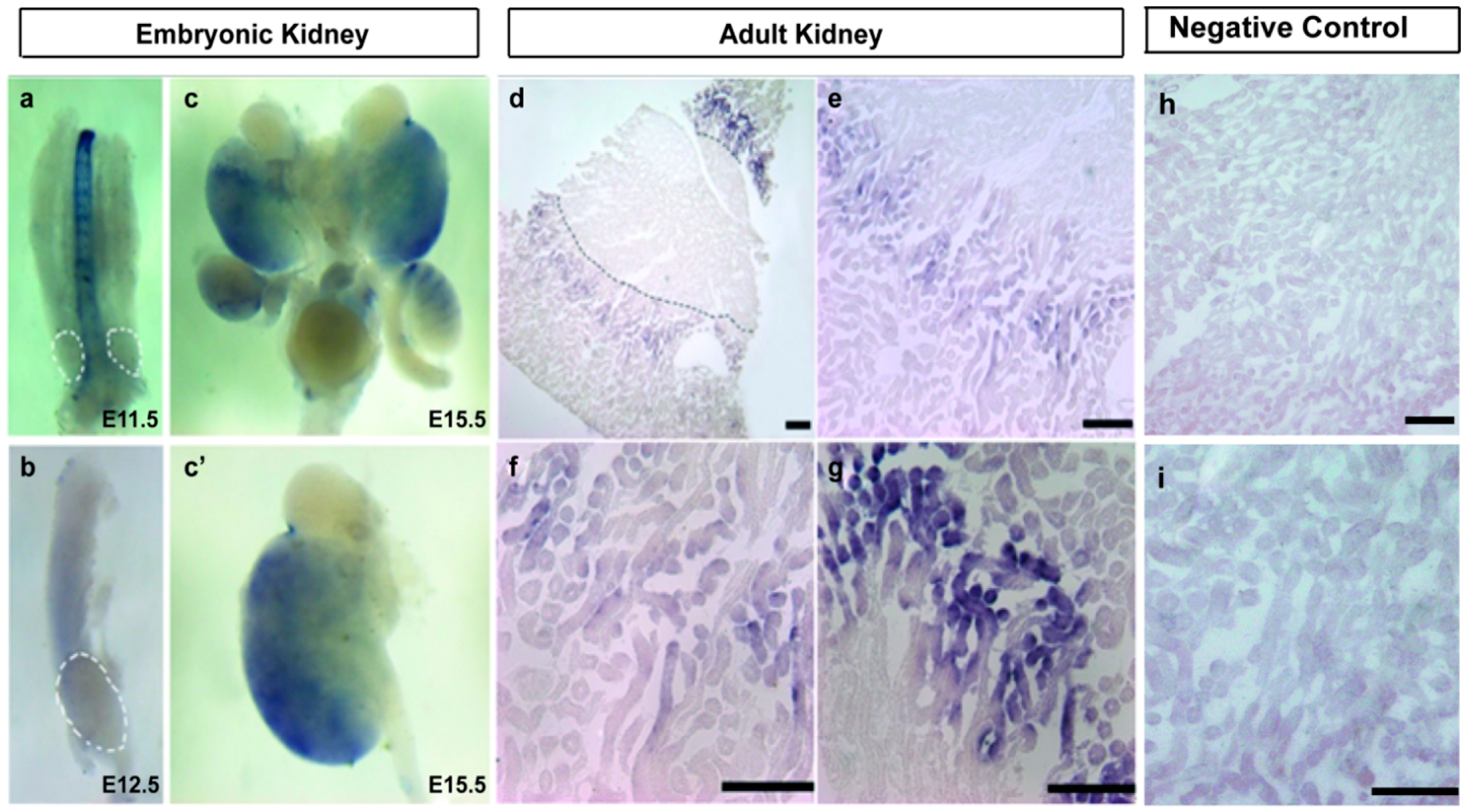
2.2. A1CF Is Highly Expressed in Kidney Tubules

2.3. A1CF Stabilizes Epithelial Character in Rat Kidney Tubular Cells


2.4. A1CF Inhibits NRK52e Cells Migration

3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transient Transfection
4.2. Plasmid Construction
4.3. RNA Interference
4.4. Western Blot
4.5. Immunofluorescence Staining
4.6. Mice and in Situ Hybridization
4.7. Scratch Wound Healing Assay
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| A1CF: | Apobec-1 complementation factor |
| hnRNP: | heterogeneous nuclear ribonucleoproteins |
| apoB: | apolipoprotein-B |
| EMT: | epithelial-to-mesenchymal transition |
| IL-6: | interleukin-6 |
| CKDS: | chronic kidney diseases |
| α-SMA: | α-smooth muscle cell actin |
| ECM: | extracellular matrix |
| RRMs: | three RNA recognition motifs |
| NLS: | the nuclear localization signal |
| ccRCC: | clear cell renal cell carcinoma |
| OCT: | opti-mum cutting temperature |
| DMEM: | Dulbecco’s modified Eagle’s medium |
References
- Mehta, A.; Driscoll, D.M. Identification of domains in apobec-1 complementation factor required for RNA binding and apolipoprotein-B mRNA editing. RNA 2002, 8, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Smith, H.C. The expression of apoB mRNA editing factors is not the sole determinant for the induction of editing in differentiating Caco-2 cells. Biochem. Biophys. Res. Commun. 2010, 391, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.O.; Shelness, G.S. Apolipoprotein B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu. Rev. Nutr. 2000, 20, 169–193. [Google Scholar] [CrossRef]
- Blanc, V.; Davidson, N.O. C-to-U RNA editing: Mechanisms leading to genetic diversity. J. Biol. Chem. 2003, 278, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Maris, C.; Masse, J.; Chester, A.; Navaratnam, N.; Allain, F.H. NMR structure of the apoB mRNA stem-loop and its interaction with the C to U editing Apobec1 complementary factor. RNA 2005, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lellek, H.; Kirsten, R.; Diehl, I.; Apostel, F.; Buck, F.; Greeve, J. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J. Biol. Chem. 2000, 275, 19848–19856. [Google Scholar] [CrossRef] [PubMed]
- Dance, G.S.; Sowden, M.P.; Cartegni, L.; Cooper, E.; Krainer, A.R.; Smith, H.C. Two proteins essential for apolipoprotein B mRNA editing are expressed from a single gene through alternative splicing. J. Biol. Chem. 2002, 277, 12703–12709. [Google Scholar] [CrossRef] [PubMed]
- Sowden, M.P.; Ballatori, N.; Jensen, K.L.; Reed, L.H.; Smith, H.C. The editosome for cytidine to uridine mRNA editing has a native complexity of 27s: Identification of intracellular domains containing active and inactive editing factors. J. Cell Sci. 2002, 115, 1027–1039. [Google Scholar] [PubMed]
- Mehta, A.; Kinter, M.T.; Sherman, N.E.; Driscoll, D.M. Molecular cloning of Apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol. Cell. Biol. 2000, 20, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Blanc, V.; Sessa, K.J.; Kennedy, S.; Luo, J.; Davidson, N.O. Apobec-1 complementation factor modulates liver regeneration by post-transcriptional regulation of interleukin-6 mRNA stability. J. Biol. Chem. 2010, 285, 19184–19192. [Google Scholar] [CrossRef] [PubMed]
- Blanc, V.; Henderson, J.O.; Newberry, E.P.; Kennedy, S.; Luo, J.; Davidson, N.O. Targeted deletion of the murine Apobec-1 complementation factor (ACF) gene results in embryonic lethality. Mol. Cell. Biol. 2005, 25, 7260–7269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the united states. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Zou, H.; Togtokh, A.; Ene-Iordache, B.; Carminati, S.; Remuzzi, A.; Wiebe, N.; Ayyalasomayajula, B.; Perico, N.; Remuzzi, G.; et al. Burden of CKD, proteinuria, and cardiovascular risk among chinese, mongolian, and nepalese participants in the international society of nephrology screening programs. Am. J. Kidney Dis. 2010, 56, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Wei, F.; Vaziri, N.D.; Cheng, X.L.; Bai, X.; Lin, R.C.; Zhao, Y.Y. Metabolomics insights into chronic kidney disease and modulatory effect of rhubarb against tubulointerstitial fibrosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Baum, O.; Vogetseder, A.; Kaissling, B.; Le Hir, M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem. Cell Biol. 2008, 130, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Kishigami, S.; Mishina, Y. In situ hybridization methods for mouse whole mounts and tissue sections with and without additional β-galactosidase staining. Methods Mol. Biol. 2014, 1092, 1–15. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Wang, H.; Zhou, Y.; Ni, D.; Hu, Y.; Long, Y.; Liu, J.; Peng, R.; Zhou, L.; Liu, Z.; et al. Apobec-1 Complementation Factor (A1CF) Inhibits Epithelial-Mesenchymal Transition and Migration of Normal Rat Kidney Proximal Tubular Epithelial Cells. Int. J. Mol. Sci. 2016, 17, 197. https://doi.org/10.3390/ijms17020197
Huang L, Wang H, Zhou Y, Ni D, Hu Y, Long Y, Liu J, Peng R, Zhou L, Liu Z, et al. Apobec-1 Complementation Factor (A1CF) Inhibits Epithelial-Mesenchymal Transition and Migration of Normal Rat Kidney Proximal Tubular Epithelial Cells. International Journal of Molecular Sciences. 2016; 17(2):197. https://doi.org/10.3390/ijms17020197
Chicago/Turabian StyleHuang, Liyuan, Honglian Wang, Yuru Zhou, Dongsheng Ni, Yanxia Hu, Yaoshui Long, Jianing Liu, Rui Peng, Li Zhou, Zhicheng Liu, and et al. 2016. "Apobec-1 Complementation Factor (A1CF) Inhibits Epithelial-Mesenchymal Transition and Migration of Normal Rat Kidney Proximal Tubular Epithelial Cells" International Journal of Molecular Sciences 17, no. 2: 197. https://doi.org/10.3390/ijms17020197