
Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells


Abstract
:1. Introduction
2. Results
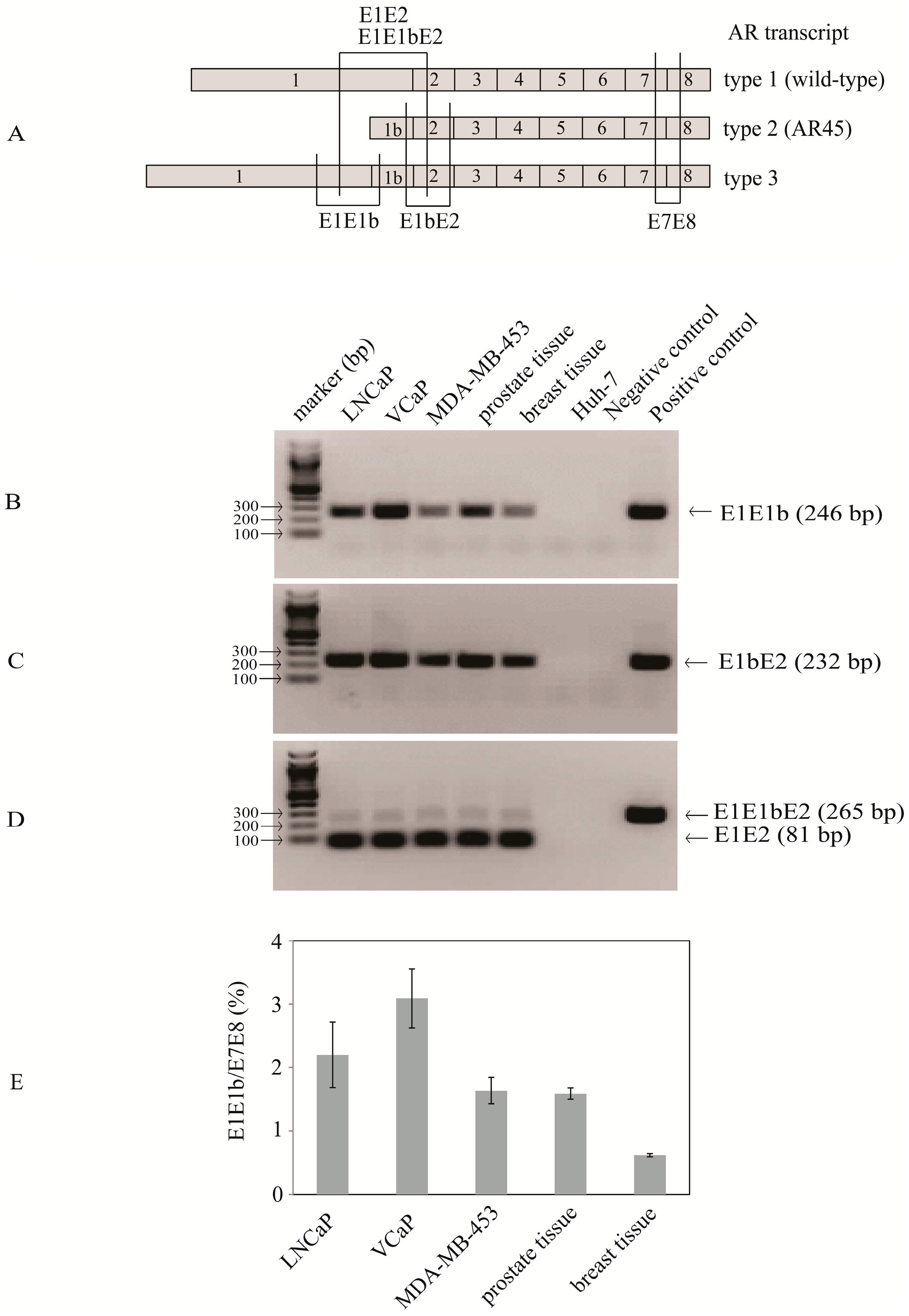
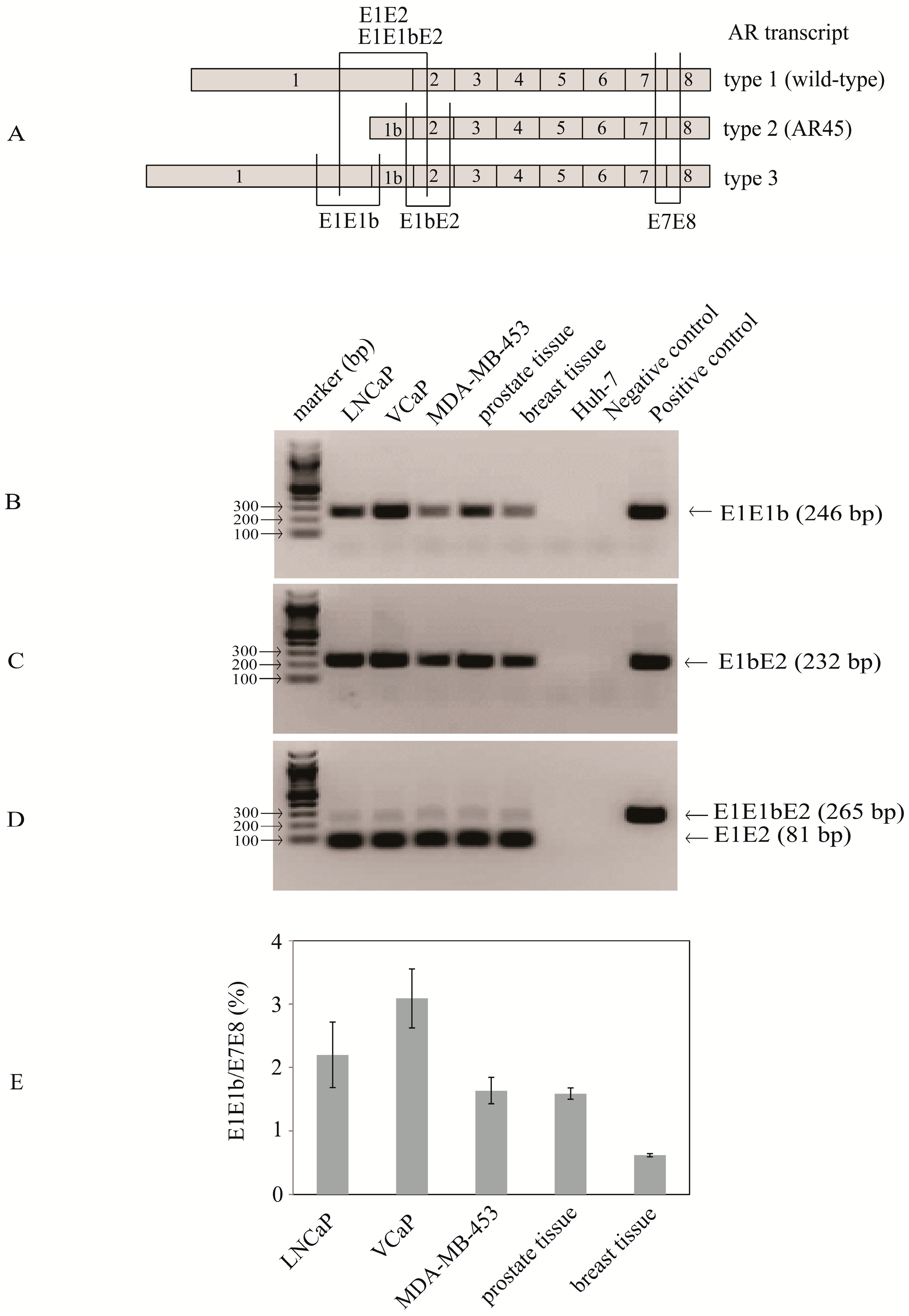
2.1. Identification of Transcripts with Exon 1b Spliced between Exon 1 and Exon 2 Suggesting the Existence of Novel Nine-Exon Androgen Receptor (AR) Variants
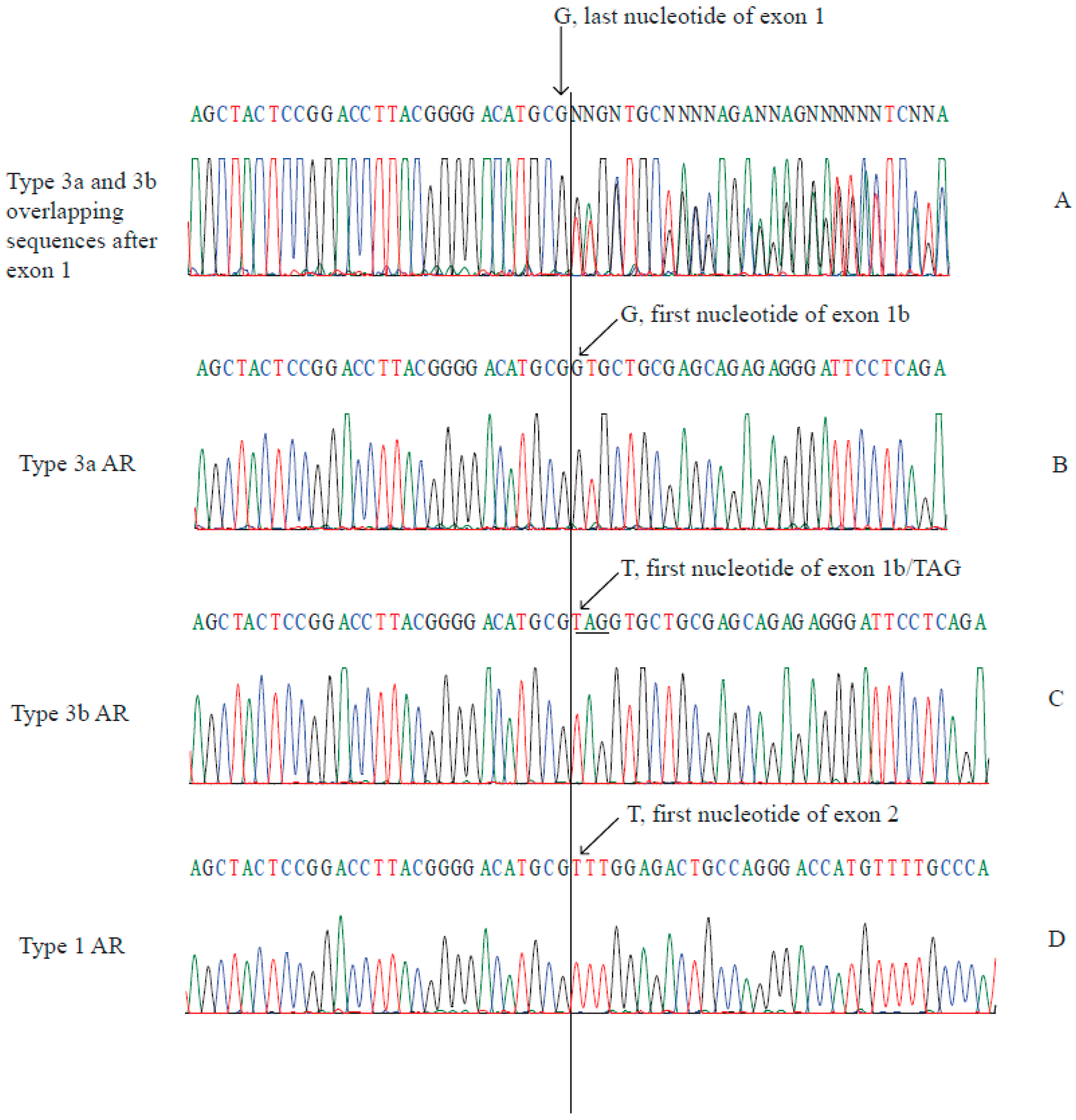
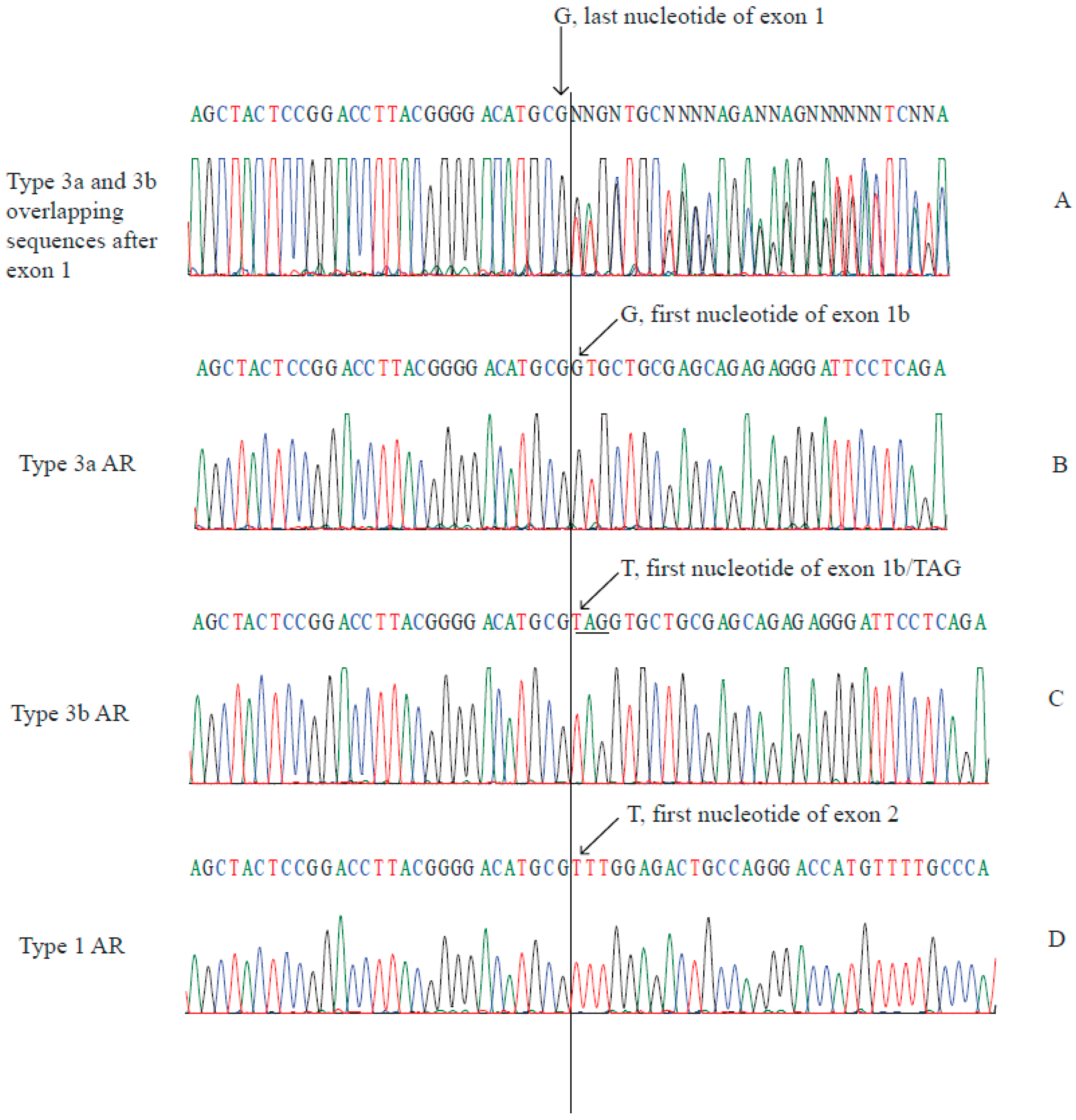
2.2. Type 3a and Type 3b AR Transcripts Are Defined by Two Different Exon 1bs
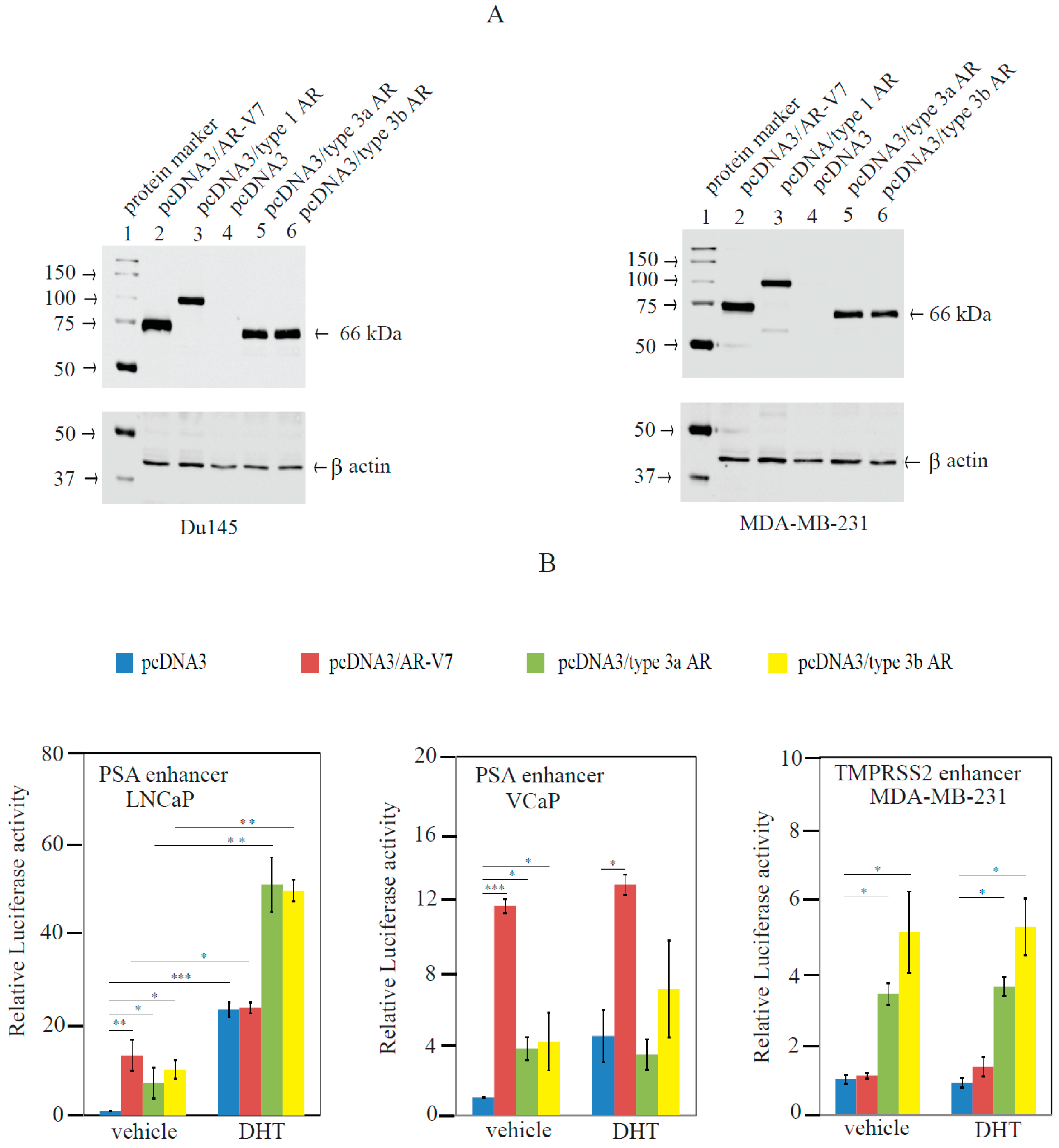
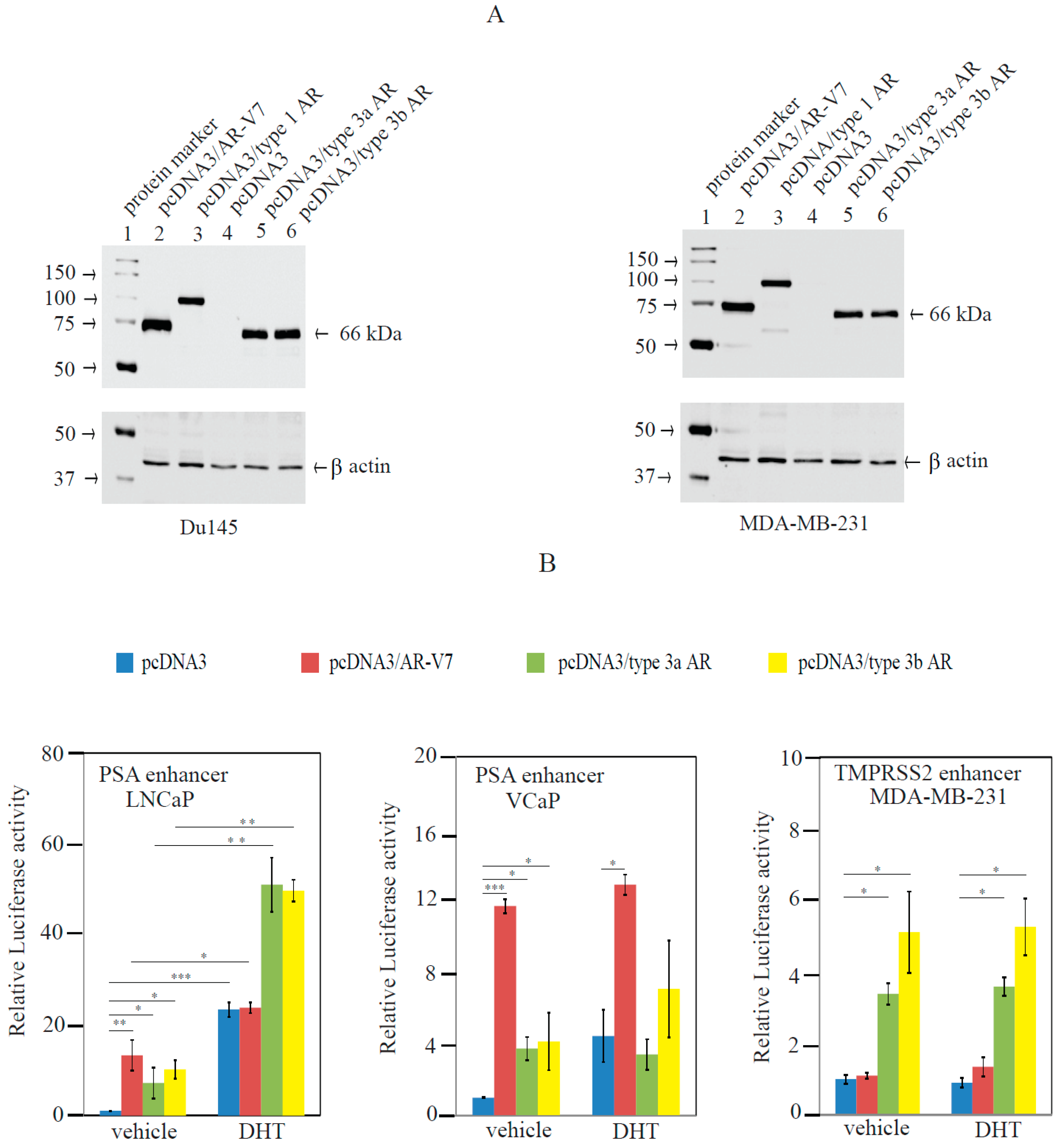
2.3. Type 3 AR Variants Stimulated AR-Responsive Reporters in Prostate and Breast Cancer Cells in an Androgen-Independent Manner
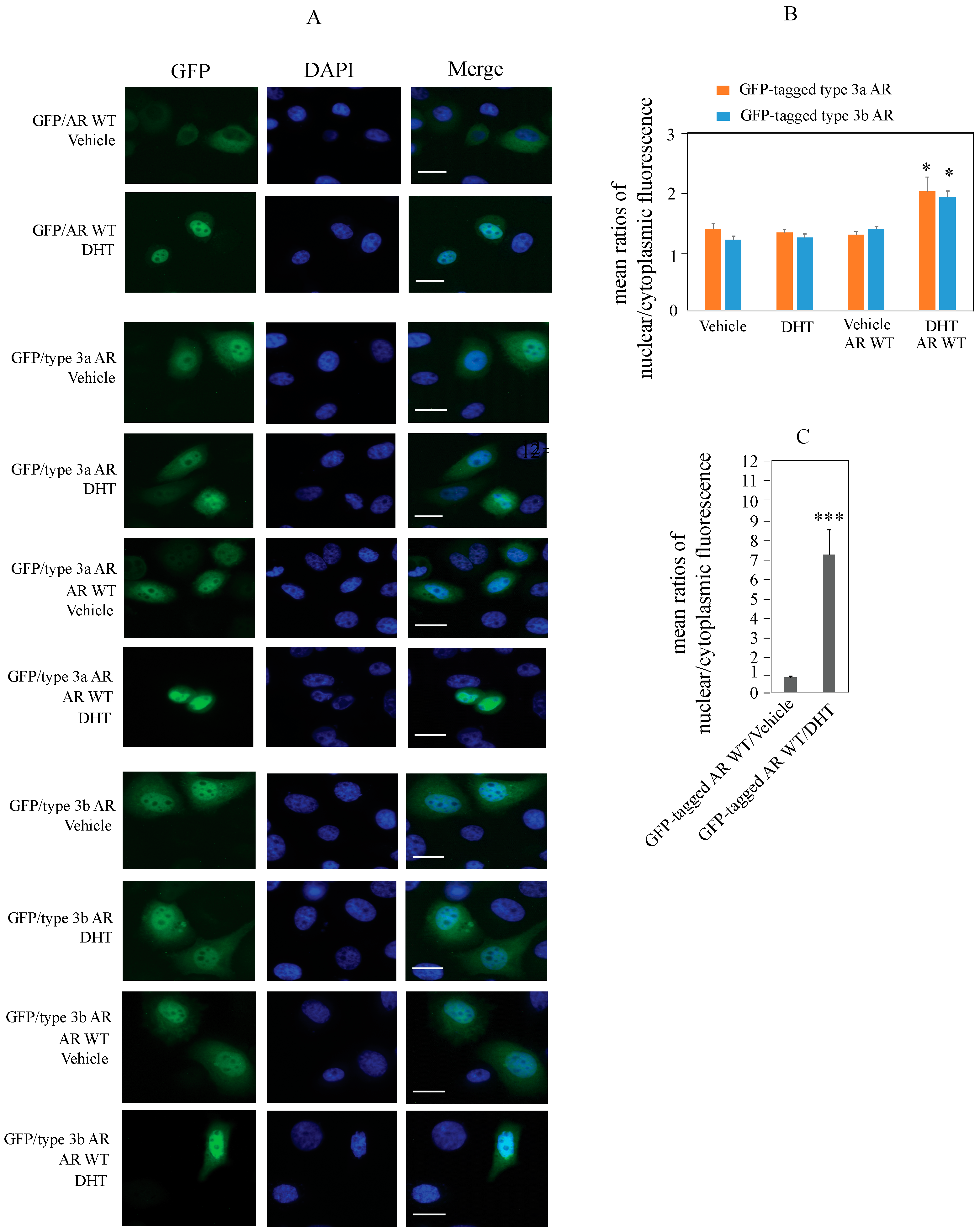
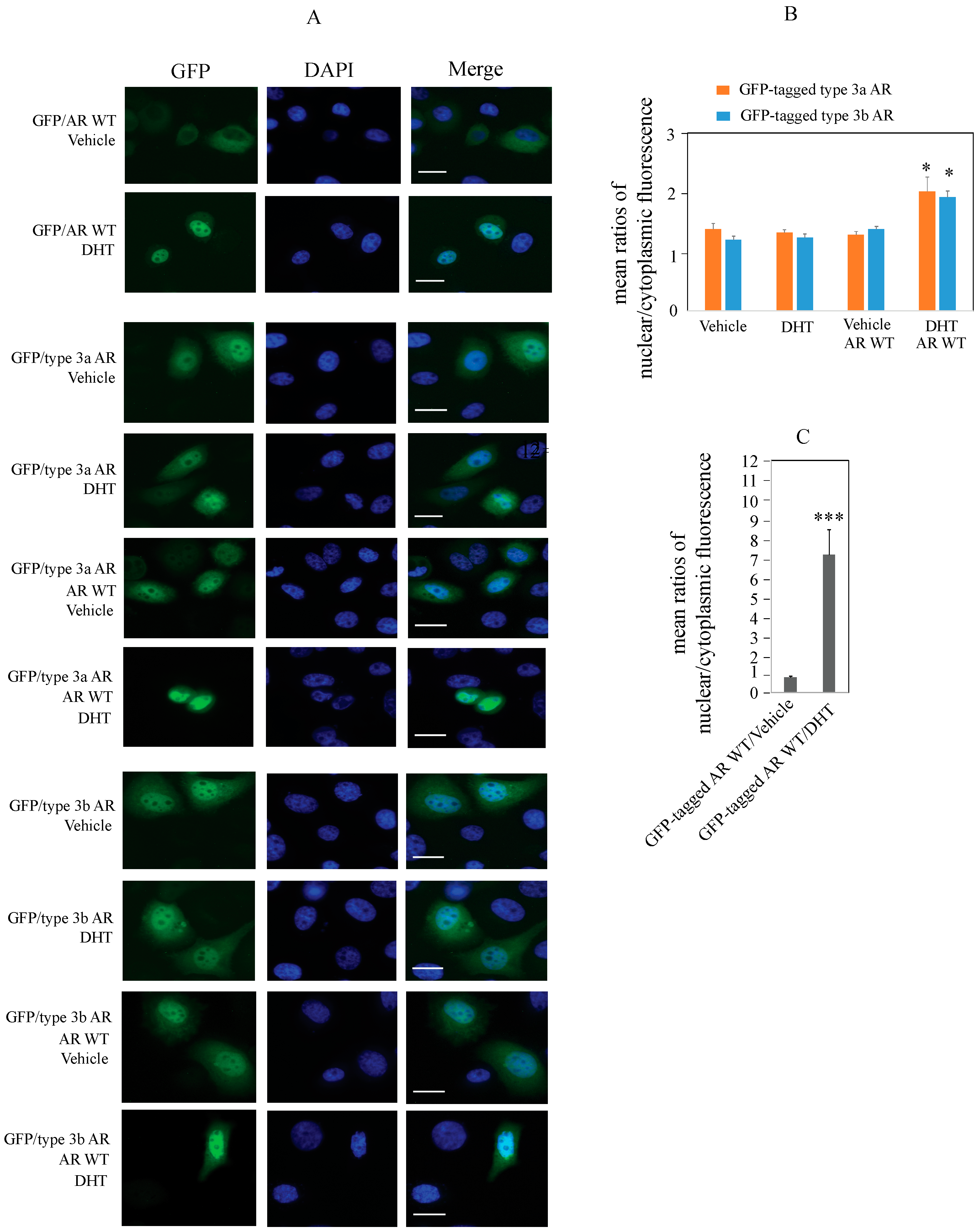
2.4. Type 3 AR Proteins Localize to Both Cytoplasm and Nuclei of PC3 Cells in Androgen-Depleted Conditions
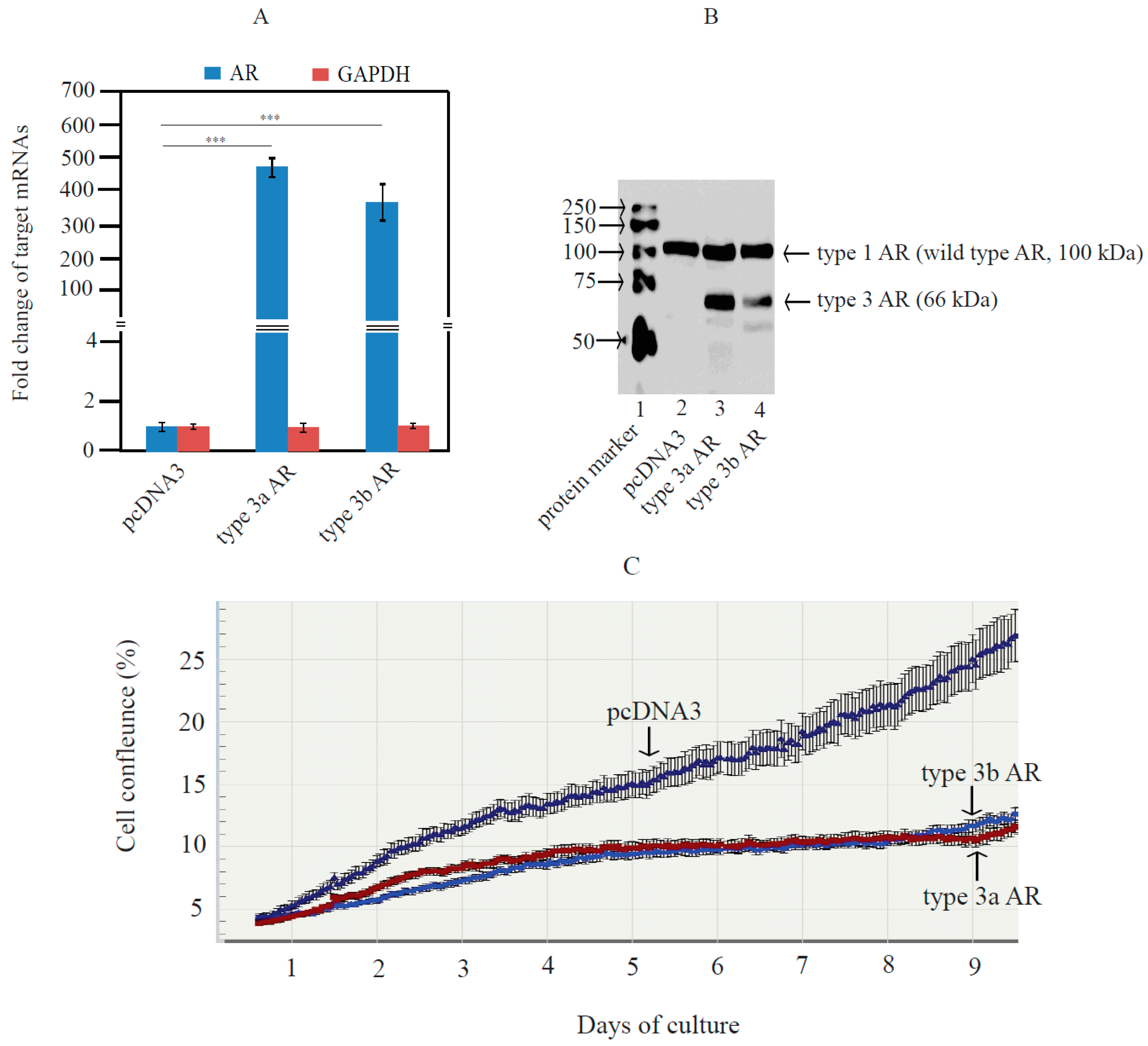
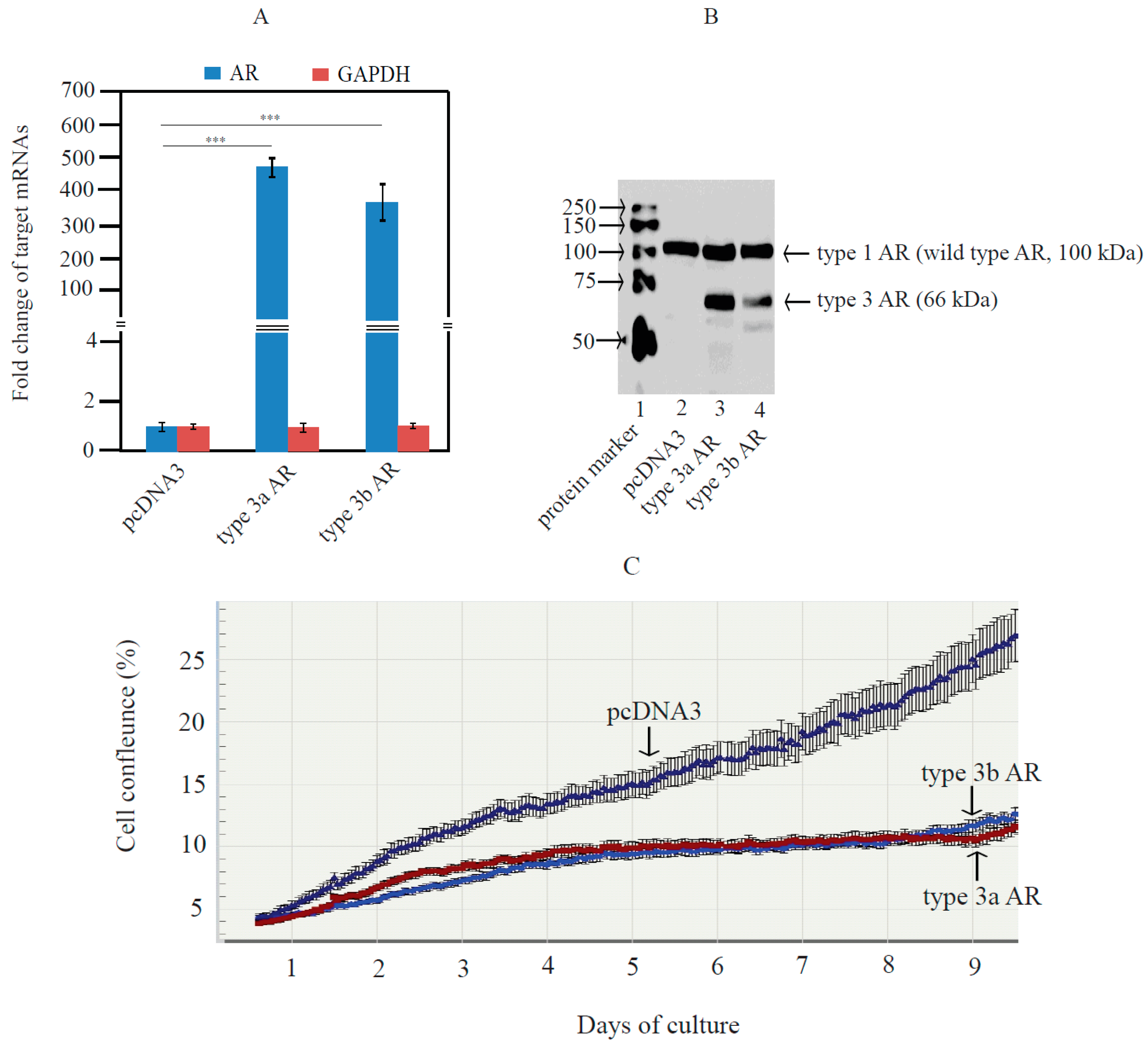
2.5. Type 3 AR Variants Inhibited Androgen-Stimulated Growth of LNCaP Cells
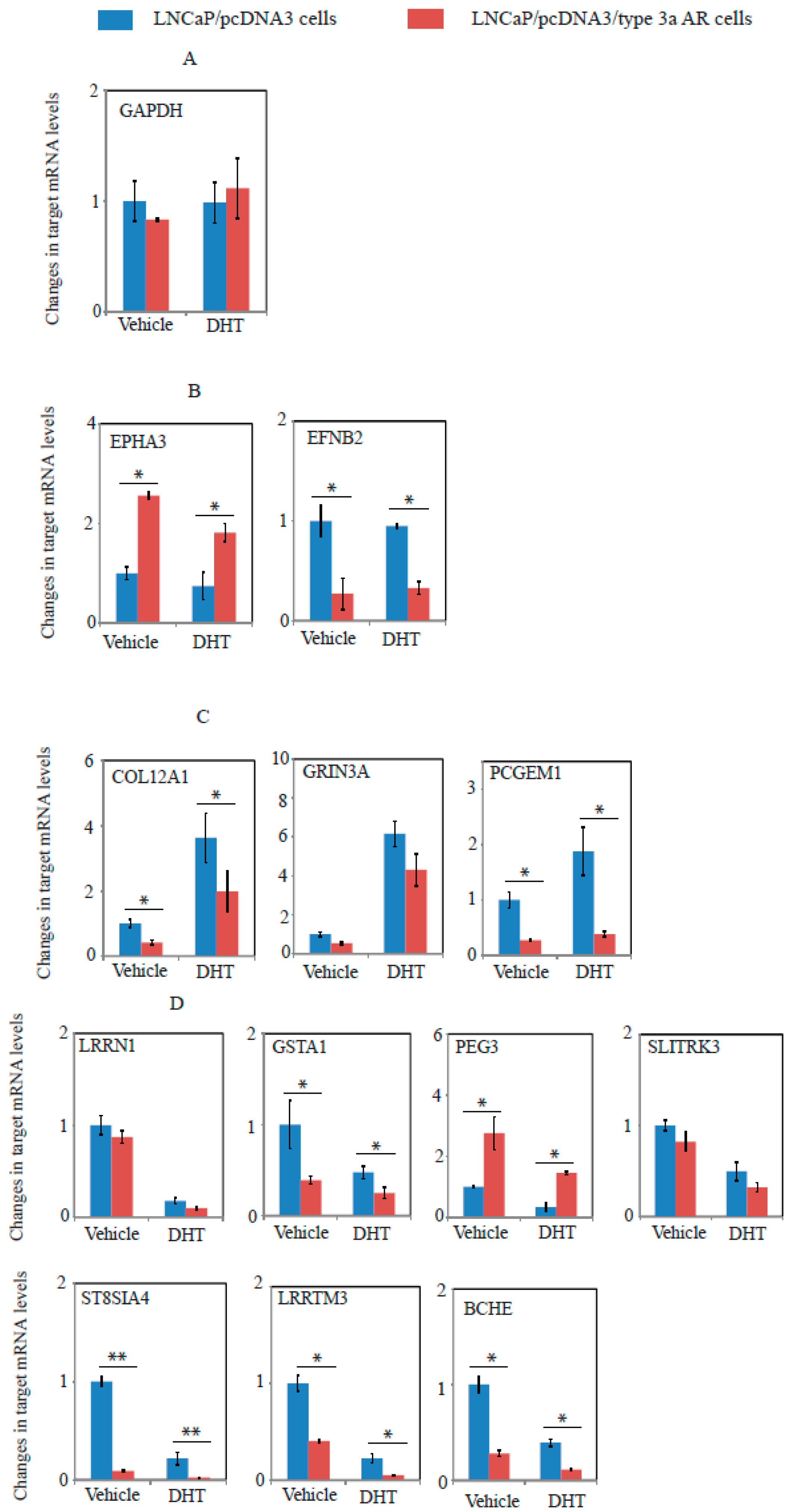
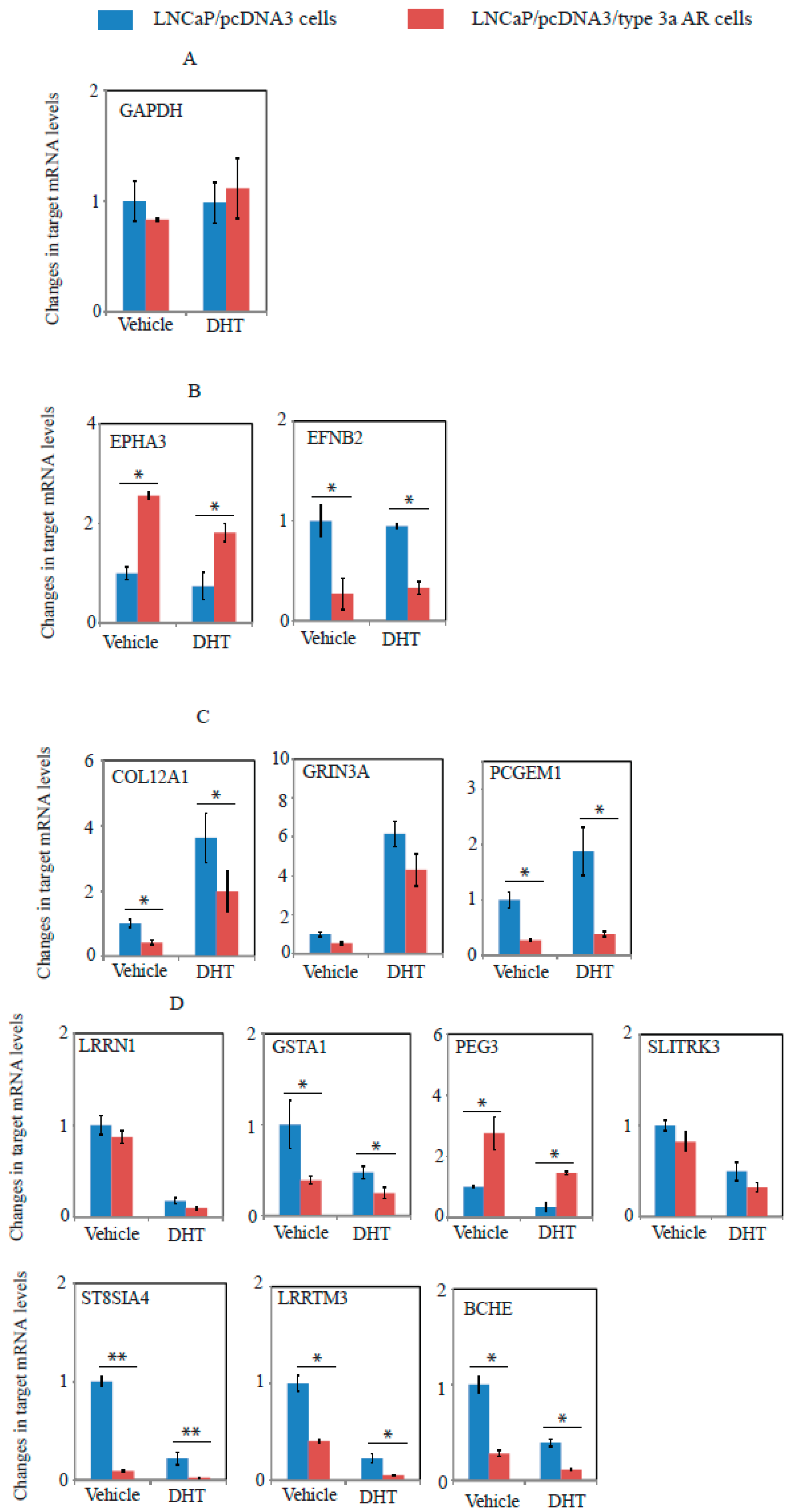
2.6. A Unique Set of Genes Regulated by Type 3a AR in LNCaP Cells as Revealed by Whole Genome Microarray Expression Profiling Assays
3. Discussion
4. Materials and Methods
4.1. Total RNA of Normal Breast and Prostate Tissues
4.2. Cell Culture, RNA Extraction, and Reverse Transcription
4.3. Cloning of RT-PCR Products Corresponding to AR Variants
4.4. Quantitative RT-PCR
4.5. Preparation of pcDNA3 Vectors Expressing Type 1 AR or V7 Variant AR
4.6. Preparation of pcDNA3 Vectors Expressing Type 3a AR or Type 3b AR
4.7. Preparation of Androgen-Responsive Luciferase Reporters
4.8. Transient Transfection and Androgen-Responsive Reporter Luciferase Activity Assays
4.9. Preparation of LNCaP Cells Stably Expressing Type 3 AR Protein and Monitoring of LNCaP Growth Using IncuCyte Imaging System
4.10. Experimental Procedure of Microarrays and Data Analysis
4.11. Western Blot Analysis of AR Variants
4.12. Determination of Cellular Localization of Type 3 AR Proteins in PC3 Cells Using GFP-Tagged Vectors and Fluorescence Imaging
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaarbo, M.; Klokk, T.I.; Saatcioglu, F. Androgen signaling and its interactions with other signaling pathways in prostate cancer. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Katzenwadel, A.; Wolf, P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer Lett. 2015, 367, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Gunter, J.H.; Lubik, A.A.; McKenzie, I.; Pollak, M.; Nelson, C.C. The Interactions between insulin and androgens in progression to castrate-resistant prostate cancer. Adv. Urol. 2012, 2012, 248607. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [PubMed]
- Hu, R.; Isaacs, W.B.; Luo, J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011, 71, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Van der Steen, T.; Tindall, D.J. Are androgen receptor variants a substitute for the full-length receptor? Nat. Rev. Urol. 2015, 12, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Hickey, T.E.; Irvine, C.; Wijayakumara, D.D.; Lu, L.; Tilley, W.D.; Selth, L.A.; Mackenzie, P.I. Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm. Cancer 2014, 5, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [PubMed]
- Li, Y.; Hwang, T.H.; Oseth, L.A.; Hauge, A.; Vessella, R.L.; Schmechel, S.C.; Hirsch, B.; Beckman, K.B.; Silverstein, K.A.; Dehm, S.M. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31, 4759–4767. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Nyquist, M.D.; Li, Y.; Hwang, T.H.; Manlove, L.S.; Vessella, R.L.; Silverstein, K.A.; Voytas, D.F.; Dehm, S.M. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17492–17497. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [PubMed]
- Hickey, T.E.; Irvine, C.M.; Dvinge, H.; Tarulli, G.A.; Hanson, A.R.; Ryan, N.K.; Pickering, M.A.; Birrell, S.N.; Hu, D.G.; Mackenzie, P.I.; et al. Expression of androgen receptor splice variants in clinical breast cancers. Oncotarget 2015, 6, 44728–44744. [Google Scholar] [PubMed]
- Weiss, B.; Faus, H.; Haendler, B. Phylogenetic conservation of the androgen receptor AR45 variant form in placental mammals. Gene 2007, 399, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed]
- Isken, O.; Maquat, L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer Basic Clin. Res. 2010, 4, 35–41. [Google Scholar]
- Srikantan, V.; Zou, Z.; Petrovics, G.; Xu, L.; Augustus, M.; Davis, L.; Livezey, J.R.; Connell, T.; Sesterhenn, I.A.; Yoshino, K.; et al. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 12216–12221. [Google Scholar] [CrossRef] [PubMed]
- Parolia, A.; Crea, F.; Xue, H.; Wang, Y.; Mo, F.; Ramnarine, V.R.; Liu, H.H.; Lin, D.; Saidy, N.R.; Clermont, P.L.; et al. The long non-coding RNA PCGEM1 is regulated by androgen receptor activity in vivo. Mol. Cancer 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Runkle, C.; Jin, H.J.; Yu, J.; Li, J.; Yang, X.; Kuzel, T.; Lee, C.; Yu, J. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene 2014, 33, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Ulrix, W.; Swinnen, J.V.; Heyns, W.; Verhoeven, G. Androgens down-regulate the expression of the human homologue of paternally expressed gene-3 in the prostatic adenocarcinoma cell line LNCaP. Mol. Cell. Endocrinol. 1999, 155, 69–76. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six classes of nuclear localization signals specific to different binding grooves of importin α. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Sar, M.; Simental, J.A.; Lane, M.V.; Wilson, E.M. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J. Biol. Chem. 1994, 269, 13115–13123. [Google Scholar] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Korlimarla, A.; Bhandary, L.; Prabhu, J.S.; Shankar, H.; Sankaranarayanan, H.; Kumar, P.; Remacle, J.; Natarajan, D.; Sridhar, T.S. Identification of a non-canonical nuclear localization signal (NLS) in BRCA1 that could mediate nuclear localization of splice variants lacking the classical NLS. Cell. Mol. Biol. Lett. 2013, 18, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed]
- Heisler, L.E.; Evangelou, A.; Lew, A.M.; Trachtenberg, J.; Elsholtz, H.P.; Brown, T.J. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol. Cell. Endocrinol. 1997, 126, 59–73. [Google Scholar] [CrossRef]
- Yuan, S.; Trachtenberg, J.; Mills, G.B.; Brown, T.J.; Xu, F.; Keating, A. Androgen-induced inhibition of cell proliferation in an androgen-insensitive prostate cancer cell line (PC-3) transfected with a human androgen receptor complementary DNA. Cancer Res. 1993, 53, 1304–1311. [Google Scholar] [PubMed]
- Urbanucci, A.; Sahu, B.; Seppala, J.; Larjo, A.; Latonen, L.M.; Waltering, K.K.; Tammela, T.L.; Vessella, R.L.; Lahdesmaki, H.; Janne, O.A.; et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene 2012, 31, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Waltering, K.K.; Helenius, M.A.; Sahu, B.; Manni, V.; Linja, M.J.; Janne, O.A.; Visakorpi, T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009, 69, 8141–8149. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Bianchi-Frias, D.; Coleman, I.; Banerji, J.; Pham, K.; Jeldres, C.; Gulati, R.; Xia, J.; Tomlins, J.; Porter, C.; Nelson, P. Novel urine markers for diagnosing and monitoring non-indolent prostate cancer. J. Clin. Oncol. 2015, 33, e16113. [Google Scholar]
- Koie, T.; Ohyama, C.; Hatakeyama, S.; Imai, A.; Yoneyama, T.; Hashimoto, Y.; Yoneyama, T.; Tobisawa, Y.; Hosogoe, S.; Yamamoto, H.; et al. Significance of preoperative butyrylcholinesterase as an independent predictor of biochemical recurrence-free survival in patients with prostate cancer treated with radical prostatectomy. Int. J. Clin. Oncol. 2016, 21, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Takahara, K.; Azuma, H.; Sakamoto, T.; Kiyama, S.; Inamoto, T.; Ibuki, N.; Nishida, T.; Nomi, H.; Ubai, T.; Segawa, N.; et al. Conversion of prostate cancer from hormone independency to dependency due to AMACR inhibition: Involvement of increased AR expression and decreased IGF1 expression. Anticancer Res. 2009, 29, 2497–2505. [Google Scholar] [PubMed]
- Parsons, J.K.; Nelson, C.P.; Gage, W.R.; Nelson, W.G.; Kensler, T.W.; de Marzo, A.M. GSTA1 expression in normal, preneoplastic, and neoplastic human prostate tissue. Prostate 2001, 49, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Cheville, J.C.; Karnes, R.J.; Therneau, T.M.; Kosari, F.; Munz, J.M.; Tillmans, L.; Basal, E.; Rangel, L.J.; Bergstralh, E.; Kovtun, I.V.; et al. Gene panel model predictive of outcome in men at high-risk of systemic progression and death from prostate cancer after radical retropubic prostatectomy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3930–3936. [Google Scholar] [CrossRef] [PubMed]
- Kosari, F.; Munz, J.M.; Savci-Heijink, C.D.; Spiro, C.; Klee, E.W.; Kube, D.M.; Tillmans, L.; Slezak, J.; Karnes, R.J.; Cheville, J.C.; et al. Identification of prognostic biomarkers for prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Sun, S. Genomic profiling screens small molecules of metastatic prostate carcinoma. Oncol. Lett. 2015, 10, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, R.; Mertens-Walker, I.; Lisle, J.E.; Herington, A.C.; Stephenson, S.A. Evidence for a dual function of EphB4 as tumor promoter and suppressor regulated by the absence or presence of the ephrin-B2 ligand. Int. J. Cancer 2012, 131, E614–E624. [Google Scholar] [CrossRef] [PubMed]
- Dozmorov, M.G.; Hurst, R.E.; Culkin, D.J.; Kropp, B.P.; Frank, M.B.; Osban, J.; Penning, T.M.; Lin, H.K. Unique patterns of molecular profiling between human prostate cancer LNCaP and PC-3 cells. Prostate 2009, 69, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, H.; Wang, J.; Wang, P.; Huang, F.; Xie, B.; Zhao, Y.; Li, S.; Zhou, J. EphA3, induced by PC-1/PrLZ, contributes to the malignant progression of prostate cancer. Oncol. Rep. 2014, 32, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Bawa, P.; Zackaria, S.; Verma, M.; Gupta, S.; Srivatsan, R.; Chaudhary, B.; Srinivasan, S. Integrative analysis of normal long intergenic non-coding RNAs in prostate cancer. PLoS ONE 2015, 10, e0122143. [Google Scholar] [CrossRef] [PubMed]
- Sharad, S.; Srivastava, A.; Ravulapalli, S.; Parker, P.; Chen, Y.; Li, H.; Petrovics, G.; Dobi, A. Prostate cancer gene expression signature of patients with high body mass index. Prostate Cancer Prostatic Dis. 2011, 14, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Petrovics, G.; Zhang, W.; Makarem, M.; Street, J.P.; Connelly, R.; Sun, L.; Sesterhenn, I.A.; Srikantan, V.; Moul, J.W.; Srivastava, S. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene 2004, 23, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, M.; Kang, M.; Wang, Q.; Wu, B.; Chu, H.; Zhong, D.; Qin, C.; Yin, C.; Zhang, Z.; et al. Association between lncrna PCGEM1 polymorphisms and prostate cancer risk. Prostate Cancer Prostatic Dis. 2013, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ravindranath, L.; Tran, N.; Petrovics, G.; Srivastava, S. Regulation of apoptosis by a prostate-specific and prostate cancer-associated noncoding gene, PCGEM1. DNA Cell Biol. 2006, 25, 135–141. [Google Scholar] [CrossRef] [PubMed]
- He, J.H.; Zhang, J.Z.; Han, Z.P.; Wang, L.; Lv, Y.B.; Li, Y.G. Reciprocal regulation of PCGEM1 and miR-145 promote proliferation of LNCaP prostate cancer cells. J. Exp. Clin. Cancer Res. CR 2014, 33, 72. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, N.; Huang, J.; Ho, T.T.; Zhu, Z.; Qiu, Z.; Zhou, X.; Bai, C.; Wu, F.; Xu, M.; et al. Regulation of androgen receptor splice variant AR3 by PCGEM1. Oncotarget 2016, 7, 15481–15491. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Wei, X.; Li, W.; Pan, J.; Lin, Y.; Bowtell, D.D.; Sassoon, D.A.; Wu, X. Pw1/Peg3 is a potential cell death mediator and cooperates with Siah1a in p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Wei, X.J.; Wu, X.; Sassoon, D.A. Peg3/Pw1 is an imprinted gene involved in the TNF-NFκB signal transduction pathway. Nat. Genet. 1998, 18, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Forse, G.J.; Uson, M.L.; Nasertorabi, F.; Kolatkar, A.; Lamberto, I.; Pasquale, E.B.; Kuhn, P. Distinctive structure of the EphA3/Ephrin-A5 complex reveals a dual mode of Eph receptor interaction for ephrin-A5. PLoS ONE 2015, 10, e0127081. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5, e14845. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Bagatini, M.D.; Maders, L.D.; Chiesa, J.; Santos, K.F.; Goncalves, J.F.; Abdalla, F.H.; Battisti, I.E.; Schetinger, M.R.; Morsch, V.M. Cholinesterase activities and biochemical determinations in patients with prostate cancer: Influence of Gleason score, treatment and bone metastasis. Biomed. Pharmacother. 2012, 66, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Watson, J.T.; Marengo, S.R.; Decker, K.S.; Coleman, I.; Nelson, P.S.; Sikes, R.A. Gene expression in the LNCaP human prostate cancer progression model: Progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer Lett. 2006, 244, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, G.; Magno, L.A.; Rios-Santos, F. Glutathione S-transferases: An overview in cancer research. Expert Opin. Drug Metab. Toxicol. 2010, 6, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; He, B.; Pan, Y.; Sun, H.; Liu, X.; Chen, J.; Ying, H.; Lin, K.; Peng, H.; Wang, S. Polymorphisms of GSTA1 contribute to elevated cancer risk: Evidence from 15 studies. J. B.U.ON. Off. J. Balk. Union Oncol. 2015, 20, 287–295. [Google Scholar]
- Komiya, Y.; Tsukino, H.; Nakao, H.; Kuroda, Y.; Imai, H.; Katoh, T. Human glutathione S-transferase A1, T1, M1, and P1 polymorphisms and susceptibility to prostate cancer in the Japanese population. J. Cancer Res. Clin. Oncol. 2005, 131, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Sa, R.A.; Moreira Ados, S.; Cabello, P.H.; Ornellas, A.A.; Costa, E.B.; Matos Cda, S.; Alves, G.; Hatagima, A. Human glutathione S-transferase polymorphisms associated with prostate cancer in the Brazilian population. Int. Braz. J. Urol. Off. J. Braz. Soc. Urol. 2014, 40, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Yokota, N.; Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003, 315, 87–94. [Google Scholar] [CrossRef]
- Milde, T.; Shmelkov, S.V.; Jensen, K.K.; Zlotchenko, G.; Petit, I.; Rafii, S. A novel family of slitrk genes is expressed on hematopoietic stem cells and leukemias. Leukemia 2007, 21, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Zhang, Z.Z.; Xu, J.; Wang, M.; Zhao, W.Y.; Tu, L.; Zhuang, C.; Liu, Q.; Shen, Y.Y.; Cao, H.; et al. SLITRK3 expression correlation to gastrointestinal stromal tumor risk rating and prognosis. World J. Gastroenterol. 2015, 21, 8398–8407. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.C.; Giehl, K.; Kastilan, C.; Hasel, C.; Muhlenhoff, M.; Adler, G.; Wedlich, D.; Menke, A. Polysialylated NCAM represses E-cadherin-mediated cell–cell adhesion in pancreatic tumor cells. Gastroenterology 2008, 134, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Z. TOX gene: A novel target for human cancer gene therapy. Am. J. Cancer Res. 2015, 5, 3516–3524. [Google Scholar] [PubMed]
- Bayraktar, S.; Thompson, P.A.; Yoo, S.Y.; Do, K.A.; Sahin, A.A.; Arun, B.K.; Bondy, M.L.; Brewster, A.M. The relationship between eight GWAS-identified single-nucleotide polymorphisms and primary breast cancer outcomes. Oncologist 2013, 18, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; Wu, X.Y.; Shen, W.; Wei, M.X.; Li, C.; Cai, B.; Tao, G.Q.; Lu, P.H. Association between polymorphisms of trinucleotide repeat containing 9 gene and breast cancer risk: Evidence from 62,005 subjects. Breast Cancer Res. Treat. 2011, 126, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Elematore, I.; Gonzalez-Hormazabal, P.; Reyes, J.M.; Blanco, R.; Bravo, T.; Peralta, O.; Gomez, F.; Waugh, E.; Margarit, S.; Ibanez, G.; et al. Association of genetic variants at TOX3, 2q35 and 8q24 with the risk of familial and early-onset breast cancer in a South-American population. Mol. Biol. Rep. 2014, 41, 3715–3722. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yao, G.; Li, F.; Li, M.; Yang, X. Risk-association of five SNPs in TOX3/LOC643714 with breast cancer in southern China. Int. J. Mol. Sci. 2014, 15, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Reeves, G.K.; Travis, R.C.; Green, J.; Bull, D.; Tipper, S.; Baker, K.; Beral, V.; Peto, R.; Bell, J.; Zelenika, D.; et al. Incidence of breast cancer and its subtypes in relation to individual and multiple low-penetrance genetic susceptibility loci. JAMA 2010, 304, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, E.T.; Barkardottir, R.B.; Arason, A.; Gunnarsson, H.; Amundadottir, L.T.; Agnarsson, B.A.; Johannsson, O.T.; Reynisdottir, I. The risk allele of SNP rs3803662 and the mRNA level of its closest genes TOX3 and LOC643714 predict adverse outcome for breast cancer patients. BMC Cancer 2012, 12, 621. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Mackenzie, P.I. Estrogen receptor α, fos-related antigen-2, and c-Jun coordinately regulate human UDP glucuronosyltransferase 2B15 and 2B17 expression in response to 17β-estradiol in MCF-7 cells. Mol. Pharmacol. 2009, 76, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, K.B.; van der Korput, H.A.; van Eekelen, C.C.; van Rooij, H.C.; Faber, P.W.; Trapman, J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol. Endocrinol. 1997, 11, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Janne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | RefSeqgene | Full Name | C/A Comparison | D/B Comparison | ||
|---|---|---|---|---|---|---|
| Fold Change | p Value | Fold Change | p Value | |||
| PCGEM1 | NR_002769 | Prostate cancer gene expression marker 1 | −2.21 | 0.00039 | −2.61 | 0.00009 |
| BCHE | NM_000055 | Butyrylcholinesterase | −2.86 | 0.00039 | −3.61 | 0.00009 |
| EFNB2 | NM_004093 | Ephrin-B2 | −4.32 | 0.00163 | −4.77 | 0.00094 |
| PEG3 | NM_006210 | Paternally expressed 3, transcript 1 | 2.21 | 0.00452 | 2.06 | 0.00838 |
| PSMD5 | NM_005047 | Proteasome (prosome, macropain) 26S subunit, non-AT | −2.61 | 0.00163 | −2.68 | 0.00134 |
| MBNL2 | NM_144778 | Muscleblind-like 2 (Drosophila) (MBNL2), transcript | −2.22 | 0.00452 | −2.43 | 0.00399 |
| GSTA1 | NM_145740 | Glutathione S-transferase α 1 | −2.21 | 0.00631 | −2.40 | 0.00572 |
| TLL1 | NM_012464 | Tolloid-like 1 | −3.13 | 0.00518 | −2.77 | 0.01127 |
| ZNF608 | NM_020747 | Zinc finger protein 608 | −2.21 | 0.02334 | −2.27 | 0.02299 |
| Has-miR-3689b | −4.23 | 0.02106 | −4.14 | 0.02301 | ||
| Has-miR-3689a | −3.58 | 0.02117 | −3.21 | 0.03057 | ||
| SYT4 | NM_020783 | Synaptotagmin IV | −2.39 | 0.00249 | ||
| ST8SIA4 | NM_005668 | ST8 α-N-acetyl-neuraminide α-2.8-sialytransferase | −6.35 | 0.00318 | ||
| LRRTM3 | NM_178011 | Leucine rich repeat transmembrane neuronal 3 | −2.74 | 0.00452 | ||
| EPHA3 | NM_005233 | EPH receptor A3, transcript variant 1 | 2.1 | 0.00012 | ||
| COL12A1 | NM_004370 | Collagen, type XII, α 1 | −2.27 | 0.01362 | ||
| GRIN3A | NM_133445 | Glutamate receptor, ionotropic, N-methyl-D-aspartat | −2.01 | 0.01362 | ||
| SLITRK3 | NM_014926 | SLIT and NTRK-like family, member 3 | −2.06 | 0.00094 | ||
| FAM198B | NM_001031700 | Family with sequence similarity 198, member B | −2.09 | 0.01362 | ||
| LRRN1 | NM_020873 | Leucine rich repeat neuronal 1 | −2.04 | 0.02305 | ||
| TOX3 | NM_001146188 | TOX high mobility group box family member 3 | −2.09 | 0.03989 | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, D.G.; McKinnon, R.A.; Hulin, J.-A.; Mackenzie, P.I.; Meech, R. Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells. Int. J. Mol. Sci. 2017, 18, 40. https://doi.org/10.3390/ijms18010040
Hu DG, McKinnon RA, Hulin J-A, Mackenzie PI, Meech R. Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells. International Journal of Molecular Sciences. 2017; 18(1):40. https://doi.org/10.3390/ijms18010040
Chicago/Turabian StyleHu, Dong Gui, Ross A. McKinnon, Julie-Ann Hulin, Peter I. Mackenzie, and Robyn Meech. 2017. "Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells" International Journal of Molecular Sciences 18, no. 1: 40. https://doi.org/10.3390/ijms18010040
APA StyleHu, D. G., McKinnon, R. A., Hulin, J.-A., Mackenzie, P. I., & Meech, R. (2017). Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells. International Journal of Molecular Sciences, 18(1), 40. https://doi.org/10.3390/ijms18010040